

Abstract
The decarbomethoxylation reaction of a substituted α-hydroxy-α-carbomethoxy pentacyclic substituted ketone, used as an advanced intermediate in the synthesis of the alkaloid aspidophytine, can be elected by heating with MgI2 in CH3CN. The reaction was shown to proceed by a novel α-hydroxy β-dicarbonyl to α-ketol ester rearrangement. It was possible to isolate a carbonate intermediate in 75% yield, thereby providing support for the proposed pathway.
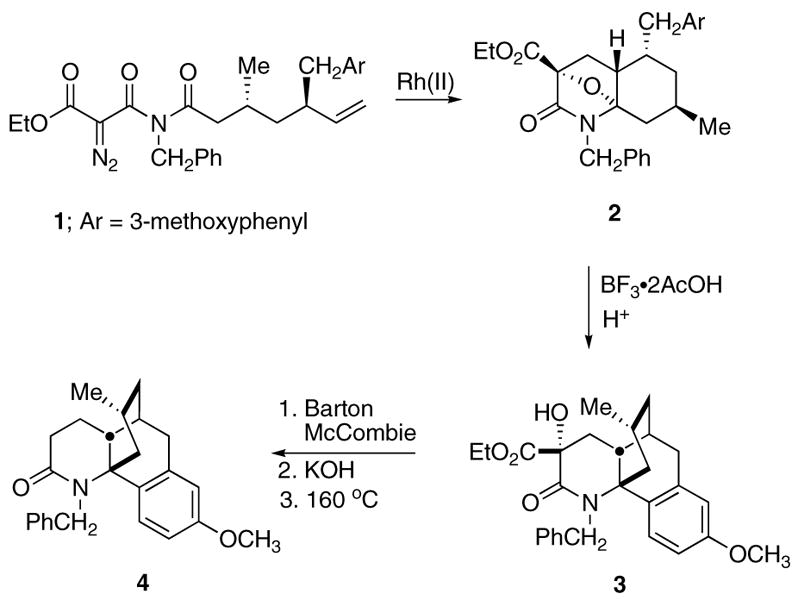
The Rh(II)-catalyzed cyclization/cycloaddition cascade of α-diazo carbonyl compounds developed in our laboratories has proven to be an extremely efficient process for the synthesis of a variety of aza polycyclic ring systems.1 Intramolecular trapping of the carbonyl ylide dipole generated from the domino Rh(II)-reaction proceeds in a highly regio and stereoselective manner and has been used in the construction of several heterocyclic natural products.2–4 For example, this domino cascade was successfully applied in our laboratory toward the synthesis of the alkaloid (±)-lycopodine (Scheme 1).5 The initially formed cycloadduct 2, obtained from the Rh(II)-cascade reaction of 1, was treated with a Lewis acid and provided the rearranged tetracyclic lactam 3 derived from a Pictet–Spengler type cyclization of a transient N-acyliminiumion.6 Although lactam 3 contains the proper ring skeleton needed for lycopodine, it is overfunctionalized. The three-step sequence that we used to remove both the hydroxyl and ester functionalities involved (i) a Barton–McCombie deoxygenation,7 (ii) ester hydrolysis, and (iii) thermal decarboxylation of the resulting carboxylic acid. Compound 4 was eventually transformed into (±)-lycopodine.
Scheme 1.
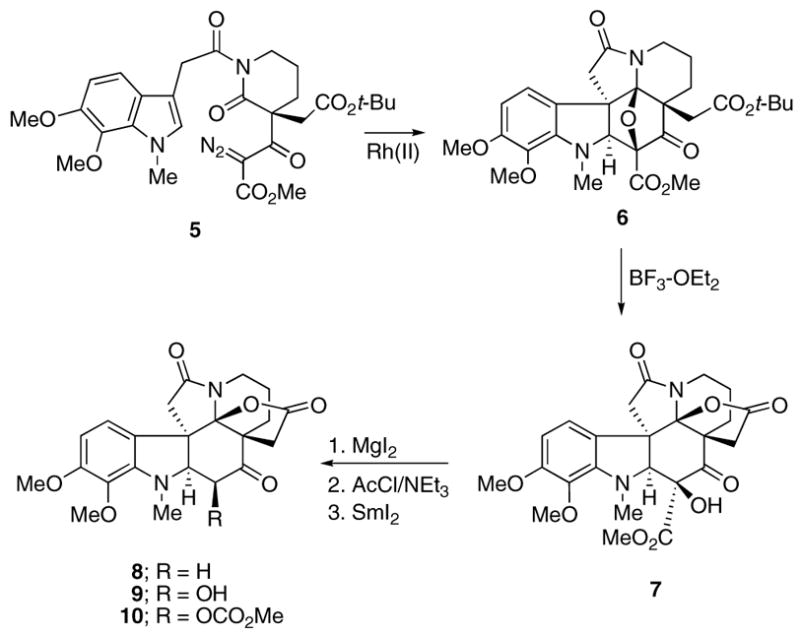
More recently, in our successful synthesis of the alkaloid (±)-aspidophytine,8 it was also necessary to remove the hydroxyl and carbomethoxy functionalities from the advanced intermediate 7, which was obtained by treating cycloadduct 6 with BF3·OEt2 (Scheme 2). In this case, the three-step protocol previously used for lycopodine resulted in only very low yields of ketone 8. During the course of preparing ketone 8 for an eventual synthesis of aspidophytine, we investigated the dealkoxycarbonylation–dehydroxylation of lactam 7 and related systems under several reaction conditions. The results that we obtained are not only useful for the future construction of molecules of the type discussed here, but also allow for the facile chemoselective removal of either (or both) the hydroxyl and/or carboalkoxy functionalities.
Scheme 2.
Dealkoxycarbonylation of activated esters via nucleophilic dealkylation9 represents a one-step process for the removal of the alkyl ester entity, particularly when structurally hindered. The reaction usually involves heating of the substrate in a dipolar aprotic solvent in the presence of a nucleophile. Following Krapcho’s original development of this approach,10 which employed NaCl in DMSO, many other nucleophiles have been found to be applicable,9 including thiolates, tert-but-oxide, thiocyanate, amines, and acetate. For malonates or β-ketoesters, this displacement is followed by rapid decarboxylation. Several accounts have also appeared describing synthetic transformations which utilize the intermediate anion generated in this reaction when run under anhydrous aprotic conditions.11 Since the introduction of this procedure by Krapcho, many applications of its synthetic utility to complex molecules have been reported.9
On the basis of the earlier decarboalkoxylation studies, our first attempts to convert hydroxyester 7 into α-hydroxyketone 9 made use of the one-pot Krapcho protocol. This involved heating 7 at reflux in DMSO in the presence of sodium chloride which indeed furnished compound 9, but only in 20% overall yield. After numerous attempts at improving the yield of the decarbomethoxylation reaction, the procedure that we eventually settled on for the aspidophytine synthesis involved heating 7 with MgI2 in refluxing acetonitrile that contained a small amount of water. Gratifyingly, this method resulted in the formation of alcohol 9 in 75% isolated yield. Acetylation of 9 with acetyl chloride/NEt3 afforded the corresponding acetate, which was subsequently reduced using SmI2 to give 8 in 90% yield for both steps. Although a cursory analysis of the conditions used suggests that a Krapcho decarboalkoxylation reaction first occurred, the isolation of small quantities of carbonate 10 from the mixture was somewhat surprising and led us to examine the MgI2 reaction in more detail using several model systems to probe the details of this process.
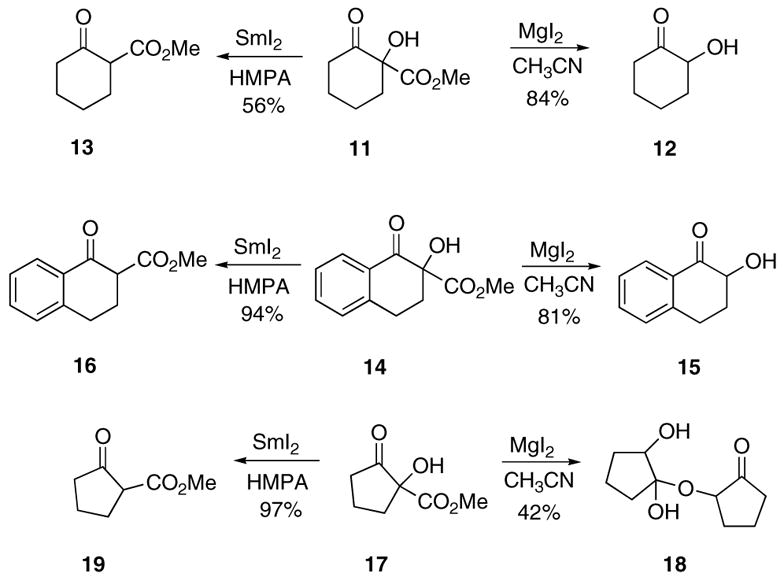
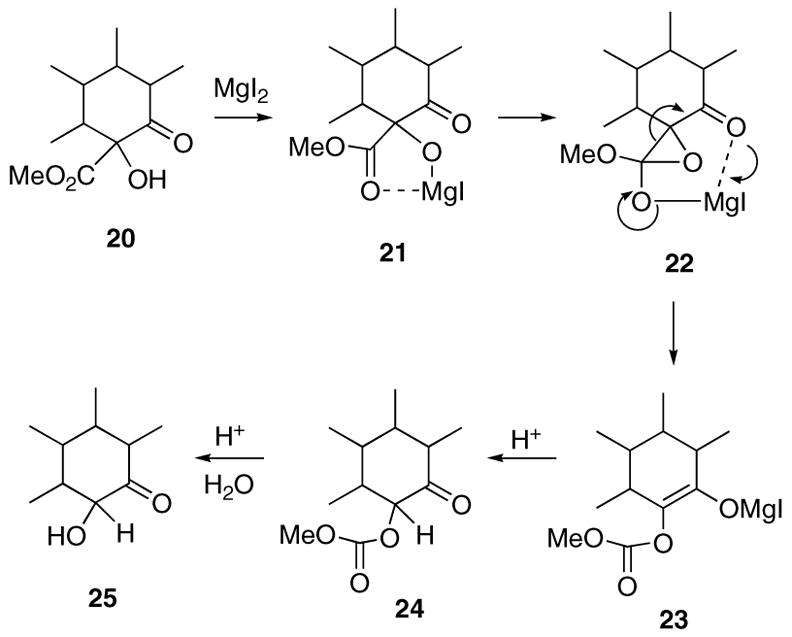
Toward this goal, we carried out experiments using α-hydroxy ketoesters 11, 14, and 17 in order to ascertain the limitations of the decarbomethoxylation process employing MgI2 and CH3CN as solvent. The optimized reaction conditions utilize 1.5 equiv of MgI2 in acetonitrile that contains a small amount of water at a temperature of 81 °C for 2 h. The reaction gives optimized yields of α-hydroxy ketones 12, 15, and 18 (Scheme 3) using substrate concentrations of ca. 0.1 M on scales up to 10 mmol. By employing this method, we were able to isolate α-hydroxy substituted cyclohexanones 12 and 15 in 84% and 81% yield, respectively. A lower yield of the hemiketal dimer 18 (42%) was obtained with the more reactive cyclopentanone system 17. During the course of these studies, we also found that it is possible to selectively remove the hydroxyl group prior to the decarbomethoxylation reaction. Thus, treatment of the starting α-hydroxy ketoesters shown in Scheme 3 with SmI2 in HMPA/THF at 0 °C produced the dehydroxylated products 13, 16, and 19 in 56%, 94%, and 97% yield, respectively. It was not necessary to convert the hydroxyl group into the corresponding acetate in order for the reduction to proceed. Most interestingly, when the ketoesters were exposed to the MgI2/CH3CN conditions previously employed, only recovered starting material was obtained, with no signs of any decarbomethoxylation. This result and the previous observation that small quantities of carbonate 10 could be isolated from the reaction of 7 with MgI2 lead us to propose an alternate mechanism for the decarbomethoxylation reaction. Thus, rather than undergoing the Krapcho reaction, we believe that the formation of 9 from 7 represents a rare example of a Lewis acid-promoted α-hydroxy β-dicarbonyl to α-ketol ester rearrangement. A plausible mechanism for this process involves coordination of the MgI2 with the more basic ester site followed by attack of the hydroxyl oxygen on the neighboring carbomethoxy group. Epoxide 22 is formed as a transient species which readily isomerizes to the magnesium enolate 23. This is followed by ketonization to furnish carbonate 24, which undergoes further hydrolysis with the H+ produced in the reaction with MgI2 to provide the final product 25. Indeed, heating an authentic sample of the carbonate derived from 12 with MgI2 in acetonitrile afforded the same decarboalkoxylation product in 96% yield.12,13 The success of this reaction is probably due to activation of the ester by chelation of the Mg(II) cation with both carbonyl groups. Analogous base-catalyzed rearrangements of α-hydroxy β-diketones to α-keto esters have been reported by Davis et al.14 and others.15–18 The pathway shown in Scheme 4 is supported by the isolation of carbonate 10 when the reaction of 7 was carried out with Cs2CO3 in acetonitrile at 81 °C for 1 h. Under these conditions, carbonate 10 was obtained in 75% yield and could be readily hydrolyzed to 9 when subjected to mild acidic conditions.
Scheme 3.
Scheme 4.
In summary, we have uncovered a novel Lewis acid-promoted α-hydroxy β-dicarbonyl to α-ketol ester rearrangement. This reaction can be used to give ready access to carbonate 10 which corresponds to a key intermediate en route to (±)-aspidophytine. Moreover, we have investigated the decarboalkoxylation of a number of related α-hydroxy carbomethoxy substituted cycloalkanones and have developed conditions for the chemo-selective removal of either (or both) the hydroxyl and/or carboalkoxy functionalities. Application of the method toward the synthesis of a number of aspidosperma alkaloids is currently underway and will be described in forthcoming publications.
Acknowledgments
We appreciate the financial support provided by the National Institutes of Health (GM 059384) and the National Science Foundation (CHE-0450779).
References
- 1.Padwa A, Hornbuckle SF. Chem Rev. 1991;91:263. (a) [Google Scholar]; Padwa A, Weingarten MD. Chem Rev. 1996;96:223. doi: 10.1021/cr950022h. (b) [DOI] [PubMed] [Google Scholar]
- 2.Padwa A. Acc ChemRes. 1991;24:22. (a) [Google Scholar]; Padwa A, Carter SP, Nimmesgern H. J Org Chem. 1986;51:1157. (b) [Google Scholar]; Padwa A, Carter SP, Nimmesgern H, Stull PD. J Am Chem Soc. 1988;110:2894. (c) [Google Scholar]; Padwa A, Fryxell GE, Zhi L. J Org Chem. 1988;53:2877. (d) [Google Scholar]; Padwa A, Chinn RL, Zhi L. Tetrahedron Lett. 1989;30:1491. (e) [Google Scholar]; Padwa A, Hornbuckle SF, Fryxell GE, Stull PD. J Org Chem. 1989;54:817. (f) [Google Scholar]; Padwa A, Sandanayaka VP, Curtis EA. J Am Chem Soc. 1994;116:2667. (g) [Google Scholar]; Curtis EA, Sandanayaka VP, Padwa A. Tetrahedron Lett. 1995;36:1989. (h) [Google Scholar]
- 3.Padwa A, Precedo L, Semones M. J Org Chem. 1999;64:4079. (a) [Google Scholar]; Padwa A, Curtis EA, Sandanayaka VP. J Org Chem. 1997;62:1317. (b) [Google Scholar]
- 4.Kinder FR, Jr, Bair KW. J Org Chem. 1994;59:6965. (a) [Google Scholar]; Chen B, Ko RYY, Yuen MSM, Cheng KF, Chiu P. J Org Chem. 2003;68:4195. doi: 10.1021/jo026399m. (b) [DOI] [PubMed] [Google Scholar]; Hodgson DM, Avery TD, Donohue AC. Org Lett. 2002;4:1809. doi: 10.1021/ol025917c. (c) [DOI] [PubMed] [Google Scholar]; Dauben WG, Dinges J, Smith TC. J Org Chem. 1993;58:7635. (d) [Google Scholar]
- 5.Padwa A, Brodney MA, Marino JP, Jr, Sheehan SM. J Org Chem. 1997;62:78. doi: 10.1021/jo960829p. [DOI] [PubMed] [Google Scholar]
- 6.Cox ED, Cook JM. Chem Rev. 1995;5:1797. (a) [Google Scholar]; Claret PA. In: Comprehensive Organic Chemistry. Barton D, Ollis WD, editors. Vol. 4. Pergamon: Oxford; 1979. p. 209. (b) [Google Scholar]
- 7.Barton DHR, McCombie SW. J Chem Soc, Perkin Trans 1. 1975:1574. [Google Scholar]
- 8.Oneto JMM, Padwa A. Org Lett. 2006;8:3275. doi: 10.1021/ol061137i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krapcho AP. Synthesis. 1982;805:893. (a) [Google Scholar]; McMurry J. Org React. 1976;101:187. (b) [Google Scholar]; Stevens RV, Lee AWM. J Am Chem Soc. 1979;101:7032. (c) [Google Scholar]
- 10.Krapcho AP, Glynn GA, Grenon BJ. Tetrahedron Lett. 1967:215. doi: 10.1016/s0040-4039(00)90519-7. (a) [DOI] [PubMed] [Google Scholar]; Krapcho AP, Weimaster JF, Eldridge JM, Jahngen EGE, Jr, Lovey AJ, Stephens WP. J Org Chem. 1978;43:138. (b) [Google Scholar]
- 11.Bunce RA, Dowdy ED, Jones PB, Holt EM. J Org Chem. 1993;58:7143. (a) [Google Scholar]; Takei S, Kowano Y. Tetrahedron Lett. 1975;16:4389. (b) [Google Scholar]; Belletire JL, Walley DR. Tetrahedron Lett. 1983;24:1475. (c) [Google Scholar]; Böhrer G, Böhrer P, Knorr R. Chem Ber. 1990;123:2167. (d) [Google Scholar]
- 12.It should be noted that heating an authentic sample of the carbonate derived from 12 in acetonitrile in the absence of MgI2 resulted in the recovery of starting material. The decarboalkoxylation of the carbonate was completely suppressed when the reaction was carried out in the presence of proton sponge. By executing the reaction of 11 in MgI2 in acetonitrile in the presence of acetic anhydride, it was possible to isolate the corresponding acetate in 78% yield.
- 13.A typical magnesium iodide decarbomethoxylation reaction. To a 0.9 g (1.9 mmol) sample of 7 in acetonitrile (150 mL) was added 1.1 g (4 mmol) of magnesium iodide and the mixture was heated at reflux for 4 h. The solution was allowed to cool to rt, concentrated under reduced pressure and the residue was taken up in methylene chloride (50 mL) and H2O (50 mL). This solution was added to a saturated aqueous solution of NaHCO3 (200 mL). The organic layer was separated and the aqueous layer was extracted twice with methylene chloride, dried over MgSO4, and concentrated under reduced pressure to give 0.6 g (75%) of 7 as a white solid: mp 210–212 °C; IR (neat) 1794, 1720, 1609, 1494, 1263, and 1193 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.30–1.45 (m, 1H), 1.58–1.81 (m, 2H), 2.27 (d, 1H, J = 17.4 Hz), 2.34 (d, 1H, J = 17.4 Hz), 2.35–2.42 (m, 1H), 2.80 (d, 1H, J = 18.0 Hz), 2.93 (dt, 1H, J = 14.0 and 6.7 Hz), 3.14 (s, 3H), 3.27 (dd, 1H, J = 18.0 and 0.8 Hz), 3.68 (d, 1H, J = 3.6 Hz), 3.69 (s, 3H), 3.83 (s, 3H), 3.85 (d, 1H, J = 3.6 Hz), 4.21–4.30 (m, 1H), 4.84 (t, 1H, J = 3.6 Hz), 6.48 (d, 1H, J = 8.6 Hz), and 6.89 (d, 1H, J = 8.6 Hz); 13C NMR (100 MHz, CDCl3) δ 20.4, 34.1, 37.3, 40.0, 41.4, 44.7, 51.7, 53.4, 56.2, 60.4, 78.0, 98.0, 105.1, 120.4, 122.9, 135.3, 146.5, 155.5, 170.4, 172.5, and 207.8; Anal. Calcd for C22H24N2O7: C, 61.67; H, 5.65; N, 6.54. Found: C, 61.18; H, 6.21; N, 5.94.
- 14.Davis FA, Clark C, Kumar A, Chen BC. J Org Chem. 1994;59:1184. (a) [Google Scholar]; Davis FA, Liu H, Chen BC, Zhou P. Tetrahedron. 1998;54:10481. (b) [Google Scholar]
- 15.Rubin MB, Inbar S. Tetrahedron Lett. 1979;20:5021. (a) [Google Scholar]; Rubin MB, Inbar S. J Org Chem. 1988;53:3355. (b) [Google Scholar]
- 16.Blatt AH, Hawkins WL. J Am Chem Soc. 1936;58:81. [Google Scholar]
- 17.House HO, Gannon WF. J Org Chem. 1958;23:879. [Google Scholar]
- 18.Charest MG, Siegel DR, Myers AG. J Am Chem Soc. 2005;127:8292. doi: 10.1021/ja052151d. [DOI] [PubMed] [Google Scholar]