

Abstract
Objectives
Understand anatomical and clinical correlatives to coarctation in right aortic arch.
Background
Coarctation of the aorta is rare in patients with a functional right aortic arch. We reviewed a single institutional experience, examining associated diagnoses, diagnostic methodology, and surgical approaches.
Methods
A retrospective study was performed of our echocardiographic, magnetic resonance imaging, catheterization, and surgical databases from 1988 to 2001.
Results
Of 240 patients with right aortic arch, 10 (4.1%) had coarctation, constituting 1.9% of all native coarctations (n = 524). Nine (90%) had long-segment hypoplasia. Six (60%) had an aberrant left subclavian artery or retroesophageal diverticulum, 3 (30%) had mirror image branching, and 1 (10%) had a double arch with an atretic left arch. Other congenital heart defects were seen in 6 (60%) comprising 3 with ventricular septal defects, and one each with double-outlet right ventricle, cor triatriatum, and pulmonary valve abnormality. No patients with long-segment hypoplasia had bicuspid aortic valve. Six (60%) had vascular rings, and 5 (50%) had other associated syndromes. Magnetic resonance imaging and/or echocardiography successfully diagnosed all of these patients. Although long-segment right aortic arch coarctation courses behind the trachea posteriorly, only 2 needed an extra-anatomic (jump) graft; the remainders were repaired with patch angioplasty.
Conclusion
Coarctation with right aortic arch is rare, constituting 4.1% of all patients with right aortic arch, compared with 5–8% of patients with left aortic arch and congenital heart disease. Nearly all had long-segment hypoplasia without bicuspid aortic valve, and half were part of other syndrome complexes. This association can be diagnosed noninvasively and can often be repaired by patch angioplasty.
Keywords: Aortic Coarctation, Right Aortic Arch, Congenital Heart Disease
Introduction
Coarctation of the aorta is one of the most common forms of congenital abnormality of the great vessels that is brought to clinical attention. It is estimated to have an incidence of approximately 400 per million live births,1 comprising about 6–8% of children with congenital heart disease.2 As many cases of coarctation are not discovered until late into childhood, or even adulthood, these are likely underestimates of the true incidence of this disorder.
Right aortic arch is by itself a rare condition, comprising approximately 0.1% of cases seen in several studies.3 These cases can be accompanied by other congenital malformations of the cardiovascular system, most notably tetralogy of Fallot and other disorders of the conotruncus.4 The combination of right aortic arch and coarctation is exceedingly rare; however, there have been a number of reports, beginning as early as the late 1960s.5–8 A more recent review of cases of right aortic arch associated with vascular obstruction noted a number of cases in the literature;3 however, all of those with coarctation were single-case reports, save 1 with 2 cases.8 More recent reports have also consisted of descriptions of 1 or 2 cases, occasionally in association with other anomalies or syndromes.9–12 Finally, an ideal surgical approach for these patients has been defined through only a small number of cases.13
We undertook the present study to review our institutional experience in this disorder. In addition, we sought to address questions raised by earlier reports, particularly regarding the etiology of this disorder, assessment of associated clinical findings, and the evaluation and treatment of these patients.
Methods
Definitions
In working to review our institutional experience, we set out to define our cases as follows. A right aortic arch was a single, patent aortic arch that crossed over the right mainstem bronchus. Coarctation was limited to a narrowing of the aortic arch that led to a clinical reduction in the pulse or blood pressure of the lower extremities prior to any clinical intervention. Narrowing in reconstructed aortic arches was excluded as well as interruption of the aortic arch that is believed to be a different disease, and was also considered separately in a recent review.3
Review of Cases
Clinical records at The Children’s Hospital of Philadelphia from 1988 to 2001 were reviewed and correlated with examinations of our databases of echocardiograms, catheterizations, and cardiac magnetic resonance imaging (MRI). The use of diagnostic modalities varied over the course of the study period, with an increased reliance on MRI and a reduction in the use of catheterization in the more recent cases.
Echocardiography
A comprehensive 2-dimentional and Doppler echocardiographic examination was performed on all patients. Hewlett Packard 77020 phased array ultrasound systems (Sonos 500, 1000, and 2500) with 2.5–7.0 megahertz transducers were used. All studies were recorded on 0.5-inch VHS format videotapes, and were available for retrospective analysis. Visualization of all parts of the heart and great vessels were attempted, utilizing subcostal, apical, parasternal, and suprasternal views.
Magnetic Resonance Imaging
Studies were performed on the Siemens SP-63 or Vision whole-body magnetic resonance systems. After obtaining localizers, contiguous, electrocardiogram-triggered, T1-weighted transverse images were obtained spanning the region of the heart and great vessels. The effective repetition time was the R–R interval (range 350–1000 milliseconds), the echo time was 15 milliseconds, the number of excitations was 3, the image matrix sizes were 128 × 256 pixels interpolated to 256 × 256 pixels, the field of view ranged from 250 to 450 mm, and the slice thickness ranged from 4 to 10 mm. These images were used to evaluate anatomy, and were the basis for 3-dimensional reconstruction. Cine 2-dimentional imaging was performed as previously described.14
Two types of 3-dimensional reconstruction were performed: multiplanar reconstruction and 3-dimensional shaded surface display. In multiplanar reconstruction, contiguous transverse images are stacked one atop the other, and oblique planes, including curved cuts, are selected to view salient parts of the anatomy. This calculation is performed in real time with interpolation, and the resulting images were saved for later analysis. For the 3-dimensional shaded surface display, images were transferred to a Sun SPARC station 10 (Sun Microsystems), and VIDA (Volumetric Image Display and Analysis) software was used to manipulate the images and create the display. Briefly, the process of creating the display involves labeling each cavity of interest on each image using threshold values of a unique pixel intensity, and then removing unlabeled pixels. These images are then stacked by the software, the edges extracted, interpolated, and smoothed, with a shading algorithm used to create the final 3-dimensional appearance of the structures in the image.
Catheterization
Cardiac catheterization was performed on patients as clinically indicated. The procedure followed was the routine for our institution, including right- and left-sided catheterization and angiography as indicated during the procedure. Images were acquired using the Toshiba biplane DPS 2000 system. Optiray350 (Mallinckrodt, St. Louis, MO, USA) was utilized for contrast angiography.
Results
Over the 13-year study period a total of 524 cases of native coarctation presented to our institution. During that same period, there were 240 cases of right aortic arches that were brought to clinical attention. Within the intersection of these groups there were a total of 10 patients that met criteria for inclusion in this study, which represented just over 1.9% of all native coarctations and 4.1% of all right aortic arches. Individual cases are listed in Table 1. Mean age at diagnosis was 25.7 ± 42.3 months (range 2 days–105/12 years), with all but 3 cases diagnosed under 18 months of age. The oldest patient (patient 1) was noted to have clinical signs of coarctation several years following repair of cor triatriatum. The other 2 patients older than 18 months at presentation (patients 3 and 6) did not have prior clinical difficulties and were diagnosed at 3⅚ years and 5⅓ years, respectively. Definitive repair of the coarctation was performed within a few days of diagnosis in all cases. The patients were evenly divided by sex.
Table 1.
Summary of Patients with Right Aortic Arch and Coarctation
| Patient | Arch Anatomy | DA | Vascular Ring | Long-segment Hypoplasia | Type of Arch Repair | Heart Disease | Other Diagnoses |
|---|---|---|---|---|---|---|---|
| 1 | Cervical RAA, ALSCA | R | No | Yes | Resection, patch angioplasty | Cor triatriatum | Azygous lobe of right lung |
| 2 | RAA, RED | L | Yes | Yes | Patch/tube graft | — | — |
| 3 | Cervical RAA, RED | R | Yes | Yes | Patch angioplasty | — | — |
| 4 | DAA, L atresia, R hypoplasia | — | Yes | Yes | Undocumented (not extra-anatomic) | Interrupted IVC, PV anomaly | Heterotaxy |
| 5 | RAA, RED | L | Yes | Yes | Patch angioplasty | Conoventricular VSD | Goldenhar |
| 6 | RAA, ALSCA | R | No | Yes | Extranatomic | — | — |
| 7 | RAA, RED | L | Yes | Yes | Extranatomic | — | Scalp hemangioma |
| 8 | RAA, MIB | L | Yes | Yes | Undocumented (not extra-anatomic) | Posterior malalignment VSD | DiGeorge |
| 9 | Cervical RAA, MIB | R | No | Yes | Patch angioplasty | DORV, valvar and subvalvar AS | Heterotaxy |
| 10 | RAA, MIB | R | No | No | Patch angioplasty | Conoventricular VSD, BAV | Goldenhar |
Patients with a dominant right aortic arch and native coarctation. The columns include precise arch anatomy (ALSCA indicates aberrant left subclavian artery; DAA, double aortic arch; MIB, mirror image branching; RAA, right aortic arch; RED, retroesophageal diverticulum), sidedness (R or L) of ductus arteriosus or ligament (DA), and presence of vascular rings or long-segment hypoplasia. It should be noted that cases with ALSCA and RED are often difficult to clearly differentiate clinically. Also listed is the type of operation, presence of additional congenital heart disease (AS indicates aortic stenosis; BAV, bicuspid aortic valve; DORV, double-outlet right ventricle; IVC, inferior vena cava; PV, pulmonary valve; VSD, ventricular septal defect), and other noncardiac diagnoses.
Branching and Arch Anatomy
All patients had “functional” right aortic arches; however, anatomically only 9 (90%) had a single, right-sided aortic arch, while 1 case had a double aortic arch with an atretic left arch and a relatively hypoplastic right-sided arch.
In terms of branching patterns, 2 patients (20%) had an aberrant (retroesophageal) left subclavian artery, where the left subclavian artery arises as the last branch off of the aorta and runs posterior to the esophagus. This is thought to occur as a result of the lack of development of the left fourth and sixth branchial arches.4 Another 4 patients (40%) had a retroesophageal (Kommerell’s) diverticulum, where the left subclavian artery arises off of a wide diverticulum, which is located posterior to the trachea and quickly narrows to the diameter of the subclavian artery as it runs to the left arm. Figure 1 shows selected transverse cuts from an MRI study of a patient with this anatomy, with projections of a 3-dimensional reconstruction of that study shown in Figure 2. Two frames of a coronal cine-MRI of this patient are shown in Figure 3, demonstrating the turbulence that resulted from the coarctation. The clinical significance of Kommerell’s diverticulum is that it forms a vascular ring completed by the remnants of a left-sided ductus arteriosus (ligamentum arteriosum). The final branching pattern observed was mirror image branching of the great vessels, seen in 3 cases (30%). This pattern is thought to result from the involution of the left sixth branchial arch, the right-sided correspondent of which is thought to disappear in normally branching left-sided aortic arches.
Figure 1.
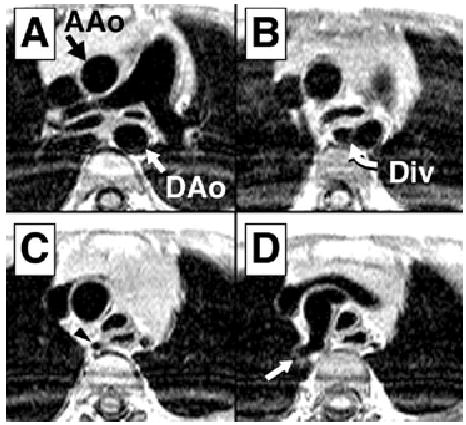
T1-weighted images from patient 7, moving caudally to cranially. In panel A, the ascending aorta (AAo) and descending aorta (DAo) are marked. Panel B demonstrates the retroesophageal diverticulum (Div). Narrowing of the aorta is shown in panel C (arrowhead) and right subclavian artery in panel D (arrow).
Figure 2.
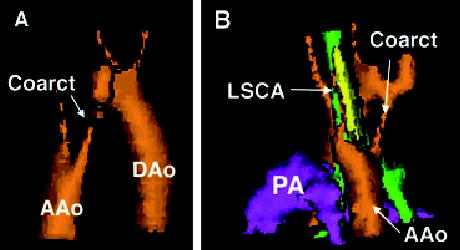
Three-dimensional reconstruction of the arch anatomy of patient 7, the most common arch we encountered. Panel A is right posterior oblique view and panel B is left posterior oblique. Both views show the coarctation (Coarct) and ascending aorta (AAo), with the descending aorta (DAo) seen well in panel A. In panel B, the left subclavian artery (LSCA) is shown arising from a retroesophageal (Kommerell’s) diverticulum. In this same panel, the trachea is green, the esophagus is yellow, and the pulmonary artery (PA) is purple.
Figure 3.
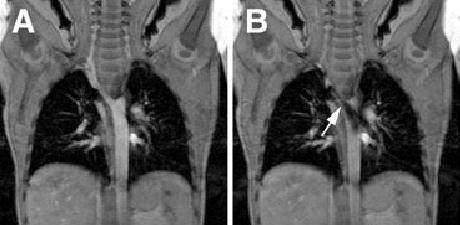
Coronal cine-MRI of patient 7. Panel A is from a frame without significant flow across the area of coarctation. Panel B is later in the cardiac cycle demonstrating significant turbulence in the posterior descending aorta, arising from the area of narrowing (arrow).
Vascular rings were noted in 6 cases (60%) and found anatomically, in the double aortic arch, in all cases of retroesophageal diverticulum, and 1 case of mirror image branching with a left-sided ductus arteriosus. Selected frames of an MRI of this latter patient are shown in Figure 4. Frames from a 3-dimensional maximum intensity projection magnetic resonance angiogram of this patient are shown in Figure 5.
Figure 4.
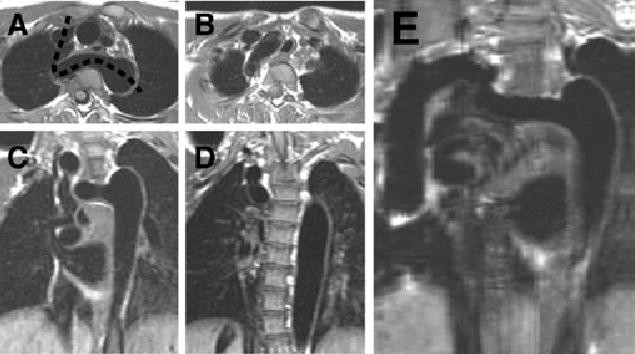
Several views of the arch of patient 8, including a curved-cut MRI. Panels A and B are selected T1-weighted axial images, moving caudally to cranially. Panels C and D are T1-weighted coronal images, moving anteriorly to posteriorly. Panel E represents the reconstruction along the plane of the dashed line in panel A. The tortuous nature of the arch, as well as the elongated area of narrowing, is well seen in this reconstruction.
Figure 5.
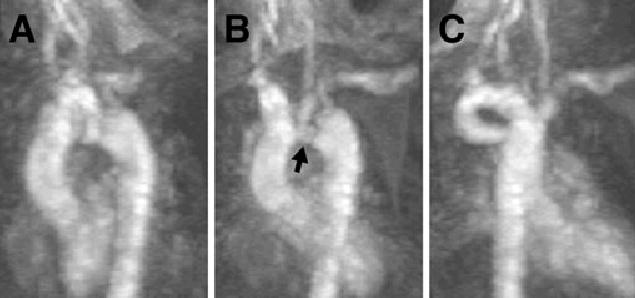
Selected frames from a 3-dimensional magnetic resonance maximum intensity projection of patient 8. Panels AC are selected frames, with a good demonstration the area of coarctation (arrow) in panel B, hidden from view in panels A and C. While difficult to appreciate in the still frames, the abnormality is easier to visualize in motion.
Long-segment hypoplasia of the transverse arch was seen in 9 (90%) and in only 1 case was there a discrete juxtaductal coarctation as is typically seen in left-sided coarctation of the aorta. This occurred in one of the cases of mirror image branching with an ipsilateral ductus arteriosus.
Associated Heart Disease and Other Diagnoses
The majority (60%) of cases in this series had additional congenital cardiac anomalies. An isolated ventricular septal defect was the most common, seen in 3 cases (30%), 2 of which were conoventricular, with the remaining defect posteriorly malaligned. In addition, 1 patient (patient 9) was noted to have double-outlet right ventricle with valvar and subvalvar aortic stenosis and patient 1 was noted to have cor triatriatum. Patient 4 with double aortic arch and atretic left arch had an abnormal pulmonary valve and an interrupted inferior vena cava. Finally, patient 10, with a discrete coarctation and a conoventricular ventricular septal defect, was the only patient noted to have a bicuspid aortic valve, making bicuspid aortic valve a rare occurrence (10%) in right-sided coarctation.
Half of our patients had associated known, well-defined syndromes. Two had heterotaxy syndrome, 2 had Goldenhar syndrome, and 1 had DiGeorge syndrome. One of our patients (patient 7) had a scalp hemangioma, which was at least evocative of a recent report by Wong and associates,12 which reported 2 patients with similar arch anomalies to our patient and multiple hemangiomas.
Diagnostic Studies and Treatment
Results of echocardiographic studies were available on all but 1 patient in this series. The proper arch sidedness was correctly diagnosed in all cases, as verified by MRI, catheterization, or operative report description. While arch branching pattern was noted on all of the studies, differentiation between a right arch with an aberrant left subclavian artery and a right arch with retroesophageal diverticulum could not be made by echocardiography; the diagnosis had to be made by MRI. Finally, the diagnosis of arch narrowing or coarctation was made by echocardiography in all but one of the patients in which echocardiograms were available. Patient 4, with double aortic arch, was not identified to have narrowing of the dominant right-sided arch by echocardiography.
Catheterization was performed on 7 patients; however, only 2 were performed after 1994, and both to evaluate issues other than the arch anatomy. The 4 most recent patients had MRI studies, of which 3 did not undergo catheterization before definitive treatment. In these latter patients, the MRI was able to evaluate the presence or absence of tracheal compression, which was not seen in catheterization or echocardiographic studies. Finally, there were no arch anatomical findings that were seen in echocardiography that were not seen in MRI. Examples of these studies for 2 of these patients are shown in Figures 1–5. Magnetic resonance imagings were performed on 4 patients. Anatomy delineated by MRI, in all cases, was confirmed at surgery.
Definitive treatment for all patients was surgery. Repair of coarctation along with other cardiac anomalies (when present) took place at the same time. Although long-segment coarctation of the right aortic arch courses behind the trachea and is often difficult to approach, in only 2 cases was an extra-anatomic (“jump”) graft required; the remainder were repaired with patch angioplasty or had other types of repairs.
Discussion
The cases described in this report are consistent with previous reports3,9,12 that included other congenital heart abnormalities along with the abnormal aortic arch architecture except that our study showed a higher frequency of congenital heart defects. Although the numbers are relatively small, our series was much larger than any of those previously reported
Because of the unusual anatomy in this type of aortic arch, ideal imaging planes are difficult to predict a priori. As such, the superior ability of MRI to reconstruct and reorient images as needed would suggest that in patients with right aortic arch and suspected coarctation, MRI is the ideal choice for initial diagnostic imaging. Given that MRI is most frequently utilized at this time for evaluation of vascular structures such as rings and coarctation,4,15–17 further recommendation for this technique in the setting of right aortic arch should not be surprising.
These results add more weight to the suggestion that these abnormalities are not simply the result of flow dynamics. Rudolph and colleagues suggest that flow dynamics may play a pivotal role in the development of aortic coarctation.18 Other mechanisms than flow, such as the role of neural crest investment of the great vessels,19 may be at play, yet there is no unified theory to account for this. If a significant role was played by neural crest, for example, more than a single case of DiGeorge syndrome should have occurred in our series. In addition, despite the fact that tetralogy of Fallot is one of the most common diagnoses with right aortic arch overall, no cases of tetralogy of Fallot, right aortic arch, and coarctation were found in our series. This is most likely due to the fact that a right-to-left shunt generally occurs at the ventricular level, increasing flow across the left ventricular outflow tract and aorta, which may decrease the susceptibility of these patients to coarctation. Coarctation, though, is not unheard of in tetralogy of Fallot.20,21
The lack of demonstration of bicuspid aortic valve in our patients with long-segment hypoplasia suggests that flow abnormalities in and of themselves may not be the cause of this aortic valvulopathy in the setting of coarctation. Similarly, alteration of flow volume by itself would not be expected to form different patterns of arch narrowing. Our data suggest that there is likely a significant patterning effect in the tissues of the right-sided aortic arches that developed long-segment hypoplasia in the setting of altered hemodynamics. Further, this patterning is different enough from the pattern of coarctation seen in left aortic arches as to suggest that they are different etiologic entities.
The combination of coarctation and right aortic arch is rare, and examination of this patient population brings to light several issues of interest to cardiologists, surgeons, and developmental biologists. Diagnosis of this clinical entity may be best accomplished through the use of MRI. The anatomical information obtained in this manner can be helpful to the operative approach. Finally, this study showed a strong relationship of this pattern of great vessel abnormality with a variety of other congenital problems in the heart. While varied in clinical phenotype, this suggests that major patterning defects of the heart and the great vessels are coupled in a complex manner, and may lead to clues regarding their regulation.
References
- 1.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 2.Beekman RH. Coarctation of aorta. In: Allen HD, Clark EB, Gutgesell HP, Driscoll DJ, editors. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents. Philadelphia, Pa: Lippincott, Williams & Wilkins; 2001. pp. 988–1010. [Google Scholar]
- 3.McElhinney DB, Tworetzky W, Hanley FL, Rudolph AM. Congenital obstructive lesions of the right aortic arch. Ann Thorac Surg. 1999;67:1194–1202. doi: 10.1016/s0003-4975(98)01325-3. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg PM. Aortic arch anomalies. In: Allen HD, Clark EB, Gutgesell HP, Driscoll DJ, editors. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents. hiladelphia, Pa: Lippincott, Williams & Wilkins; 2001. pp. 707–735. [Google Scholar]
- 5.Grossman LM, Jacoby WJ., Jr Right aortic arch and coarctation of the aorta. Dis Chest. 1969;56:158–160. doi: 10.1378/chest.56.2.158. [DOI] [PubMed] [Google Scholar]
- 6.Merin G, Borman JB, Aviad I, Maddock CR, Stern S. Double aortic arch associated with coarctation, ventricular septal defect and right ventricular out-flow tract obstruction. Successful surgical repair. Am J Cardiol. 1972;29:564–567. doi: 10.1016/0002-9149(72)90451-1. [DOI] [PubMed] [Google Scholar]
- 7.Price HL, Schieken RM. Right aortic arch with coarctation of the aorta. Chest. 1974;65:110–112. doi: 10.1378/chest.65.1.110. [DOI] [PubMed] [Google Scholar]
- 8.Honey M, Lincoln JC, Osborne MP, de Bono DP. Coarctation of aorta with right aortic arch. Report of surgical correction in 2 cases: one with associated anomalous origin of left circumflex coronary artery from the right pulmonary artery. Br Heart J. 1975;37:937–945. doi: 10.1136/hrt.37.9.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gil-Jaurena JM, Murtra M, Goncalves A, Miro L. Aortic coarctation, vascular ring, and right aortic arch with aberrant subclavian artery. Ann Thorac Surg. 2002;73:1640–1642. doi: 10.1016/s0003-4975(01)03370-7. [DOI] [PubMed] [Google Scholar]
- 10.Pettitt TW, Caspi J, Sandhu SK, Ward KJ, Hixon RL. Repair of coarctation in a right-sided circumflex retroesophageal aortic arch. Ann Thorac Surg. 2001;72:611–613. doi: 10.1016/s0003-4975(00)02407-3. [DOI] [PubMed] [Google Scholar]
- 11.McMahon CJ, Vick GW, 3rd, Nihill MR. Right aortic arch and coarctation: delineation by three dimensional magnetic resonance angiogram. Heart. 2001;85:492. doi: 10.1136/heart.85.5.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong CH, Wright JG, Silove ED, Willetts R, Brawn WJ. A new syndrome of multiple hemangiomas, right dominant double aortic arch, and coarctation. J Thorac Cardiovasc Surg. 2001;121:1207–1209. doi: 10.1067/mtc.2001.112627. [DOI] [PubMed] [Google Scholar]
- 13.Ad N, Vidne BA. Coarctation of the aorta with right aortic arch: surgical technique and new classification. Ann Thorac Surg. 1999;67:1125–1129. doi: 10.1016/s0003-4975(99)00047-8. [DOI] [PubMed] [Google Scholar]
- 14.Fogel MA, Hubbard A, Weinberg PM. A simplified approach for assessment of intracardiac baffles and extracardiac conduits in congenital heart surgery with two- and three-dimensional magnetic resonance imaging. Am Heart J. 2001;142:1028–1036. doi: 10.1067/mhj.2001.118469. [DOI] [PubMed] [Google Scholar]
- 15.Beekman RP, Hazekamp MG, Sobotka MA, et al. A new diagnostic approach to vascular rings and pulmonary slings: the role of MRI. Magn Reson Imaging. 1998;16:137–145. doi: 10.1016/s0730-725x(97)00245-2. [DOI] [PubMed] [Google Scholar]
- 16.van Son JA, Julsrud PR, Hagler DJ, et al. Imaging strategies for vascular rings. Ann Thorac Surg. 1994;57:604–610. doi: 10.1016/0003-4975(94)90552-5. [DOI] [PubMed] [Google Scholar]
- 17.Ho VB, Prince MR. Thoracic MR aortography: imaging techniques and strategies. Radiographics. 1998;18:287–309. doi: 10.1148/radiographics.18.2.9536478. [DOI] [PubMed] [Google Scholar]
- 18.Rudolph AM, Heymann MA, Spitznas U. Hemodynamic considerations in the development of narrowing of the aorta. Am J Cardiol. 1972;30:514–525. doi: 10.1016/0002-9149(72)90042-2. [DOI] [PubMed] [Google Scholar]
- 19.Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ Res. 1995;77:211–215. doi: 10.1161/01.res.77.2.211. [DOI] [PubMed] [Google Scholar]
- 20.Elami A, Rein AJ, Preminger TJ, Milgalter E. Tetralogy of Fallot, absent pulmonary valve, partial anomalous pulmonary venous return and coarctation of the aorta. Int J Cardiol. 1995;52:203–206. doi: 10.1016/0167-5273(95)02458-1. [DOI] [PubMed] [Google Scholar]
- 21.Gunthard J, Murdison KA, Wagner HR, Norwood WI., Jr Tetralogy of Fallot and coarctation of the aorta: a rare combination and its clinical implications. Pediatr Cardiol. 1992;13:37–40. doi: 10.1007/BF00788228. [DOI] [PubMed] [Google Scholar]