

Abstract
Background
Previous studies have demonstrated that alterations in myocardial structure, consistent with tissue and sarcomere disruption as well as myofibril dissociation, occur after myocardial ischemia and reperfusion. In this study we determine the onset of these structural changes and their contribution to electrical conduction.
Methods
Langendorff perfused rabbit hearts (n = 47) were subjected to 0, 5, 10, 15, 20, 25, and 30 minutes global ischemia, followed by 120 minutes reperfusion. Hemodynamics were recorded and tissue samples were collected for histochemical and immunohistochemical studies. Orthogonal epicardial conduction velocities were measured, with temperature controlled, in a separate group of 10 hearts subjected to 0 or 30 minutes of global ischemia, followed by 120 minutes of reperfusion.
Results
Histochemical and quantitative light microscopy spatial analysis showed significantly increased longitudinal and transverse interfibrillar separation after 15 minutes or more of ischemia (p < 0.05 versus control). Confocal immunohistochemistry and Western blot analysis demonstrated significant reductions (p < .05 versus control) of the intercellular adherens junction protein, N-cadherin, and the active phosphorylated isoform of the principal gap junction protein, connexin 43 at more than 15 minutes of ischemia. Cellular redistribution of connexin 43 was also evidenced on immunohistochemistry. No change in integrin-β1, an extracellular matrix attachment protein, or in epicardial conduction velocity anisotropy was observed.
Conclusions
These data indicate that there are significant alterations in the structural integrity of the myocardium as well as gap and adherens junction protein expression with increasing global ischemia time. The changes occur coincident with previously observed significant decreases in postischemic functional recovery, but are not associated with altered expression of matrix binding proteins or electrical anisotropic conduction.
In previous studies we have shown that there are alterations in myocardial structure after ischemia and reperfusion [1, 2]. These morphologic alterations occurred coincident with significant decreases in postischemic functional recovery [2]. The modulation of postischemic functional recovery may occur at many levels; however, recent data suggest that changes occurring at the intercalated disc may play an important role [3, 4].
The intercalated disc functions as the site of electromechanical coupling within the myocardium. Principal components of the intercalated disc include the gap junctions that allow for direct communication between adjacent cells, and the adherens junctions that provide cell–cell adhesion [5, 6]. N-cadherin is the primary cell adhesion molecule found in the adherens junction and appears critical in maintaining the integrity of the intercalated disc structure, while connexin 43-α1 (Cx43[α1]) is the principal connexin expressed in the ventricle [5–8]. Cx43[α1] exists primarily in the phosphorylated active state; however, it has been shown in animal models that after ischemic injury, Cx43[α1] becomes dephosphorylated and nonfunctional [6, 9].
The appropriate organization of each of these junctional complexes within the intercalated disc is essential for myocardial tissue development as well as the coordinated contractile function of the heart [5, 6]. Disruption of the intercalated disc and subsequent alterations in cell–cell interactions have been implicated in several human cardiovascular diseases states, including ischemic and hypertrophic cardiomyopathy, atrial fibrillation, and congestive heart failure [8, 10, 11].
Apart from intercellular junctions, the interaction of cardiomyocytes with the extracellular matrix also serves an important role in both the structural and functional integrity of the myocardium and in the stabilization of force transmission during contraction [12]. Alterations in extracellular matrix proteins such as integrin-β1 during ischemia and reperfusion may also play a role in the modulation of postischemic functional recovery.
Whether changes in these gap junction, adherens junction, or matrix binding proteins occur during ischemia and reperfusion is unknown. In this study we examine the time course to the onset of morphologic changes in the myocardium after global ischemia and reperfusion by using histochemical and immunologic analysis to identify whether alterations in gap junction, adherens junction, and extracellular matrix proteins occur. We have also used electrophysiology to determine the impact of alterations occurring at the intercalated disc after ischemia and reperfusion on conduction velocity anisotropy.
Material and Methods
Animals
New Zealand white rabbits (n = 57; 20 weeks; 3 to 4 kg) were obtained from Millbrook Farm (Amherst, MA). All experiments were approved by the Beth Israel Deaconess Medical Center Animal Care and Use Committee and the Harvard Medical Area Standing Committee on Animals (Institutional Animal Care and Use Committee) and conformed to the US National Institutes of Health Guidelines Regulating the Care and Use of Laboratory Animals (NIH publication 5377-3, 1996).
Langendorff Perfusion
All rabbits were anesthetized with acepromazine (0.5mg/kg intramuscularly), followed by intravenous (IV) pentobarbital (50 mg/kg). They received heparin (200 U/kg IV) in a marginal ear vein. Langendorff retrograde perfusion was performed as previously described [1]. Hearts were paced through the right atrium so that heart rate was maintained at 180 ± 3 beats/min throughout the experiment using a Medtronic Model 5330 stimulator (Medtronic, Minneapolis, MN). Hemodynamic variables were acquired using the PO-NE-MAH digital data acquisition system with an Acquire Plus processor board, and left ventricular pressure analysis software (Gould, Valley View, OH) [1].
Experimental Protocol
Hearts were perfused for 30 minutes to establish equilibrium hemodynamics. Equilibrium was achieved when heart rate, coronary flow, left ventricular peak developed pressure, and end-diastolic pressure were maintained at the same level for three continuous measurement periods timed 5 minutes apart. Control hearts (n = 7) were perfused without global ischemia (GI) for 180 minutes. Rabbit hearts were subjected to 5 (GI-5; n = 7), 10 (GI-10; n = 6), 15 (GI-15; n = 7), 20 (GI-20; n = 8), 25 (GI-25; n = 6), or 30 minutes (GI-30; n = 6) of global ischemia, achieved by cross-clamping the perfusion line, followed by 120 minutes of reperfusion [1].
Determination of Interfibrillar Space
Myocardial tissue samples from eight predesignated, standardized regions within the left and right ventricles were obtained at the end of the experimental protocol and immediately fixed in 4% paraformaldehyde in phosphate-buffered saline (pH 8.0) overnight at 4°C. After embedding, tissue samples were sectioned (5 μm thickness). Thirty sections from each specimen were mounted on glass slides and divided sequentially into 10 groups of three and used for hematoxylin and eosin (H&E), and Masson’s Trichrome staining [1].
Interfibrillar space was determined using a Zeiss Axiovert 200M inverted microscope (Carl Zeiss, Thornwood, NY) fitted for bright-field, phase contrast, differential interference contrast, and fluorescence. Quantitative light microscopy spatial analysis was performed at low magnification (×100) covering the full specimen area using Metamorph Imaging Analysis 6.2 software (Universal Imaging, Downingtown, PA). All analysis was performed by a pathologist using blinded samples.
Immunohistochemistry
After tissue samples were embedded and sectioned, slides were baked overnight at 65°C, deparaffinized in xylene, rehydrated through a graded ethanol series, and subjected to antigen retrieval by heating three times for 5 minutes in 1 mM ethylene diamine tetraacetic acid (pH 8.0) using a 700-W microwave oven set to high.
Slides were immunohistochemically stained with one of the following primary antibodies: anti-Cx43[α1] (monoclonal antibodies [MAB] MAB3067, 1:100 dilution; Chemicon Internationals, Temecula, CA), anti-N-cadherin (CADHNabmX, 1:100 dilution, RDI, Concord, MA), or anti-integrin-β1 (MAB 1951, MAB 1965, and MAB 1981, 1:100 dilution, Chemicon Internationals; and integrin-β1 mouse monoclonal immunoglobulin [Ig] G1 antibody, 1:100 dilution, Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibodies were detected with species-appropriate Alexa488-conjugated secondary antibodies (Molecular Probes, Invitrogen, Carlsbad, CA) mixed with Alexa568-phalloidin and 4,’6-diamidino-2-phenylindole, dihydrochloride (DAPI, Molecular Probes; Invitrogen).
Slides were then mounted and visualized on a multi-point spinning disk confocal system (Atto, BD Biosciences, Rockville, MD) attached to a Zeiss Axiovert 200M microscope [13]. The confocal system and microscope were each illuminated with an X-Cite 120 mercury-halide light source and images were acquired by using either a CoolSNAP HQ (Photometrics, Huntington Beach, CA) or MicroMAX 1300YHS CCD camera (Prince-ton Instruments, Trenton, NJ) controlled with MetaMorph 6.2 software. Image processing was accomplished with MetaMorph 6.2 and Photoshop CS (Adobe, San Jose, CA).
Western Blot Analysis
Total protein was isolated from pooled tissue samples from apex, left, and right ventricular tissue samples obtained after 120 minutes of reperfusion using sodium deoxycholate, Nonidet P40 lysis buffer [14]. All protein lysis buffers contained the multiprotease inhibitor, Complete (2 mM; Boehringer, Mannheim, Germany) and the cell permeable caspase inhibitors z-DEVD.fmk, IETD.fmk and z-LEHD.fmk, 10 mM each, and the nonselective irreversible caspase inhibitor, Z-VAD.fmk, 50 mM (Kamiya Biomedical Co, Seattle, WA). Protein samples (25 μg) were fractionated on 12% Novex Tris-Glycine gels (Invitrogen) and then electroblotted to nitrocellulose membranes (Invitrogen). Protein equivalency, transfer efficiency, and membrane blocking were performed as previously described [1].
Immunoblotting was performed using one of the following antibodies: anti-N-cadherin (monoclonal antibody 610921, 1:1000 dilution, BD Biosciences, San Jose, CA); anti-integrin-β1 (MAB 1951, MAB 1965, and MAB 1981, each at 1:2000 dilution, Chemicon Internationals, Temecula, CA; and integrin-β1 mouse monoclonal IgG1 antibody, 1:2000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA); and anti-Cx43[α1] (monoclonal antibody 35–5000; or polyclonal antibody 71–0700, 1:1000 dilution each, Zymed Laboratories, San Francisco, CA). The Cx43[α1] antibodies used detect phosphorylated isoforms of Cx43[α1], migrating at approximately 46 kDa, and dephosphorylated isoforms, migrating at 41 kDa [3, 15]. Blots were detected using ECL Plus (Amersham Pharmacia Biotech, Piscataway, NJ.) with species-appropriate secondary antibodies. Densitometric analysis was performed using the SigmaGel gel analysis software (Jandel Scientific, San Rafael, CA).
Conduction Velocity Measurements
Epicardial conduction velocities were measured at 30, 60, 70, 90, 120, 150, and 180 minutes of perfusion using a simplified method of Schalij and colleagues [16]. Hearts were exposed to either zero (control) or 30 minutes of global ischemia. The temperature-controlled perfusion system resulted in a mean ± SD average epicardial temperature among experiments of 35.3° ± 0.7°C with a maximum allowable temperature deviation of ±0.5°C within an experiment. A small electrode plaque consisting of a single 1.3-mm-diameter coaxial stimulation electrode (Harvard Apparatus, Holliston, MA) and three 0.8-mm-diameter silver-wire recording electrodes arranged in a square array with an interelectrode distance of 5 mm was placed into light contact with the anterior left ventricle, with one edge containing the stimulation electrode aligned parallel to the left anterior descending artery (LAD).
The heart was continuously epicardially paced at 300-millisecond cycle length (200 beats/min) to ensure continuous capture above the sinus rate, with pulse strength 2 × threshold at 1 millisecond duration. Pseudounipolar electrograms were recorded at the three epicardial recording electrodes, with a remote aortic band electrode serving as the indifferent electrode. Signals were amplified (10× gain, CyberAmp 380, Molecular Devices, Sunnyvale, CA), A/D was converted at 10 kHz and 16-bit resolution (PowerLab SP8, ADInstruments, Colorado Springs, CO) and continuously displayed and stored to computer disk (Chart 5 software 5.1, ADInstruments).
For offline analysis, unipolar signals were low-pass filtered at 100 Hz and their first derivative calculated. Conduction times to each recording electrode were measured between the onset of the stimulus artifact and time of minimum derivative by means of electronic calipers; five consecutive conduction times were averaged at each designated time-point and divided by the stimulating-to-recording electrode distance to calculate conduction velocity along each vector. Conduction velocities perpendicular to the LAD were faster than parallel to it and were taken to approximate conduction velocities along the longitudinal (CVL) and transverse (CVT) axes of epicardial myocardial fiber orientation, respectively. Conduction velocity at a 45° angle between these was consistently intermediate. Anisotropic ratios were calculated as CVL/CVT.
Wet Weight/Dry Weight Ratios
Determination of wet weight-to-dry weight ratios was performed as previously described [1].
Statistical Analysis
Statistical analysis was performed using the SAS 6.12 software package (SAS Institute, Cary, NC). The mean ± the standard error of the mean for all data was calculated for all variables. Statistical significance was assessed using repeated measures analysis of variance (ANOVA) with group as a between-subjects factor and time as a within-subjects factor. Dunnett’s test was used for comparisons between control and other groups to adjust for the multiplicity of tests. Bonferroni correction was used for comparisons between groups other than control. A one-way ANOVA was used for interfibrillar space and protein expression analysis. Statistical significance was claimed at p < 0.05.
Results
Histochemical Analysis and Interfibrillar Space Determination
Masson Trichrome and H&E stained myocardial sections of left ventricular tissue sections from control, GI-5, and GI-10 hearts showed preserved myofilament structure with minimal evidence of contraction bands or interfibrillar separation (Fig 1A). An increasing number of contraction bands were evident as global ischemia time increased, with the most prominent bands being seen in GI-30 hearts. After GI-15, tissue disruption and longitudinal and transverse interfibrillar separation were evident, as well as complete fiber disruption in some areas (Fig 1A). Metamorph analysis indicated that longitudinal and transverse interfibrillar space was progressively and significantly increased from GI-15 to GI-30 (p < 0.001 vs. control, Fig 1B), such that GI-30 > GI-25 > GI-20 > GI-15 (p < 0.001 for all comparisons).
Fig 1.
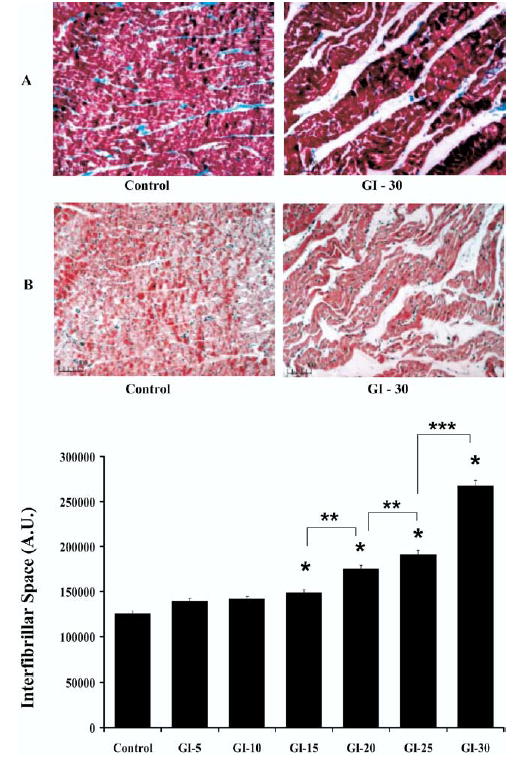
(A) Histochemical analysis. Representative Masson Trichrome (Panel A) and hematoxylin and eosin (Panel B) stained myocardial sections (×100 original magnification) of left ventricular tissue sections from control and hearts after 30 minutes of global ischemia (GI-30). Scale bars are shown in microns. Histochemistry shows control hearts with preserved myofilament structure and GI-30 hearts with longitudinal and transverse interfibrillar separation. (B) Interfibrillar space. Quantitative light microscopy spatial analysis was performed at low magnification (×100 original magnification) covering the full microscopic slide specimen area using Metamorph Imaging Analysis software and expressed as interfibrillar space in arbitrary units (AU). All results are shown as mean ± SEM for n = 30 for each sample. Significant differences are shown as *p < 0.001 versus control; **p < 0.001 versus GI-20; and ***p < 0.001 versus GI-25.
Expression of N-cadherin, Integrin-β1 and Cx43[α1]
Immunohistochemistry using confocal microscopy demonstrated qualitative reductions in expression of N-cadherin and Cx43[α1] after more than 15 minutes ischemia (Fig 2A,C). In control hearts, Cx43[α1] was localized predominantly to the intercalated disc but was redistributed to the periphery of the myocyte after more than 15 minutes of global ischemia (Fig 2C). Additionally, immunohistochemistry revealed increasingly fragmented Cx43[α1] in GI-20, GI-25, and GI-30 hearts compared with the intact Cx43[α1] seen in control hearts. There was no observed alteration in the expression of integrin-β1 (Fig 2B).
Fig 2.
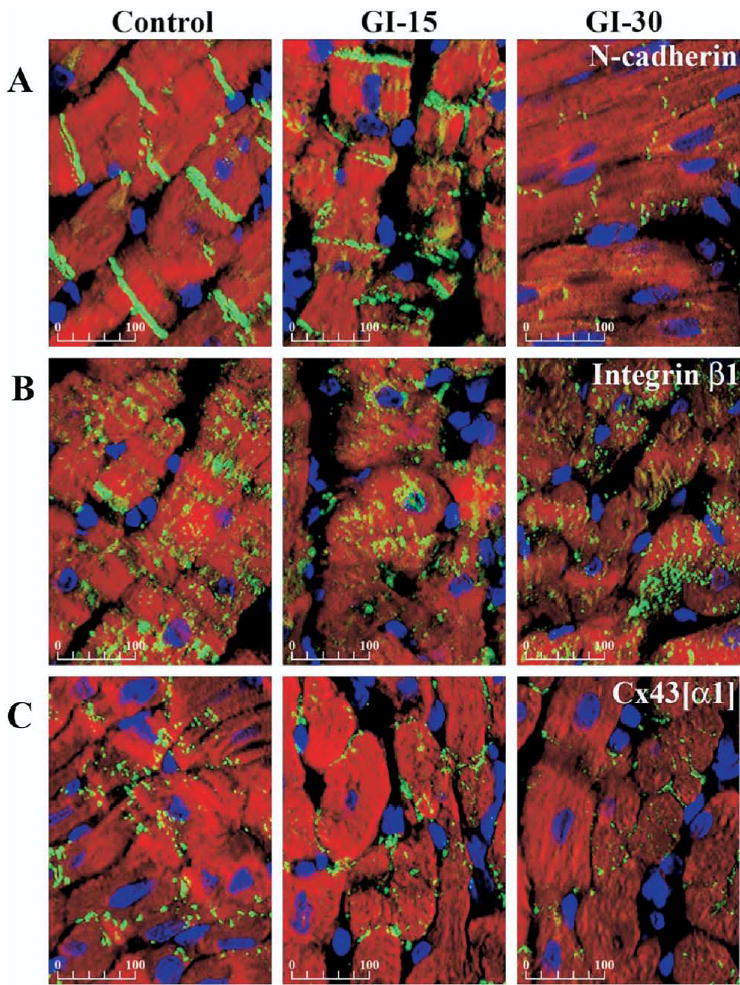
Immunohistochemical analysis. Representative confocal photomicrographs (×40 original magnification) of immunohistochemically stained left ventricular free wall sections from control and hearts after 15 and 30 minutes of global ischemia (GI). Overlay shows nuclei as blue, myocytes as red (F-actin stained with Alexa568-phalloidin), and N-cadherin (A), integrin-β1, (B) and Cx43[α1] (C) as green. Scale bars are shown in microns. Photomicrographs show a qualitative decrease in N-cadherin and Cx43[α1] at GI-15 and GI-30. Cx43[α1] was localized predominantly at the intercalated disc in control, but was redistributed to the periphery of the myocyte after more than 15 minutes global ischemia. There was no change in integrin-β1.
Western blot analysis revealed progressive and significant reductions of N-cadherin (p < 0.001 versus control) after more than 15 minutes of ischemia and in Cx43[α1] (p < 0.001 versus control) after more than 20 minutes of ischemia (Fig 3). Pooled samples of tissue from left and right ventricle as well as apex were used for study, because the entire heart is at risk for ischemic injury in our global ischemia model. Analysis with a monoclonal anti-Cx43[α1] antibody revealed a single band illustrating significant reductions in Cx43[α1] after more than 20 minutes of global ischemia (p < 0.001 versus control; Fig 4). Analysis with a polyclonal anti-Cx43[α1] antibody showed significant decreases in the phosphorylated form of Cx43[α1] after more than 20 minutes of global ischemia (p < 0.001 versus control; Fig 3).The dephosphorylated form of Cx43[α1] decreased with increasing ischemia time; however, this change did not approach statistical significance (p = 0.075 versus control; Fig 3). No significant change in integrin-β1 was detected (p = 0.410; Fig 3), in agreement with histologic analysis.
Fig 3.
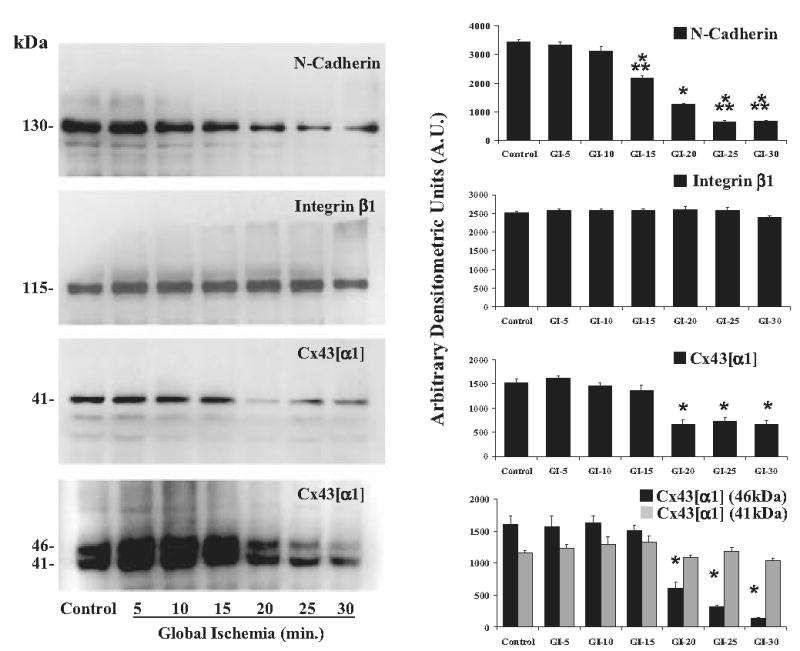
Western blot analysis. Representative Western blots for N-cadherin, integrin-β1, and Cx43[α1] and Cx43[α1] phosphorylated Cx43[α1] (46 kDa) and dephosphorylated Cx43[α1] (41 kDa). Molecular weights (kDa) for each band are shown. Densitometric analysis for n = 4 blots each is shown on the right. Results show a significant decrease in N-cadherin at 15 minutes of global ischemia (GI) and Cx43[α1] and phosphorylated Cx43[α1] (46kDa) at GI-20 (p < 0.0001 versus control). There were no significant changes in integrin-β1 All results are shown as mean ± SEM. Significant differences are shown as *p < 0.001 versus control, and **p < 0.001 versus GI-20.
Fig 4.
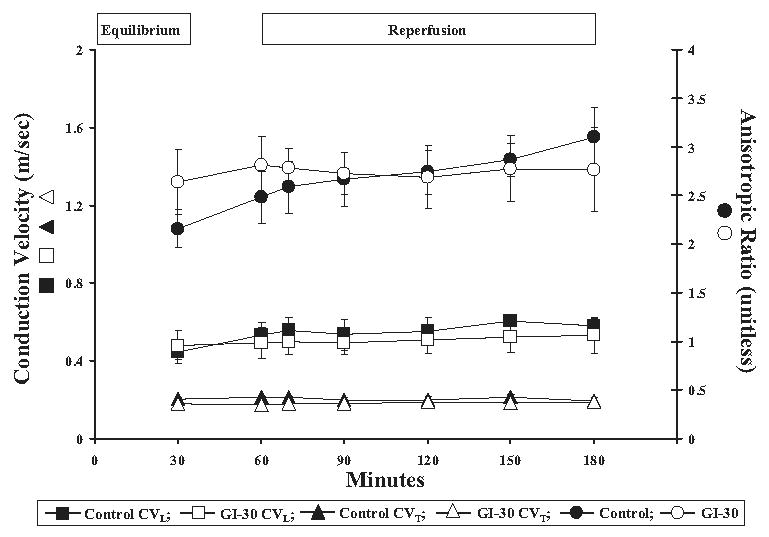
Conduction velocity (CV) analysis. Epicardial conduction velocities were measured at 30 (equilibrium), 60, 70, 90, 120, and 180 minutes of perfusion along the longitudinal (CVL) and transverse (CVT) axes of epicardial myocardial fiber orientation. Anisotropic ratios (AR) were calculated as CVL/CVT. No significant change was seen in CVL, CVT or AR between global ischemia (GI) after 30 minutes and control groups at equilibrium or for the duration of perfusion time (p = 0.903 for CVL; p = 1.0 for CVL; p = 0.586 for AR). All results are shown as mean ± SEM for n = 5 in each group.
Anisotropic Conduction
Conduction velocities along transverse and longitudinal axes did not differ between control and GI-30 hearts at equilibrium or for the duration of perfusion time (p = 1.0 for GI-30 CVT versus control CVT; p = 0.903 for GI-30 CVL versus control CVL; Fig 4). Similarly, there was no difference in the anisotropic ratio found between GI-30 and control hearts either at equilibrium or throughout reperfusion (p = 0.586; Fig 4).
Wet Weight/Dry Weight Ratios
No significant difference in the wet weight/dry weight ratio was observed within or between groups (p = 0.541; results not shown).
Comment
Our data indicate that after global ischemia and reperfusion there is a significant increase in the myocardial interfibrillar space and cellular redistribution and a significant decrease in the active phosphorylated form of the principal gap junction protein, Cx43[α1], and in N-cadherin, the principal adherens junction protein. No change was detected in integrin-β1, an extracellular matrix attachment protein. These structural changes are not associated with alterations in conduction velocity anisotropy or in tissue edema but occur coincident with previously observed significant decreases in postischemic functional recovery as well as increases in myocardial apoptosis and necrosis [1].
In our studies we have used a variety of commercial antibodies. Using a monoclonal anti-Cx43[α1] antibody, we were unable to detect the phosphorylated and dephosphorylated isoforms of Cx43[α1] and were only able to detect a single band migrating at approximately 41kDa that was significantly decreased after 20 minutes of ischemia (Fig 3). Additional Western blot analysis using a polyclonal anti-Cx43[α1] antibody did allow for identification of both the phosphorylated and dephosphorylated isoforms of Cx43[α1]. Our results indicate that with increasing ischemia there is a transition in Cx43[α1] from the active phosphorylated isoform to the inactive dephosphorylated isoform, in agreement with previous reports by others [3, 9, 17]. This change in phosphorylation status would be expected to limit or inhibit gap junction function and significantly impact postischemic functional recovery after ischemia and reperfusion.
Supporting this hypothesis is the work of Beardslee and colleagues [3], who have shown that as little as 15 minutes of ischemia is associated with dephosphorylation of Cx43[α1]. These authors demonstrated that the dephosphorylation of Cx43[α1] occurs coincident with increased tissue resistance attributed to gap junction uncoupling and proposed that Cx43[α1] was dephosphorylated with ischemia and was partially rephosphorylated with reperfusion. It is important to note that only a partial rephosphorylation of Cx43[α1], occurring in two of four hearts as evidenced by Western blotting performed on individual tissue samples, was reported [3]. In our studies, the level of phosphorylated Cx43[α1] was significantly decreased after ischemia and reperfusion. It is possible that there were greater decreases after ischemia only and that during reperfusion some partial rephosphorylation did occur, however, overall levels of phosphorylated Cx43[α1] isoform remain significantly decreased in our model.
We also show a significant decrease in N-cadherin, the principal adherens junction protein [5, 6]. This is in agreement with others who have also shown, in animal models, that ischemia results in a decrease in the number of adherens junctions, and thus N-cadherin, in the heart [4, 18]. As the adherens junction is involved in maintaining attachments between adjacent cardiomyocytes, the decreased expression of N-cadherin has implications on altering cell–cell attachments and, subsequently, altering mechanical coupling between cardiomyocytes (6). Our data are in agreement with Kostetskii and colleagues [5] who used a conditional N-cadherin knock-out model within the adult mouse myocardium to show that complete loss of N-cadherin expression resulted in the subsequent decreased expression of intercalated disc components, such as Cx43[α1], and the loss of any visible intercalated disc structures [5]. We have shown in our study that the decrease in N-cadherin precedes the decrease in expression of Cx43[α1], thus supporting the contention that Cx43[α1] expression is, to some extent, dependent on N-cadherin expression [5, 19].
Because connexins are fundamentally involved in the propagation of electrical activity from cell to cell, we sought to quantify whether the changes in Cx43[α1] phosphorylation and cellular redistribution after ischemia and reperfusion resulted in altered electrical conduction on a macroscopic scale. Measurement of conduction velocity using extracellular epicardial electrograms did not demonstrate statistically different conduction velocity anisotropy in GI-30 rabbits compared with controls.
These findings may be attributed to the relatively large physiologic reserve of gap junctions that exists the myocardium [20, 21]. It is possible that although our reduction in Cx43[α1] was statistically significant, it was insufficient to result in a functional conduction deficit. Although the lack of change in anisotropy seems at odds with the peripheral redistribution of Cx43[α1] that we observed after ischemia, it is possible that the expected reduction in anisotropy was countered by an increase in discontinuous, or zig-zag, conduction between myofibrils imposed by the concomitant increase in interfibrillar spaces, which we also observed.
One limitation to our study is that we examined total protein extracts from transmural samples of myocardium. Poelzing and colleagues [22] have shown that there is heterogeneous expression of Cx43[α1] within transmural wedge sections of canine myocardium, with subepicardial layers expressing a lesser amount of Cx43[α1] than deeper layers [22]. Using optical mapping, they found that this differential expression of Cx43[α1] translated into slowed conduction velocities within the subepicardium compared with deeper layers [22]. It is possible that the reductions in Cx43[α1] that we have detected occur as a result of differential reductions in different muscle layers of the myocardium. Additionally, we have only measured epicardial electrophysiologic recordings, and it is possible that had we been able to investigate conduction in each layer individually, we may have detected more significant electrophysiologic changes.
It is important to note that our results indicate that there was no alteration of integrin-β1, an extracellular matrix attachment protein. The maintenance of cell–matrix interactions during ischemia is clinically relevant because it affords the potential for full functional recovery after cardiac surgery. If extracellular matrix attachment were compromised, an irreversible end point of cell death would occur, with no possibility of restoration of normal cell structure and function [23].
Our studies were performed in an isolated perfused heart model, and further studies using an in situ blood perfused model are required to validate findings; however, our data provide important insight regarding the functional impact of alterations in intercalated disc components resulting from ischemia and reperfusion. In this study, significant changes in key gap junction and adherens junction proteins as well as in structural integrity together were insufficient to result in a parallel deterioration in electrical conduction, allowing for the possibility of pharmacologic or surgical intervention.
Footnotes
Presented at the Poster Session of the Forty-second Annual Meeting of The Society of Thoracic Surgeons, Chicago, IL, Jan 30-Feb 1, 2006.
References
- 1.McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia/reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:H1923–35. doi: 10.1152/ajpheart.00935.2003. [DOI] [PubMed] [Google Scholar]
- 2.McCully JD, Wakiyama H, Cowan DB, Federman M, Parker RA, Levitsky S. Diazoxide amelioration of myocardial injury and mitochondrial damage during cardiac surgery. Ann Thorac Surg. 2002;74:2138–46. doi: 10.1016/s0003-4975(02)04348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beardslee MA, Lerner DL, Tadros PN, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–62. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 4.Matsushita T, Oyamada M, Fujimoto K, et al. Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ Res. 1999;85:1046–55. doi: 10.1161/01.res.85.11.1046. [DOI] [PubMed] [Google Scholar]
- 5.Kostetskii I, Li J, Xiong Y, et al. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96:346–54. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- 6.Zuppinger C, Eppenberger-Eberhardt M, Eppenberger HM. N-cadherin: structure, function and importance in the formation of new intercalated disc-like cell contacts in cardiomyocytes. Heart Fail Rev. 2000;5:251–7. doi: 10.1023/A:1009809520194. [DOI] [PubMed] [Google Scholar]
- 7.Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. 2000;384:205–15. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- 8.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic hearts. Circulation. 1993;88:864–75. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- 9.Huang XD, Sandusky GE, Zipes DP. Heterogeneous loss of connexin43 protein in ischemic dog hearts. J Cardiovasc Electrophysiol. 1999;10:79–91. doi: 10.1111/j.1540-8167.1999.tb00645.x. [DOI] [PubMed] [Google Scholar]
- 10.Dupont E, Matsushita T, Kaba RA, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–71. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 11.Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002;54:361–79. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- 12.Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. 2004;279:12001–4. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- 13.Cowan DB, Lye SJ, Langille BL. Regulation of vascular connexin43 gene expression by mechanical loads. Circ Res. 1998;82:786–3. doi: 10.1161/01.res.82.7.786. [DOI] [PubMed] [Google Scholar]
- 14.McCully JD, Lotz MM, Krukenkamp IB, Levitsky S. A brief period of retrograde hyperthermic perfusion enhances myocardial protection from global ischemia: association with accumulation of Hsp 70 mRNA and protein. J Mol Cell Cardiol. 1996;28:231–41. doi: 10.1006/jmcc.1996.0022. [DOI] [PubMed] [Google Scholar]
- 15.Nagy JI, Ionescu AV, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: implications from normal and connexin32 knockout mice. Glia. 2003;44:205–18. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schalij MJ, Lammers WJ, Rensma PL, Allessie MA. Anisotropic conduction and reentry in perfused epicardium of rabbit left ventricle. Am J Physiol. 1992;263:H1466–78. doi: 10.1152/ajpheart.1992.263.5.H1466. [DOI] [PubMed] [Google Scholar]
- 17.de Groot JR, Coronel R. Acute ischemia-induced gap junctional uncoupling and arrhythmogenesis. Cardiovasc Res. 2004;62:323–34. doi: 10.1016/j.cardiores.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi C, Araujo EG, Sato K, Selke FW. Biochemical and structural evidence for pig myocardium adherens junction disruption by cardiopulmonary bypass. Circulation. 2001;104:I319–24. doi: 10.1161/hc37t1.094519. [DOI] [PubMed] [Google Scholar]
- 19.Wei CJ, Francis R, Xu X, Lo CW. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J Biol Chem. 2005;280:19925–36. doi: 10.1074/jbc.M412921200. [DOI] [PubMed] [Google Scholar]
- 20.van Rijen HV, Eckardt D, Degen J, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–55. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- 21.Danik SB, Liu F, Zhang J, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–41. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poelzing S, Akar FG, Baron E, Rosenbaum DS. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am J Physiol Heart Circ Physiol. 2004;286:H2001–9. doi: 10.1152/ajpheart.00987.2003. [DOI] [PubMed] [Google Scholar]
- 23.Meredith JE, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–61. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]