

Abstract
Obesity has been implicated in several diseases including cancer, however, the relationship of obesity and susceptibility to ultraviolet (UV) radiation-caused skin diseases has not been investigated. As UV-induced oxidative stress has been implicated in several skin diseases, we assessed the role of obesity on UVB-induced oxidative stress in genetically obese Lepob/Lepob (leptin-deficient) mice. Here, we report that chronic exposure to UVB (120 mJ/cm2) resulted in greater oxidative stress in the skin of obese mice in terms of higher levels of H2O2 and NO production, photo-oxidative damage of lipids and proteins, and greater depletion of antioxidant defense enzymes, like, glutathione, glutathione peroxidase and catalase. As UV-induced oxidative stress mediates activation of MAPK and NF-κB signaling pathways, we determined the effect of UVB on these pathways in obese mice. Exposure of obese mice to UVB resulted in phosphorylation of ERK1/2, JNK and p38 proteins of the MAPK family. Compared to wild-type mice, the obese mice exhibited higher levels of phosphorylation of these proteins, greater activation of NF-κB/p65, and higher levels of circulating proinflammatory cytokines, including TNF-α, IL-1-β and IL-6, on UVB irradiation. Together, our study suggests for the first time that obesity in mice is associated with greater susceptibility to UVB-induced oxidative stress, and therefore may be a risk factor for skin diseases associated with UVB-induced oxidative stress.
Keywords: Skin, oxidative stress, hydrogen peroxide, glutathione peroxidase, mitogen-activated protein kinase, NF-κB, cytokines, ultraviolet radiation
Introduction
The prevalence of obesity has increased dramatically over the past 20 years in the United States [1], and 64% of adults were classified as overweight and 30.5% as obese between 1999 and 2000 [2,3]. Obesity has been shown to be associated with many diseases, including cardiovascular diseases, type 2 diabetes and cancers of various organs, which collectively accounted for approximately 65% of all deaths in the year 2000 [4]. The recent results of a large prospective study illustrate that obesity is associated with increased death rates from all cancers combined and for cancers at multiple specific sites in both men and women [5]. Excess body weight has been implicated in ∼20% of all cancers deaths in women and 14% in men and an estimated 90,000 cancer deaths yearly in the US [5].
Data from the National Health and Nutrition Examination Survey (NHANES) reveal dramatic increase in the prevalence of overweight and obesity in the adult population in the US [1,2,6]. Over an 11-year period (1988-1999), the prevalence of overweight and obesity increased from 55.9 to 64.5% and from 22.9 to 30.5%, respectively [2]. It should be noted that this increase in weight is not limited to adults; a similar trend is evident in children. The prevalence of overweight in children tripled between 1980 and 2000 to 16% [1,6]. There is strong evidence to support the role of obesity in cancer risk and mortality. In a cohort study of 900,000 individuals, those men and women with a body mass index (BMI; body weight measured kilograms divided by the square of height measured in meters) of >40 had death rates from all cancers that were 52 and 62% greater than men and women in the normal range. Traditionally, the cancers that are linked with obesity are breast, colon, pancreas liver, cervix, stomach and kidney [1,6]. Little information is available regarding the potential link between obesity and melanoma and nonmelanoma skin cancers. Nonmelanoma skin cancers, including basal and squamous cell carcinoma, represent the most common malignant neoplasms in humans especially in Caucasians. It has been determined that chronic exposure of solar ultraviolet (UV) radiation is an important etiologic agent in both melanoma and nonmelanoma skin cancers, which account for approximately 1.3 million new cases of cancer diagnosed each year in the USA.
UV-induced oxidative stress and inflammatory responses have been implicated in skin diseases and skin disorders, including cancers and premature aging of the skin (i.e. photoaging). It has been observed that in obese mice, the levels of oxidative stress are greater in the adipose tissue than other tissues, and is accompanied by augmented expression of NADPH oxidase and decreased levels of anti-oxidant enzymes [7]. Weisberg et al [8] and Xu et al [9] reported that macrophages that infiltrate the obese adipose tissues are an important source of inflammatory responses. Since, UV-induced infiltrating macrophages in the skin are both a potential source of oxidative stress and inflammation and have been implicated in the increased incidence of photocarcinogenesis and premature aging of the skin [10,11], we formulated the hypothesis that obese mice which have higher amount of adipose tissues would be at greater risk of UV-induced oxidative stress and inflammatory responses than non-obese mice. As high-fat feeding can complicate the interpretation of results [12-14], we chose to use a mouse model of genetic obesity rather than diet-induced obesity to test our hypothesis. The genetically obese Lepob/Lepob mouse strain was used as it is well characterized model in which the genetic defect leads to a deficiency in leptin, a cytokine-like protein made in adipose tissue and released into the circulation [15]. Here we report that, upon UVB exposure, leptin-deficient obese mice exhibit higher levels of oxidative stress or oxidative damage in the skin than their wild-type counterparts (C57BL/6). The UVB-induced oxidative stress in this mouse model was determined in terms of: (i) increased hydrogen peroxide (H2O2) and nitric oxide production, lipid and protein oxidation, (ii) depletion of endogenous antioxidant defense enzymes, and (iii) activation of the mitogen-activated protein kinases (MAPK) and NF-κB signaling pathways. We also found that UVB radiation resulted in enhanced induction of pro-inflammatory cytokines in the obese mice.
Materials and methods
Chemicals and antibodies
OxyBlot™ Protein Oxidation Detection Kit was purchased from INTERGEN Company (Purchase, NY). Phosphorylated ERK1/2 (Thr202/Tyr204), JNK (Thr183/Tyr185), p38 (Thr180/Tyr182) and non-phosphorylated ERK1/2, JNK and p38 antibodies and the anti-β-actin were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Antibodies for NF-κB, IκBα, IKKα, and ERK1/2, JNK and p38 MAP kinase phosphatases (MKP-1, V-15), and the anti-mouse IgG HRP-linked and anti-rabbit IgG HRP-linked secondary antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The protein assay kit was obtained from Bio-Rad (Hercules, CA) and the enhanced chemiluminescence western blotting detection reagents were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). ELISA kits for TNF-α, IL-1-β, IL-6 and IL-10 were procured from BioSource International (Camarillo, CA). All other chemicals employed in this study were of analytical grade and purchased from Sigma Chemical Co. (St. Louis, MO).
Animals
Leptin-deficient obese female mice (Lepob/Lepob) and wild-type C57BL/6 mice of 4-5 weeks of age were purchased from Jackson Labs (Bar Harbor, ME). All mice were housed in the Animal Resource Facility of the University of Alabama at Birmingham under the following conditions: 12 h dark/12 h light cycle, 24±2°C temperature and 50± 10% relative humidity. The mice were given control Purina chow diet and drinking water ad libitum throughout the experiment. The animal protocol used in this study was approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
In vivo analyses of body weight, lean and fat mass in mice by dual-energy X-ray absorptiometry (DXA)
The body weight, bone-free soft-lean tissue mass and fat mass in mice of 22 weeks of age were measured using DXA analysis technique as described previously [18]. For DXA analysis, the mice were sacrificed and kept at −80°C until they were analyzed. The head part of the mouse was not included in lean and fat mass measurements, and the DXA analysis was performed on thawed animals. In addition, the kinetics of normal gain in body weight of each mouse was measured on a weekly basis using a digital balance.
UVB irradiation of mice
Mice were UVB-irradiated as described previously [16,17]. Briefly, the clipper-shaved dorsal skin was exposed to UV radiation from a band of four FS24T1 UVB lamps (Daavlin, UVA/UVB Research Irradiation Unit, Bryan, OH) equipped with an electronic controller to regulate UV dosage. The UV lamps emit UVB (290-320 nm; ≈80% of total energy) and UVA (320-375 nm; ≈20% of total energy), with UVC emission being insignificant. The majority of the resulting wavelengths of UV radiation are in the UVB (290-320 nm) range with peak emission at 313 nm [16,17]. The Daavlin UV radiation unit consists of a fixture mounted on fixed legs. Mounted within the exposure unit are four UVB lamps and the exposure is controlled using Daavlin Flex Control Integrating Dosimeters. In this system, the UVB dose can be entered as millijoules per square centimeter and the unit automatically compensates for variations in energy output such that the desired UV dose is delivered. The UVB emission was monitored regularly using an IL-1700 phototherapy radiometer (International Light, Newburyport, MA). Mice were subjected to either acute UVB exposure of 120 mJ/cm2 or chronic UVB exposure involving exposure to UVB (120 mJ/cm2) on alternate days for one month.
Histologic evaluation of the skin
Mice were sacrificed 24 h after the last UVB exposure and dorsal skin samples were obtained, fixed in 10% buffered formalin and processed for routine H & E staining for microscopic evaluation.
Preparation of cytosols and microsomal fractions
Twenty-four h after the last UVB exposure, mice were sacrificed and skin samples were collected. Sub-cutaneous fatty tissues were removed and then cytosolic and microsomal fractions were prepared as described previously [19]. Briefly, skin samples were homogenized with a Polytron System, PT 3100 (Kinematica, Switzerland) in PBS buffer containing potassium chloride (1.1%, w/v) and centrifuged at 18 000 g for 15 min at 4°C to prepare cytosolic and microsomal fractions [19]. Cytosols were used to determine the levels of endogenous antioxidant enzymes, including glutathione (GSH), glutathione peroxidase (GPx), catalase (CAT) and superoxide dismutase (SOD) whereas microsomal fraction was used to determine the lipid peroxidation content.
Assays for antioxidant enzymes
The levels of GPx, GSH, CAT and SOD were measured in cytosolic fractions following the standard analytical methods of Flohe and Gunzler [20], Akerboom and Sies [21], Nelson and Kiesow [22] and Misra and Fridovich [23], respectively. The experiments were repeated at least three times.
Immunohistochemical detection of H2O2-producing cells
Immunohistochemical detection of H2O2 in normal as well as in UV-irradiated skin was performed following a procedure described earlier [24]. Briefly, 6 μm thick frozen skin sections were incubated with 0.1 M Tris-HCl buffer, pH 7.5, containing 1 mg/ml glucose and 1 mg/ml diaminobenzidine for 6 h at 37°C. Sections were then washed in distilled water and counterstained with methyl green.
Assay for nitric oxide
The levels of nitric oxide were determined by measuring their stable degradation products, nitrate and nitrite, using a colorimetric Nitric Oxide Assay Kit (Oxford Biomedical Research, Inc., Oxford, MI) following the manufacturer's protocol, as described previously [24]. In this method, nitrate is enzymatically converted into nitrite by the enzyme nitrate reductase followed by determination of nitrite using Griess reagent.
Quantitative analysis of protein carbonyls or oxidized proteins
The quantitative analysis of protein carbonyls was performed using 2,4-dinitrophenylhydrazine (DNPH) as described previously [25]. Briefly, the nucleic acids present in the skin lysates that contain carbonyl groups reactive with DNPH were initially precipitated out using streptomycin sulfate followed by dialysis against water for 2.5 h. To 1 mL of the above solution (containing ∼1.0-1.5 mg/mL protein) 4 mL of 12.5 mM DNPH in 2.5 M HCl or 2.5 M HCl alone (blank) was added and incubated at room temperature for 1 h. The protein was precipitated with 10% trichloroacetic acid. The pellet was washed 3-5 times by breaking the pellet with a glass rod to remove the free DNPH with 4 mL of ethanol:ethyl acetate (1:1, v/v). The pellets were dissolved in 6 M Guanidine-HCl at 37°C for 20 min with frequent vortexing. Insoluble materials were removed by centrifugation and absorbance was measured at 370 nm. The protein carbonyl content was calculated from the molar absorption coefficient (ε) of 22,000 M−1 cm−1. The protein content was determined by using BioRad protein detection kit following the manufacturer's protocol.
Assay for protein oxidation following western blot analysis
The levels of oxidized proteins were detected using the OxyBlot™ Protein Oxidation Detection kit (Intergen Company, Purchase, NY) following the manufacturer's protocol as described previously [25]. Briefly, samples of 10 μg proteins were subjected to DNPH derivatization. Incubation of equal aliquots with a control solution lacking DNPH served as negative control. The dinitrophenylhydrazone-derivatized protein samples were separated by 10% SDS–PAGE gel electrophoresis, and blotted onto nitrocellulose membranes. After blocking the non-specific binding, the membranes were incubated with a rabbit anti-dinitrophenylhydrazone antibody for 1 h and then with a peroxidase-coupled goat anti-rabbit IgG antibody. The membrane was then treated with ECL (Amersham Life Sciences, Arlington, IL) to visualize protein bands. To ensure equal protein loading, the membranes were stripped and reprobed with anti-β-actin antibodies.
Assay for lipid peroxidation (LPO)
Lipid peroxidation (LPO) was determined in microsomal fraction of the skin samples using the thiobarbituric acid reaction method, as described previously [26]. Briefly, 0.2 ml of the microsomal fraction was treated with 0.2 ml of 8.1% SDS and 3 ml of thiobarbituric acid. Total volume was made up to 4 mL with distilled water and kept at 95°C in a water bath for 1 h. Color was extracted with n-butanol and pyridine (15:1 v/v). The absorbance was measured at 530 nm, and the resultant lipid peroxidation was expressed in terms of nmol/mg protein.
Assay for myeloperoxidase (MPO)
MPO was determined as a marker of tissue infiltration following the procedure of Bradley [27] and as modified by us [28]. In brief, the skin samples were homogenized in 50 mM potassium phosphate buffer, pH 6.0, containing 0.5% hexadecyltrimethyl-ammonium bromide followed by sonication of the homogenate at 4°C for three 10 s bursts with a heat system sonicator equipped with a microprobe. For complete extraction of MPO from infiltrating neutrophils, the tissue homogenates were frozen and thawed three times, and each time sonication was repeated. The tissue homogenate thus obtained was centrifuged at 40,000 g for 15 min at 4° C, and the resulting supernatants were used for MPO estimation. MPO activity in the supernatant (0.1 ml) was assayed by mixing with 50 mM phosphate buffer (2.9 ml), pH 6.0, containing 0.167 mg/ml ortho-dianisidine dihydrochloride and 0.0005% hydrogen peroxide. The change in absorbance resulting from decomposition of H2O2 in the presence of ortho-dianisidine was measured at 460nm utilizing Beckman Coulter™ DU 530 spectrophotometer. The results are expressed as the mean MPO unit/mg protein.
Western blot analysis and densitometry
Whole skin lysates or nuclear fractions were prepared as described previously [25]. The proteins (25–50 μg) were resolved over 8-12% SDS-PAGE gels and transferred onto a nitrocellulose membrane, as described previously [29]. After blocking the non-specific binding sites in blocking buffer (5% non-fat dry milk, 1% Tween 20 in 20 mM TBS, pH 7.6), the blots were then incubated overnight with primary antibodies specific for the protein to be assessed. The blot was washed and incubated again with HRP-conjugated secondary antibody. After washing, the protein expression was detected and visualized using chemiluminescence ECL detection system (Amersham Life Sciences, Arlington, IL).
The density of each band in an immunoblot was analyzed using the Scion Image Program (NIH, Bethesda, MD). The numerical values are shown under each immunoblot. In case of MAPK and pMAPK, the results of quantitation of band densities are expressed in terms of the ratio of pMAPK/MAPK. For this purpose the band density of each MAPK protein (i.e, ERK, JNK and p38) was arbitrarily taken as “1” and comparison was then made with the band densities of pMAPK (i.e., pERK, pJNK and pP38). In the case of the NF-κB family, the values for the control group (non-UVB-exposed wild-type) were used as “1” (arbitrary unit) and comparison was then made with densitometry values of other treatment groups. To ensure equal protein loading, the membranes were stripped and re-probed with anti-β-actin antibodies using the protocol described above.
ELISA for cytokine assay
At the termination of the experiments, mice were sacrificed, blood samples were collected from each mouse and allowed to clot for 2 h at room temperature before centrifuging for 20 min at 2000 xg. Serum supernatants were collected and assayed for TNF-α, IL-1β, IL-6 and IL-10 using commercially available ELISA kits (BioSource International, Camarillo, CA) following the manufacturer's protocol.
Statistical analysis
The results of the antioxidant enzymes and markers of oxidative stress are expressed as the mean ± standard deviation. Statistical analysis was done using ANOVA followed by post hoc tests. The difference in the experimental groups was considered significant if p<0.05.
Results
Leptin-deficient obese mice have a greater weight and fat mass than their wild-type counterparts
As shown in Figure 1A, the obese mice gained weight faster than the wild-type mice, and at the termination of the experiment at 22 weeks, the weight of obese mice (60±5 g) was significantly greater than that of the wild-type mice (22±2 g) (p<0.001; Table 1). The consumption of food per animal/day was recorded on a daily basis for 30 days, and it was found that the diet consumption by obese mice was 4.9±0.5 g/day/animal as compared to 3.0±0.4 g/day/animal (Fig. 1C). The significantly higher consumption of diet by obese mice on per day basis (p<0.01) is compatible with the reports that leptin-deficiency stimulates uncontrolled consumption of food and that this is the basis for the obesity of Lepob/Lepob mice.
Figure 1.
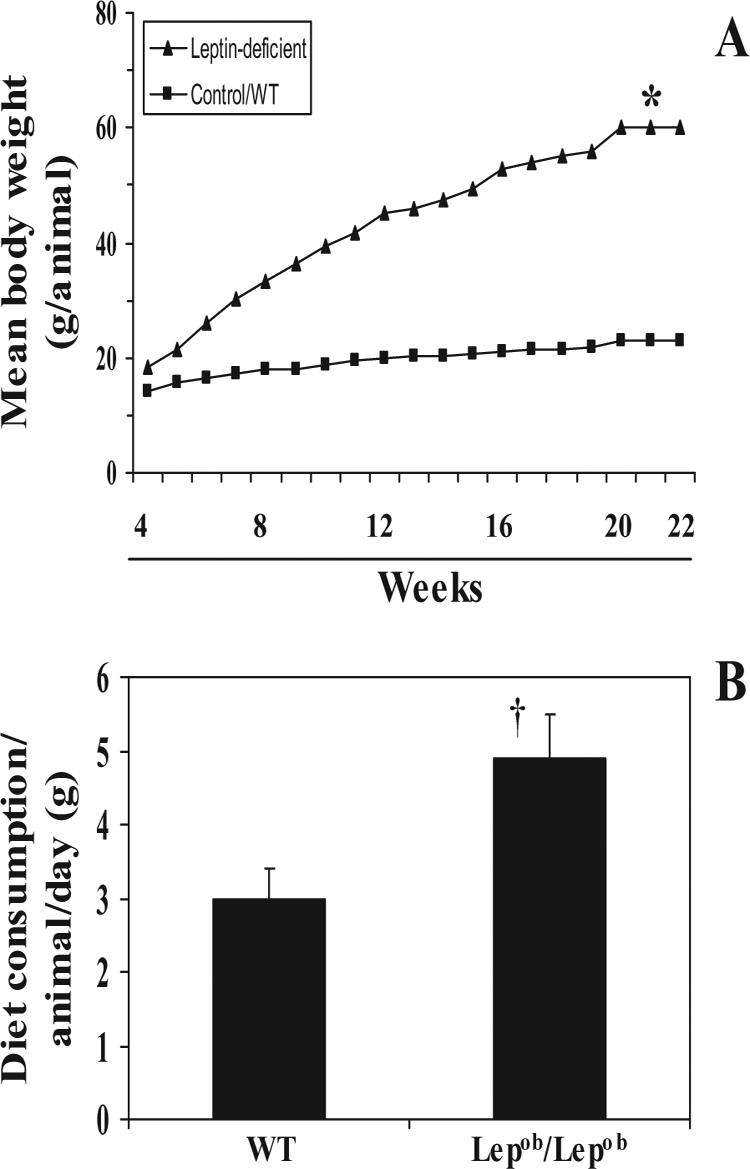
Comparative differences in mean body weight and consumption of diet/animal/day between leptin-deficient (obese mice) and their wild-types (WT). A, Body weight was measured on weekly basis and plotted against the period in weeks as mean values from 20 mice in each treatment group. B, Diet consumption in grams per animal per day was recorded in all the treatment groups for at least 30 days. Data are presented as the mean of diet consumption in g/animal/day ± SD. n=20. Significant difference versus wild-types, *p<0.001; †p<0.01
Table 1.
The comparative analysis of body weight, lean and fat mass in obese (Lepob/Lepob) and wild-type (C57BL/6) micea.
| Treatment group | Body weight | Lean (g) | Fat mass (g) | % Body fat |
|---|---|---|---|---|
| Wild-type | 22±2 | 17.9±0.8 | 4.1±0.5 | 18.6 |
| Lepob/Lepob | 60±5b | 24.1±2.4c | 35.3±3.1 | 58.8b |
Mice were given Purina chow diet and water ad libitum for 22 weeks. At the end of the experiment, mice were sacrificed and subjected to dual-energy X-ray absorptiometry analysis. The resultant data were compared between obese and their wild-type counterparts. n=20.
Highly significant difference versus wild-types, p<0.001
Significant difference versus wild-types, p<0.01
DXA analysis, which was performed at the termination of the experiment at 22 weeks, revealed that the fat mass deposition was significantly higher in obese mice (35.3±3.1 g fat/animal) than wild-type mice (4.1±0.5 g/animal) (p<0.001; Table 1). The lean mass also was significantly higher (p<0.01) in the obese mice than wild-type mice.
Obese mice have lower levels of antioxidant defense enzymes than wild-type mice
The levels of antioxidant defense enzyme were determined by analysis of the levels in the cytosolic fractions of skin samples. The levels of antioxidant defense enzymes in the skin of control mice that had not been exposed to the UVB irradiation protocol indicated a lower basal level of GSH, GPx and catalase enzymes in the obese mice than the wild-type mice; however, the difference was not statistically significant (Fig. 2). The basal levels of SOD were similar in the obese and wild-type mice.
Figure 2.
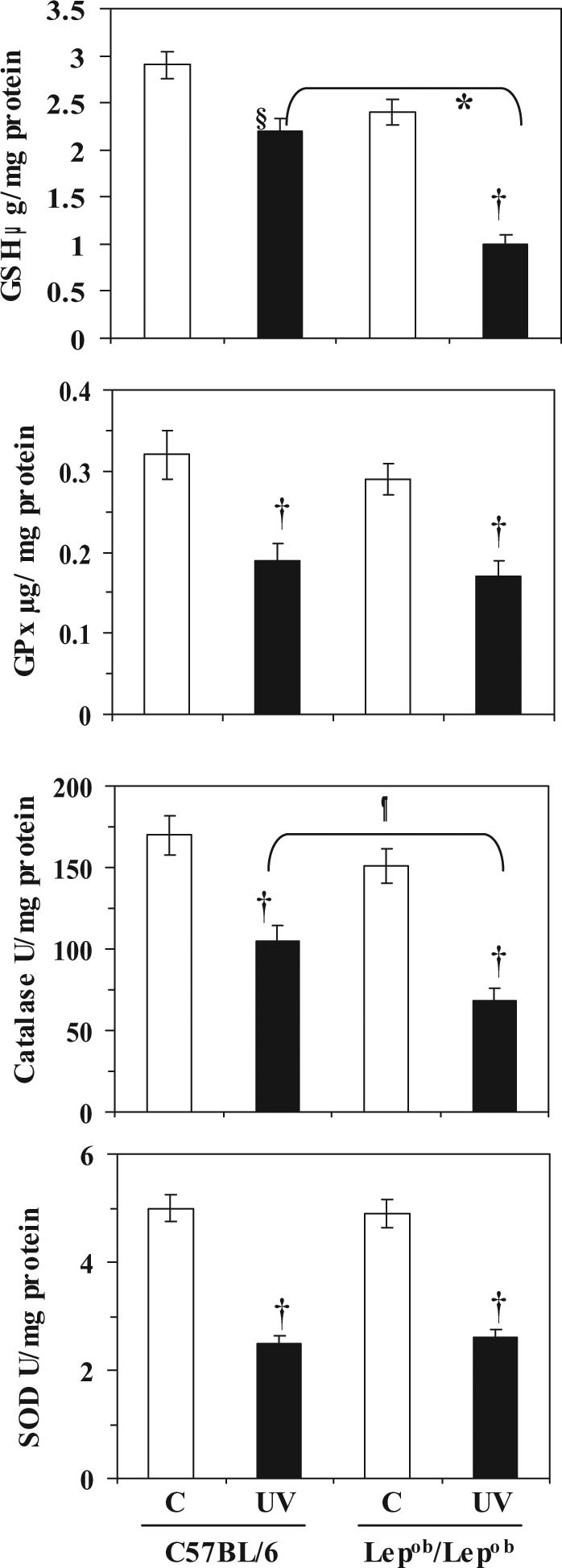
Chronic exposure of mice to UVB irradiation induces depletion of antioxidant defense enzymes in the skin. Mice were irradiated with UVB (120 mJ/cm2) on alternate days for one month, and sacrificed 24 h after the last UVB exposure. Skin samples were collected and cytosolic fractions were used for the analysis of antioxidant enzymes, as described in Materials and methods. Data are presented as mean ± SD, n=5.
Significant decrease compared to wild-types, *p<0.005; ¶p<0.01
Significant difference versus non-UVB-exposed control animals, †p<0.001; §p<0.01.
To compare the effects of chronic UVB irradiation, the obese and wild-type mice were exposed to UVB radiation (120 mJ/cm2) on alternate days for one month. Comparison of the irradiated mice with their non-irradiated counterparts indicated that chronic UVB irradiation led to a significant reduction in the levels of GSH, GPx, catalase and SOD in both obese and wild-type mice (p<0.005-0.01) (Fig. 2). Comparison of the UVB-irradiated obese and wild-type mice indicated a significantly greater UVB-induced depletion of GSH (58%, p<0.005) and catalase (55%, p<0.005) in the chronically exposed obese mice than in the chronically exposed wild-type mice. There was no significant difference in the pattern of SOD and GPx in obese and wild-type mice after chronic UVB exposure for one month (Fig. 2).
The effect of a single UVB (120 mJ/cm2) exposure on the levels of antioxidant enzymes was determined using cytosolic fractions of skin samples obtained from the mice 24 h after UV exposure. In this case, UVB-induced depletion of antioxidant defense enzymes was noticed but this effect was significantly lower than the effect of chronic exposure of UVB in both the obese and wild-type mice (data not shown). These results, together with the fact that most skin diseases are initiated after chronic or multiple exposures to UVB radiation, led us to focus on determining the effects of chronic exposure to UVB.
UVB exposure-induces higher amount of leukocyte infiltration in obese mice than wild-type mice
UVB-induced skin infiltration of leukocytes was analyzed 24 h after the last UVB exposure. Comparison of the irradiated mice with their non-irradiated counterparts indicated that chronic UVB irradiation resulted in infiltration of leukocytes in the skin (Fig. 3A). Histologic evaluation of the skin of the UVB-irradiated obese and wild-type mice indicated that the density of UVB-induced infiltrating leukocytes was higher in the irradiated obese mice than the irradiated wild-type mice. In addition to the leukocyte infiltration, epidermal edema and a hyperplastic response were evident with the epidermal hyperplastic response being quantifiable in terms of the thickness of epidermis in terms of cellular layers. Both the epidermal edema and the hyperplastic response were greater in the irradiated obese mice than the irradiated wild-type mice.
Figure 3.
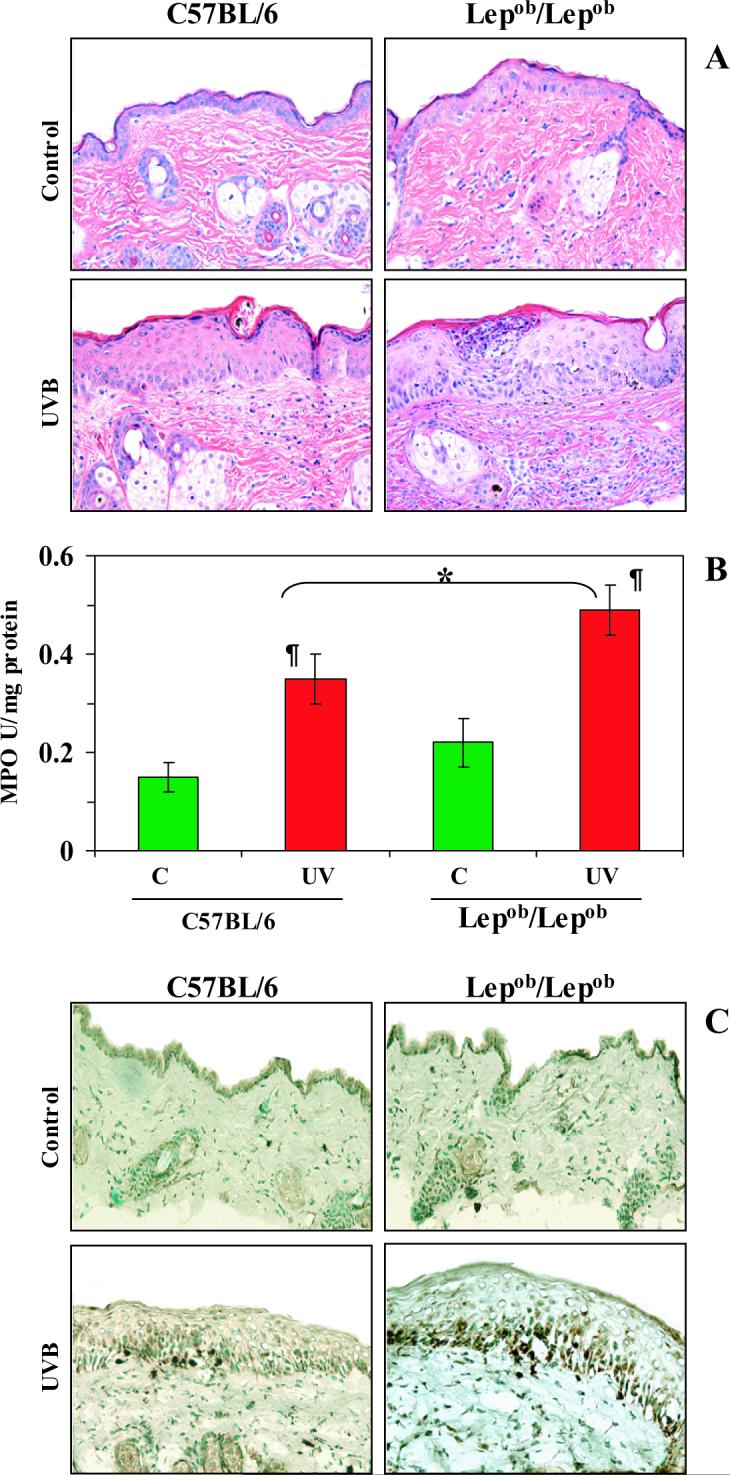
Chronic exposure of Lepob/Lepob mice to UVB results in greater leukocyte infiltration, and higher levels of MPO and hydrogen peroxide production than observed in UVB-irradiated wild-type (C57BL/6) mice. Mice were exposed to UVB on alternate days for one month and sacrificed 24 h after the last UVB exposure. Skin samples were collected and frozen in OCT under liquid nitrogen. A, UVB-induced edema and leukocyte infiltration was higher in Lepob/Lepob mice than wild-type mice. The paraffin-embedded skin samples (5 μm thick) were processed for routine H & E staining following a standard protocol. Magnification, x40; n=5. B, MPO was determined as a marker of UVB-induced tissue infiltration. The levels of UVB-induced MPO were greater in Lepob/Lepob mice compared to their wild-type counterparts. Data were reported as mean ± SD, n=5. Significant difference versus controls (non-UVB exposed), ¶p<0.01. Significant difference versus UVB-exposed wild-type, *p<0.05. C, The 5 μm thick sections of frozen skin samples were analyzed for UVB-induced H2O2 production following immunohistochemistry, as described in Materials and methods. The immunostaining intensity and the number of UVB-induced H2O2-positive cells appear dark brown. n=5, magnification, x40. Representative examples of micrographs of staining figures are shown from experiments conducted in skin samples from 5 mice that had identical patterns.
The infiltration of leukocytes was further determined by analysis of the levels of MPO activity in the cytosolic fractions of the skin. MPO is commonly used as a marker of infiltrating leukocytes (monocytes/macrophages and neutrophils) in the skin after UVB-irradiation. We found an increase in MPO activity in both obese and wild-type mice after chronic UVB exposure but the levels of activity were significantly higher in the obese mice than the wild-type mice (p<0.01, Fig. 3B).
UVB exposure-induces higher amount of H2O2 and nitric oxide in obese mice than wild-type mice
We used H2O2 production as a marker of oxidative stress in the mouse skin. The H2O2-producing cells were detected and localized by immunostaining (Fig. 3C). H2O2-positive cells were not detected in the skin of the control mice that were not exposed to UVB irradiation. As shown by immunostaining, chronic UVB-irradiation resulted in higher numbers of H2O2-producing cells than were found in non-UVB-exposed control skin in both obese and wild-type mice. However, the intensity of the staining as well as the numbers of H2O2-producing cells after UVB-irradiation was higher in the obese mice than their wild-type counterparts (Fig. 3C).
UVB-induced infiltrating leukocytes in the skin, notably activated macrophages and neutrophils, are an important source of NO production/generation and the production of NO is central to the induction of inflammatory responses. Therefore, quantitative analysis of NO was performed in the form of its stable product nitrite, which indicated that UVB-exposure induced higher levels of NO in wild-type mice (5-fold) and obese mice (8-fold) than in their non-UVB-irradiated counterparts (Fig. 4A).
Figure 4.
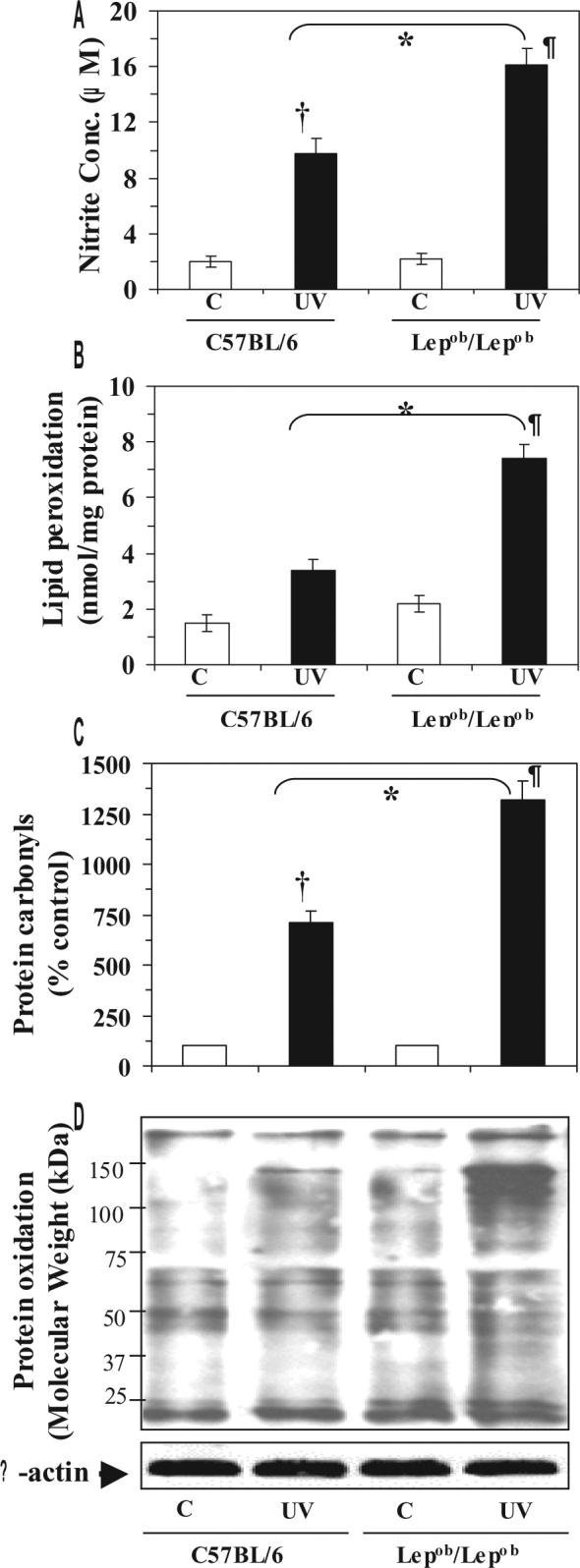
Obese mice are susceptible to UVB-induced oxidative stress. Obese and wild-type mice were exposed to UVB as described in Figure 2, sacrificed and skin samples were collected. Cytosolic fractions were used for determination of nitric oxide in the form of stable nitrite, and the microsomal fraction was used for the analysis of LPO. Skin lysates were used for the analysis of protein oxidation using OxyBlot™ Protein Oxidation Detection Kit. A, The concentration of UVB-induced nitrite was higher in the skin of obese mice than wild-type (C57BL/6) mice. The data are expressed as mean± SD, n=5. B, LPO is expressed in terms of nmol/mg protein and the data are presented as the mean ± SD from three independent experiments, n=5. C, UVB exposure increases the formation of protein carbonyls, a marker of oxidized proteins. Data are expressed in terms of per cent of control and as mean ± SD. D, UVB-irradiation increases the oxidation of proteins compared to non-UVB-exposed mice. Relative intensity of various oxidized proteins in different treatment groups is visible by the thickness or darkness of the bands. Experiments were conducted and repeated separately in five animals, and a representative blot is shown with similar pattern. The equivalent protein loading was confirmed by probing stripped blots for β-actin.
*Significant difference versus UVB-exposed WT mice, p<0.01
†Significant difference versus non-UVB exposed control WT mice, p<0.005
¶Significant difference versus non-UVB-exposed control Lepob/Lepob mice, p<0.001
UVB exposure-induces higher photo-oxidative damage of lipids and proteins in obese mice than wild-type mice
UVB-induced oxidation to lipids and proteins is the hallmark of photo-oxidative damage of the skin that results in age-related disorders of the skin. We found that chronic exposure to UVB enhanced the peroxidation of lipids in the skin of both obese (3-fold, p<0.001) and wild-type (2-fold, p<0.01) mice as compared with the levels in their non-UVB-exposed control counterparts (Fig. 4B). The lipid peroxidation in the skin of UVB-exposed obese mice was significantly higher (p<0.01) than that in the skin of UVB-exposed wild-type mice.
Oxidation of amino acid constituent of proteins, such as lysine, arginine and proline, leads to the formation of carbonyl derivatives that affects the biologic activity of the native proteins [30]. The presence of carbonyl groups has been widely used to measure oxidative damage of proteins under conditions of oxidative stress in an assay in which the carbonyl groups are reacted with DNPH to form stable hydrazone derivatives [31]. We found that chronic UV exposure resulted in a 7-fold higher level of protein carbonyls in the skin of wild-type mice and a 12-fold higher level in the skin of obese mice, as compared to the levels in the skin of their nonUVB-exposed control counterparts (Fig 4C). The UVB-induced photo-oxidative damage of proteins was further confirmed by specialized western blot analysis (Fig 4D). The UVB-induced protein oxidation is evident from the darker and/or the presence of new protein bands compared to the protein bands in non-UVB-exposed control mice.
UVB exposure-induces greater phosphorylation of ERK1/2, JNK and p38 proteins of the MAPK family in obese mice than their wild-type counterparts
UVB-induced phosphorylation of MAPK proteins has been implicated in various skin diseases, including skin cancer and photoaging. The phosphorylation of the MAPK proteins, namely, ERK1/2, JNK and p38, has been shown to be mediated through UVB-induced oxidative stress [17,32,33]. We examined UVB-induced phosphorylation of MAPK proteins using densitometric analysis of western blots and expressed the data in terms of the ratio of the levels of phosphorylated MAPK to the levels of the unphosphorylated form. Western blot analysis revealed that the basal levels of phosphorylated ERK1/2, JNK and p38 proteins were higher in the obese mice that were not irradiated than the wild-type mice that were not irradiated (Fig. 5). Chronic exposure to UVB resulted in enhanced phosphorylation of ERK1/2, JNK and p38 in both the obese and wild-type mice compared to their non-UVB-exposed counterparts. However, the levels of phosphorylated ERK1/2, JNK and p38 proteins were markedly higher in the chronically irradiated obese mice than the chronically irradiated wild-type mice (Fig. 5A, 5B and 5C). These changes were not due to differences in the amounts of proteins loaded on the gels as equivalent protein loading was confirmed by probing stripped blots for β-actin (Fig. 5).
Figure 5.
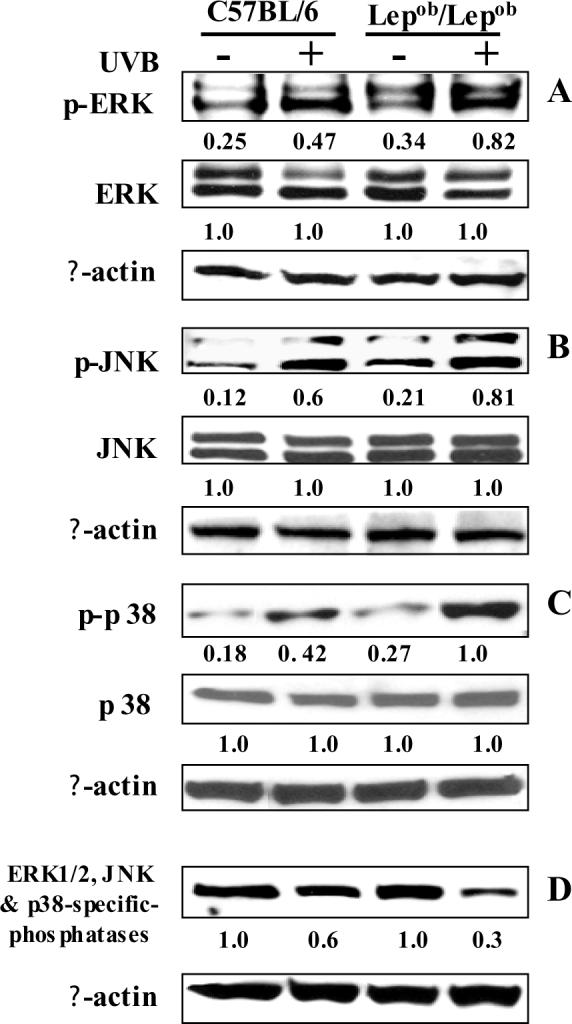
Obese mice are more susceptible to UVB-induced phosphorylation of MAPK protein than wild-type mice. Chronic exposure of mice to UVB induces phosphorylation of ERK1/2 (A), JNK (B) and p38 (C) proteins of MAPK family, and induces inactivation of ERK1/2, JNK and p38 MAP kinase phosphatases (D). Mice were treated as described in Figure 2. Skin lysates were prepared to determine the phosphorylated and total protein levels of ERK1/2, JNK and p38, and the levels of ERK, JNK and p38 MAP kinase phosphatases using western blot analysis. A representative blot is shown from three independent experiments with identical observations, and equivalent protein loading was confirmed by probing stripped blots for β-actin as shown. The relative density of each band in pMAPK immunoblots are shown in terms of the ratio of the band densities of respective MAPK immunoblots. The densitometry value in case of total MAPK protein was assigned the value 1 (arbitrary unit).
To determine the negative regulation of MAP kinase phosphatases on UVB-induced increases in the phosphorylation of MAPK proteins, we determined the levels of ERK1/2, JNK and p38-specific MAP kinase phosphatases in skin lysate samples using western blot analysis. The antibodies against MAP kinase phosphatases (MKP-1) have dual-specificity directed to phosphothreonine and phosphotyrosine residues within MAP kinases. Western blot analysis revealed that UVB exposure suppressed the levels of MAP kinase phosphatases (e.g., JNK, p38 and ERK1/2) in the skin of both obese and wild-type mice; however, the UVB-induced suppression or inactivation of MAP kinase phosphatases was greater in the skin of obese mice than the skin of wild-type mice (Fig. 5D).
UVB exposure-induces activation of NF-κBp65 and IKKα and the degradation of IκBα is greater in obese mice than wild-type counterparts
NF-κB/p65 is a downstream target of the MAPK signal transduction pathways. Comparison of the levels of NF-κB/p65 in the nuclear fraction of skin samples obtained from control mice that were not irradiated indicated that the basal level of NF-κB/p65 in the nuclear fraction was higher in the obese mice than the wild-type mice. This finding was consistent with our data that indicate that the basal level of oxidative stress is higher in obese mice than wild-type mice. Western blotting and densitometric analysis of the intensity of bands relative to β-actin indicated that chronic exposure to UVB resulted in markedly greater activation of NF-κB/p65 and its translocation to the nucleus in both obese and wild-type mice. However, the UVB-induced activation and translocation of NF-κB/p65 to the nucleus in the skin of obese mice was greater than the skin of UVB-exposed wild-type mice (Fig. 6A).
Figure 6.
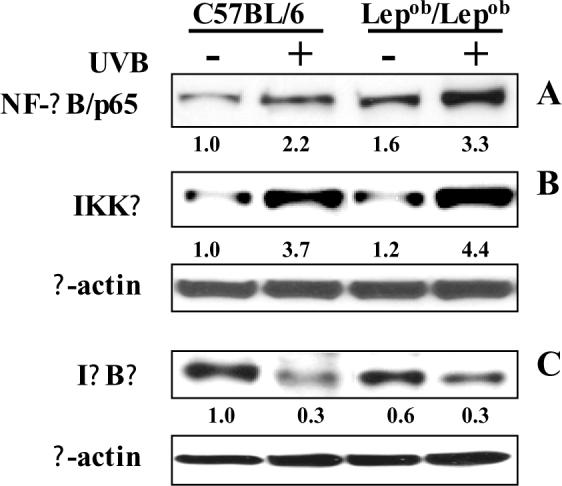
Chronic exposure of mice to UVB induces activation of NF-κB and IKKα, and degradation of IκBα. The activation of NF-κB (A), IKKα (B) or degradation of IκBα (C) was determined using western blot analysis. A representative blot is shown from three independent experiments with almost identical observations. Equivalent protein loading was confirmed by probing stripped blots for β-actin as shown. The relative density (arbitrary) of each band after normalization for β-actin is shown under each immunoblot.
Chronic UVB exposure also resulted in activation of IKKα in the skin of both obese and wild-type mice. The induction of IKKα has been shown to be essential for UVB-induced phosphorylation and degradation of IκBα. Western blot analysis and subsequent measurement of intensity of bands relative to β-actin indicated that activation level of IKKα was higher in the skin of UVB-irradiated obese mice than in the skin of UVB-irradiated wild-type mice (Fig. 6B). Greater degradation of IκBα was noted in both UVB-irradiated obese and UVB-irradiated wild-type mice as compared to their respective non-irradiated counterparts (Fig. 6C).
UVB exposure-induces higher levels of secretion of pro-inflammatory cytokines in obese mice than wild-type counterparts
It has been suggested that the increased oxidant stress that is associated with obesity may be due to the presence of excessive adiposity per se because adipocytes and preadipocytes have been identified as sources of inflammatory cytokines, including TNF-α, interleukin (IL)-1-β and IL-6 [34]. Cytokines are potent stimuli for the production of ROS/RNS by macrophages and monocytes [35]; therefore, we examined the effect of chronic UVB exposure on the secretion of pro-inflammatory cytokines. For this purpose, the levels of cytokines in blood serum were determined using ELISA. As shown in Fig. 7, chronic exposure to UVB resulted in significantly higher levels of TNF-α, IL-1-β and IL-6 in both obese and wild-type mice as compared with non-UVB-exposed control mice. However, the levels of these cytokines were significantly higher (p<0.01-0.005) in the sera of the UVB-irradiated obese mice than the sera of the UVB-irradiated wild-type mice. There was no significant difference in the levels of the immunosuppressive cytokine IL-10 in the sera of UVB-irradiated obese and UVB-irradiated wild-type mice.
Figure 7.
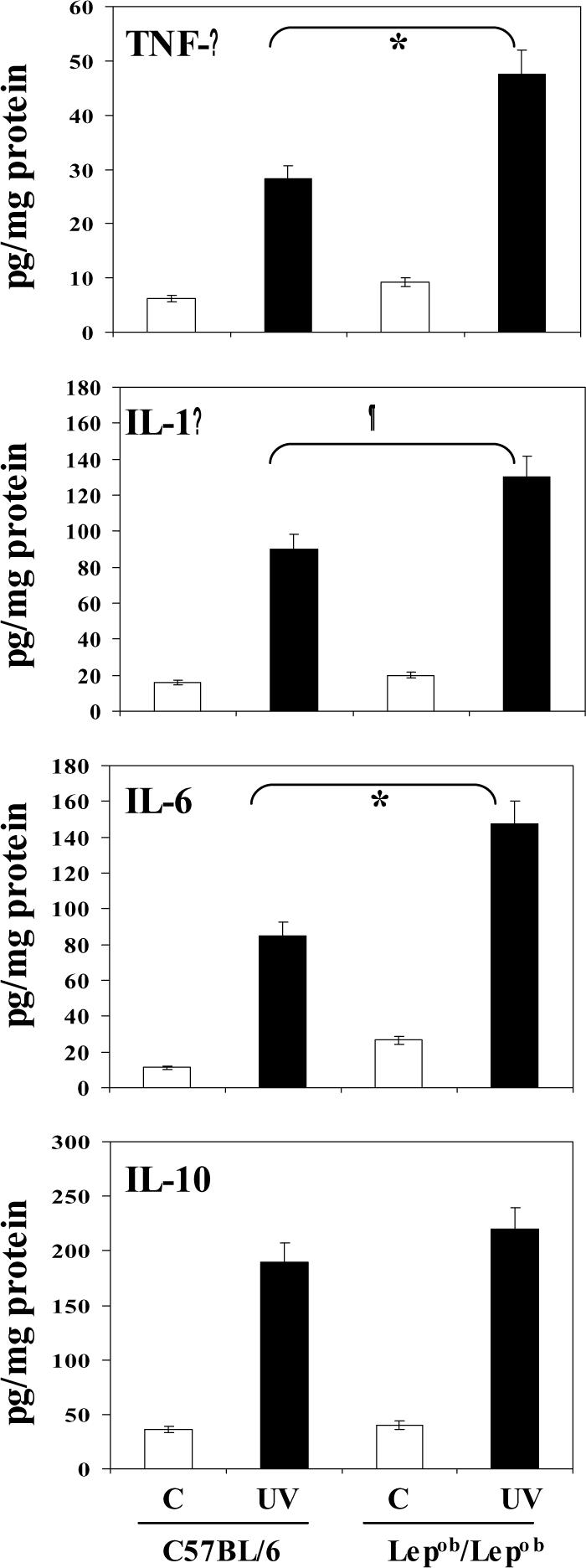
Chronic exposure of mice to UVB increases the levels of pro-inflammatory cytokines in blood serum of mice. Mice were exposed to UVB as detailed in Figure 2. At the termination of the experiment, mice were sacrificed, blood serum obtained and the levels of TNF-α, IL-1β, IL-6 and IL-10 determined using ELISA. Multi-fold increases in these cytokines were observed after UVB irradiation compared to non-UVB-exposed animals. The concentration of each cytokine is reported in terms of pg/mg protein as a mean ±SD, n=5. Significant increases in TNF-α (*p<0.005), IL-6 (*p<0.005) and IL-1β (¶p<0.01) in UVB-exposed Lepob/Lepob versus UVB-exposed wild-type counterparts.
Discussion
Obesity is associated with traditional cardiovascular diseases and diabetes but relatively little is known regarding the relationship between obesity and oxidative stress and, to our knowledge, no information is available on the effects of obesity on UV radiation-induced oxidative stress in an in vivo skin system. It has been established that UVB-induced oxidative stress contributes to several adverse biologic effects including skin cancers and premature aging of the skin in an in vivo system [11,36,37]. We observed that chronic exposure to UVB radiation induced higher levels of oxidative stress in the skin of obese mice than wild-type mice when measured in terms of depletion of antioxidant defense enzymes, such as GSH, GPx and catalase, and generation of H2O2 and nitric oxide. Antioxidant enzymes function cooperatively, and a change in any one of them may affect the equilibrium state of oxidative stress or the levels of reactive oxygen species (ROS). If the ROS remain, without being scavenged in the biological system, they can induce biochemical alterations, including inflammation, oxidation of lipid and protein, DNA damage and activation or inactivation of certain enzymes [38,39,40-43]. Thus, the generation of chronic and more intense oxidative stress and inflammatory responses in the obese mice on repeated exposure to UVB irradiation would be anticipated to render them more susceptible to oxidative stress-mediated skin diseases than wild-type mice.
The UVB-induced oxidation of macromolecules, including lipids and proteins, also was analyzed as a marker of oxidative stress in the present system. The occurrence of LPO in the biological membrane is a free radical-mediated event and is regulated by the availability of substrates in the form of polyunsaturated fatty acids, pro-oxidants which promote peroxidation. Elevated levels of LPO have been linked to injurious effects such as loss of fluidity, inactivation of membrane enzymes, and increases in permeability of ions which may lead to disruption of cell membrane potential [42-44]. Peroxidation products also can damage DNA [39,42,43]. Thus, the higher levels of LPO in the skin of obese mice compared to wild-type mice will make them more susceptible to skin disorders. The UV-induced oxidative stress-mediated protein oxidation may also be a contributing factor for skin diseases. The presence of higher amount of fat mass in obese mice could be responsible for the enhanced inflammatory responses and generation of oxidative stress. As we observed in this study that UVB-induced leukocyte infiltration was higher in obese mice, and as inflammatory leukocytes are the major source of H2O2 and NO production in the skin [10,24], ROS may play an important role in induction of skin diseases. Thus obesity appears to be an important contributing factor in UVB radiation-induced oxidative stress-mediated diseases.
Chronic exposure to UV radiation promotes the development of various skin diseases, including tumor development, by activating various intracellular signaling cascades that play a major role in cell growth, differentiation and proliferation and lead to clonal expansion of UVB-initiated cells into skin tumors [45]. Members of the MAPK family are important upstream regulators of transcription factor activities and their signaling is critical to the transduction of a wide variety of extracellular stimuli into intracellular events. The phosphorylation of MAPK molecules controls the activities of various downstream transcription factors implicated in proliferation, differentiation and tumor promotion [46]. ERK1/2 has been shown to be strongly activated by tumor promoters and UV radiation, and has an important role in transmitting signals initiated by them [47]. Activation of JNK regulates activator protein-1 (AP-1) transcription in response to environmental stress such as UV radiation [48]. Increased AP-1 activity has been implicated in inflammation, metastasis and angiogenesis, and also in the promotion and progression of various types of cancers [49-51]. Therefore, higher levels of ERK1/2, JNK and p38 activation may promote skin diseases in obese mice. Our present data confirmed our previous observation that UVB radiation induces H2O2 production in the target cells [52], which in turn initiates phosphorylation of MAPK proteins and activation of downstream signals and expression of genes having direct relevance in skin carcinogenesis [52]. Therefore, it can be suggested that higher levels of phosphorylation of MAPK proteins in obese mice are due to enhanced levels of UVB-induced oxidative stress. The UVB-induced phosphorylation of MAPK proteins may activate down stream events, such as activation of NF-κB, which would lead to the stimulation of events which are associated with the skin diseases in vivo. ERK and p38 proteins of MAPK family have been shown to modulate NF-κB activation [53,54]. We suggest that UVB-induced phosphorylation of MAPK proteins in obese mice might be responsible for the activation of transcription factor NF-κB. Therefore, the activation of MAPK and NF-κB could be the potential signaling pathways in obese mice that activate oxidant-responsive element-dependent genes. We also observed that enhanced phosphorylation of MAPK proteins in UVB-exposed skin of obese and wild-type mice were associated with the inhibition or inactivation of MAP kinase phosphatases. It has been shown that reactive oxygen species promotes the activation of MAPK proteins by inhibiting MAP kinase phosphatases [55]. These phosphatases can dephosphorylate both phosphorylated threonine and phosphorylated tyrosine (dual specificity) residues and inactivate MAPK signaling [56]. It has been shown that MKP-1 (a family of dual-specificity protein phosphatases) can inactivate all three major MAPKs, including ERK, JNK and p38 [57]. Therefore, it is possible that UVB-induced oxidative stress induces the phosphorylation of MAPK proteins in the mouse skin through inactivation of MAP kinase phosphatases; however, further studies are required to confirm this observation.
The activation and translocation of NF-κB to the nucleus has an important regulatory role in inflammation, cellular proliferation and carcinogenesis [58-60]. Therefore, investigation of the signaling pathways leading to the regulation of NF-κB activity is crucial. The activation of NF-κB by the extracellular inducers depends on the phosphorylation and subsequent degradation of IκB proteins. Activation of NF-κB is mediated through the action of a family of serine/threonine kinases known as IκB kinase (IKK). The IKK (IKKα and/or IKKβ) phosphorylates IκB proteins and the members of the NF-κB family. NF-κB is commonly activated by oxidants including H2O2 [61] and by agents that generate ROS/H2O2, such as UV radiation [62,63]. In our current study, we observed that NF-κB/p65 is activated in the skin of obese mice following UVB exposure and subsequently translocated to the nucleus, and its activation level was higher in the obese mice than the wild-type mice. UVB exposure also resulted in an increased degradation of IκBα protein. This suggests that the activation of NF-κB/p65 in obese mice is mediated through the inhibition of proteolysis of IκBα protein. It is well documented that IκBα is bound to NF-κB/p65 through a protein-protein interaction and thus prevents migration of NF-κB/p65 into the nucleus [60]. Additionally, the IKK complex is an important site for integrative signals that regulate the NF-κB pathway. We observed that chronic UVB exposure resulted in an increase in IKKα protein expression in mice, and that the UVB-induced expression level of IKKα was markedly higher in UVB-exposed obese mice than UVB-exposed wild-type mice. These data suggest that leptin-deficient obese mice are more susceptible to UVB-induced activation and nuclear translocation of NF-κB/p65 through greater activation of IKKα and degradation of IκBα proteins.
We were also interested in the effects of UVB radiation on proinflammatory cytokine secretion and whether it has any association with obesity. Our rationale was based on the fact that cytokines stimulate monocytes/macrophages and neutrophils to produce reactive oxygen/nitrogen species [35]. It was observed that obese mice were more sensitive to UVB-induced increases in the synthesis or release of proinflammatory cytokines, such as TNF-α, IL-1-β and IL-6, in the blood serum than the wild-type mice. The presence of higher concentrations of proinflammatory cytokines in UVB-exposed obese mice may be responsible for the increased oxidative stress in these mice; however, further studies are required to address this issue.
In summary, our data suggest that obesity in mice makes the skin more susceptible to UVB-induced: (i) oxidative stress, (ii) photo-oxidative damage of macromolecules, such as lipids and proteins, (iii) activation of MAPK and NF-κB signaling pathways, and that (iv) UVB-induced enhanced phosphorylation of MAPK proteins and NF-κB activation in mice appears to be mediated through the inhibition or inactivation of MAP kinase phosphatases in the skin. To the best of our knowledge, it is the first study that reports that obese mice are susceptible to UVB-induced oxidative stress. Further in vivo mechanism-based studies are required to examine whether obese mice are also more susceptible to photocarcinogenesis and/or photoaging than non-obese mice.
Acknowledgments
This work was supported by the funds from USPHS Grants CA104428, AT002536, the Merit Review Award from the Veterans Administration (SKK), and dual-energy X-ray absorptiometry analysis from the Core-Facility in the Department of Nutritional Sciences (P30DK56336, T. Nagy).
Abbreviations used
- DXA
dual-energy X-ray absorptiometry
- DNPH
dinitrophenylhydrazine
- GSH
glutathione
- SOD
superoxide dismutase
- GPx
glutathione peroxidase
- LPO
lipid peroxidation
- MPO
myeloperoxidase
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- NF-κB
nuclear factor κB
- UV
ultraviolet
- IL
interleukin
- H2O2
hydrogen peroxide
References
- 1.Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999-2002. JAMA. 2004;291:2847–2850. doi: 10.1001/jama.291.23.2847. [DOI] [PubMed] [Google Scholar]
- 2.Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000. JAMA. 2002;288:1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 3.Flegal KM, Graubard BI, Williamson DF, Gail MH. Excess deaths associated with underweight, overweight, and obesity. JAMA. 2005;293:1861–1867. doi: 10.1001/jama.293.15.1861. [DOI] [PubMed] [Google Scholar]
- 4.Eyre H, Kahn R, Robertson RM. ACS/ADA/AHA Collaborative Writing Committee. Preventing cancer, cardiovascular disease, and diabetes: a common agenda for the American Cancer Society, the American Diabetes Association, and the American Heart Association. CA Cancer J. Clin. 2004;54:190–207. doi: 10.3322/canjclin.54.4.190. [DOI] [PubMed] [Google Scholar]
- 5.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 6.Ogden CL, Flegal KM, Carroll MD, Johnson CL. Prevalence and trends in overweight among US children and adolescents, 1999-2000. JAMA. 2002;288:1728–1732. doi: 10.1001/jama.288.14.1728. [DOI] [PubMed] [Google Scholar]
- 7.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittal A, Elmets CA, Katiyar SK. CD11b+ cells are the major source of oxidative stress in UV radiation-irradiated skin: Possible role in photoaging and photocarcinogenesis. Photochem. Photobiol. 2003;77:259–264. doi: 10.1562/0031-8655(2003)077<0259:ccatms>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Katiyar SK. Oxidative stress and photocarcinogenesis: Strategies for prevention. In: Singh Keshav K., editor. Oxidative Stress, Disease and Cancer. Imperial College Press; London: 2006. pp. 933–964. Ph.D. [Google Scholar]
- 12.Welsch CW, House JL, Herr BL, Eliasberg SJ, Welsch MA. Enhancement of mammary carcinogenesis by high levels of dietary fat: a phenomenon dependent on ad libitum feeding. J. Natl. Cancer Inst. 1990;82:1615–1620. doi: 10.1093/jnci/82.20.1615. [DOI] [PubMed] [Google Scholar]
- 13.Ip C. Controversial issues of dietary fat and experimental mammary carcinogenesis. Prev. Med. 1993;22:728–737. doi: 10.1006/pmed.1993.1067. [DOI] [PubMed] [Google Scholar]
- 14.Willett WC. Dietary fat intake and cancer risk: a controversial and instructive story. Semin. Cancer Biol. 1998;8:245–253. doi: 10.1006/scbi.1998.0076. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 16.Mittal A, Piyathilake C, Hara Y, Katiyar SK. Exceptionally high protection of photocarcinogenesis by topical application of (−)-epigallocatechin-3-gallate in hydrophilic cream in SKH-1 hairless mouse model: Relationship to inhibition of UVB-induced global DNA hypomethylation. Neoplasia. 2003;5:555–565. doi: 10.1016/s1476-5586(03)80039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vayalil PK, Elmets CA, Katiyar SK. Treatment of green tea polyphenols in hydrophilic cream prevents UVB-induced oxidation of lipids and proteins, depletion of antioxidant enzymes and phosphorylation of MAPK proteins in SKH-1 hairless mouse skin. Carcinogenesis. 2003;24:927–936. doi: 10.1093/carcin/bgg025. [DOI] [PubMed] [Google Scholar]
- 18.Nagy TR, Clair A-L. Precision and accuracy of dual-energy X-ray absorptiometry for determining in vivo body composition of mice. Obesity Res. 2000;8:392–398. doi: 10.1038/oby.2000.47. [DOI] [PubMed] [Google Scholar]
- 19.Katiyar SK, Afaq F, Perez A, Mukhtar H. Green tea polyphenol (−)-epigallocatechin-3-gallate treatment of human skin inhibits ultraviolet radiation-induced oxidative stress. Carcinogenesis. 2001;22:287–294. doi: 10.1093/carcin/22.2.287. [DOI] [PubMed] [Google Scholar]
- 20.Flohe L, Gunzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114–121. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 21.Akerboom TP, Sies H. Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol. 1981;77:373–382. doi: 10.1016/s0076-6879(81)77050-2. [DOI] [PubMed] [Google Scholar]
- 22.Nelson DP, Kiesow LA. Enthalpy of decomposition of hydrogen peroxide by catalase at 25°C (with molar extinction coefficients of H2O2 solutions in the UV) Anal. Biochem. 1972;49:474–478. doi: 10.1016/0003-2697(72)90451-4. [DOI] [PubMed] [Google Scholar]
- 23.Misra HP, Fridovich I. The role of superoxide anion in the autooxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 24.Katiyar SK, Mukhtar H. Green tea polyphenol (−)-epigallocatechin-3-gallate treatment to mouse skin prevents UVB-induced infiltration of leukocytes, depletion of antigen presenting cells and oxidative stress. J. Leukoc. Biol. 2001;69:719–726. [PubMed] [Google Scholar]
- 25.Vayalil PK, Mittal A, Hara Y, Elmets CA, Katiyar SK. Green tea polyphenols prevent ultraviolet light-induced oxidative damage and matrix metalloproteinases expression in mouse skin. J. Invest. Dermatol. 2004;122:1480–1487. doi: 10.1111/j.0022-202X.2004.22622.x. [DOI] [PubMed] [Google Scholar]
- 26.Mittal A, Elmets CA, Katiyar SK. Dietary feeding of proanthocyanidins from grape seeds prevents photocarcinogenesis in SKH-1 hairless mice: Relationship to decreased fat and lipid peroxidation. Carcinogenesis. 2003;24:1379–1388. doi: 10.1093/carcin/bgg095. [DOI] [PubMed] [Google Scholar]
- 27.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J. Invest. Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 28.Katiyar SK, Matsui MS, Elmets CA, Mukhtar H. (1999) Polyphenolic antioxidant (−)-epigallocatechin-3-gallate from green tea reduces UVB-induced inflammatory responses and infiltration of leukocytes in human skin. Photochem. Photobiol. 1999;69:148–153. [PubMed] [Google Scholar]
- 29.Roy AM, Baliga MS, Elmets CA, Katiyar SK. Grape seed proanthocyanidins induce apoptosis through p53, Bax and caspase 3 pathways. Neoplasia. 2005;7:24–36. doi: 10.1593/neo.04412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stadtman ER. Protein oxidation in aging and age-related diseases. Ann. N.Y. Acad. Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 31.Levine RL. Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic. Biol. Med. 2002;32:790–796. doi: 10.1016/s0891-5849(02)00765-7. [DOI] [PubMed] [Google Scholar]
- 32.Peus D, Vasa RA, Beyerle A, Meves A, Krautmacher C, Pittelkow MR. UVB activates ERK1/2 and p38 signaling pathways via reactive oxygen species in cultured keratinocytes. J. Invest. Dermatol. 1999;112:751–756. doi: 10.1046/j.1523-1747.1999.00584.x. [DOI] [PubMed] [Google Scholar]
- 33.Guyton KZ, Gorospe M, Kensler TW, Holbrook NJ. Mitogen-activated protein kinase (MAPK) activation by butylated hydroxytoluene hydroperoxide: implications for cellular survival and tumor promotion. Cancer Res. 1996;56:3480–3485. [PubMed] [Google Scholar]
- 34.Coppack SW. Pro-inflammatory cytokines and adipose tissue. Proc. Nutr. Soc. 2001;60:349–356. doi: 10.1079/pns2001110. [DOI] [PubMed] [Google Scholar]
- 35.Fenster CP, Weinsier RL, Darley-Usmar VM, Patel RP. Obesity, aerobic exercise, and vascular disease: The role of oxidant stress. Obes. Res. 2002;10:964–968. doi: 10.1038/oby.2002.131. [DOI] [PubMed] [Google Scholar]
- 36.Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J. Am. Acad. Dermatol. 1994;30:774–778. doi: 10.1016/s0190-9622(08)81509-5. [DOI] [PubMed] [Google Scholar]
- 37.Kligman LH. Photoaging. Manifestations, prevention, and treatment. Dermatol. Clin. 1986;4:517–528. [PubMed] [Google Scholar]
- 38.Shindo Y, Witt E, Packer L. Antioxidant defense mechanisms in murine epidermis and dermis and their responses to ultraviolet light. J. Invest. Dermatol. 1993;100:260–265. doi: 10.1111/1523-1747.ep12469048. [DOI] [PubMed] [Google Scholar]
- 39.Mukhtar H, Elmets CA. Photocarcinogenesis: Mechanisms, models and human health implications. Photochem. Photobiol. 1996;63:355–447. doi: 10.1111/j.1751-1097.1996.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 40.Shindo Y, Witt E, Han D, Epstein W, Packer L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Invest. Dermatol. 1994;102:122–124. doi: 10.1111/1523-1747.ep12371744. [DOI] [PubMed] [Google Scholar]
- 41.Punnonen K, Autio P, Kiistala U, Ahotupa M. In-vivo effects of solar-simulated ultraviolet irradiation on antioxidant enzymes and lipid peroxidation in human epidermis. Br. J. Dermatol. 1991;125:18–20. doi: 10.1111/j.1365-2133.1991.tb06032.x. [DOI] [PubMed] [Google Scholar]
- 42.Sun Y. Free radical antioxidant enzymes and carcinogenesis. Free Radic. Biol. Med. 1990;8:583–599. doi: 10.1016/0891-5849(90)90156-d. [DOI] [PubMed] [Google Scholar]
- 43.Girotti AW. Photodynamic lipid peroxidation in biological systems. Photochem. Photobiol. 1990;51:497–509. doi: 10.1111/j.1751-1097.1990.tb01744.x. [DOI] [PubMed] [Google Scholar]
- 44.Bus JS, Gibson JE. Lipid peroxidation and its role in toxicology. Rev. Biochem. Toxicol. 1979;1:125–149. [Google Scholar]
- 45.Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 1999;11:2063–2073. doi: 10.1093/carcin/20.11.2063. [DOI] [PubMed] [Google Scholar]
- 46.Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci. STKE. 2003;2003:re2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 47.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 48.Dunn C, Wiltshire C, MacLaren A, Gillespie DA. Molecular mechanism and biological functions of c-Jun N-terminal kinase signaling via the c-Jun transcription factor. Cell Signal. 2002;4:585–593. doi: 10.1016/s0898-6568(01)00275-3. [DOI] [PubMed] [Google Scholar]
- 49.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 50.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 51.McCarty MF. Polyphenol-mediated inhibition of AP-1 transactivating activity may slow cancer growth by impeding angiogenesis and tumor invasiveness. Med. Hypotheses. 1998;50:511–514. doi: 10.1016/s0306-9877(98)90273-0. [DOI] [PubMed] [Google Scholar]
- 52.Mantena SK, Katiyar SK. Grape seed proanthocyanidins inhibit UV radiation-induced oxidative stress and activation of MAPK and NF-κB signaling in human epidermal keratinocytes. Free Rad. Biol. Med. 2006;40:1603–1614. doi: 10.1016/j.freeradbiomed.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 53.Bonvin C, Guillon A, van Bemmelen MX, Gerwins P, Johnson GL, Widmann C. Role of the amino-terminal domains of MEKKs in the activation of NF kappa B and MAPK pathways and in the regulation of cell proliferation and apoptosis. Cell Signal. 2002;14:123–131. doi: 10.1016/s0898-6568(01)00219-4. [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Richmond A. Nuclear factor-kappa B activation by the CXC chemokine melanoma growth-stimulatory activity/growth-regulated protein involves the MEKK1/p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 2001;276:3650–3659. doi: 10.1074/jbc.M006115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 56.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 57.Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 58.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 59.Maniatis T. Catalysis by a multiprotein IkappaB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- 60.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 61.Ikeda M, Hirose Y, Miyoshi K, Kodama H. Nuclear factor kappaB (NF-kappaB) activation by hydrogen peroxide in human epidermal keratinocytes and the restorative effect of interleukin-10. Dermatol. Sci. 2002;28:159–170. doi: 10.1016/s0923-1811(01)00164-5. [DOI] [PubMed] [Google Scholar]
- 62.Helenius M, Makelainen L, Salminen A. Attenuation of NF-kappaB signaling response to UVB light during cellular senescence. Exp. Cell Res. 1999;248:194–202. doi: 10.1006/excr.1999.4393. [DOI] [PubMed] [Google Scholar]
- 63.Vile GF, Tanew-Ilitschew A, Tyrrell RM. Activation of NF-kappa B in human skin fibroblasts by the oxidative stress generated by UVA radiation. Photochem. Photobiol. 1995;62:463–468. doi: 10.1111/j.1751-1097.1995.tb02369.x. [DOI] [PubMed] [Google Scholar]