

Abstract
The memory loss in Alzheimer’s disease (AD) has been linked to cholinergic hypoactivity. Mutations in presenilin-1 (PS-1) may regulate cholinergic signaling, although their precise roles in cholinergic neurotransmission in AD are unsettled. Neuronal uptake of choline via the high affinity choline transporter (CHT1) is essential for cholinergic neurotransmission. CHT1 is a Na+-dependent, hemicholinium-3 (HC-3) sensitive choline transporter. Although cholinergic neurons in the nucleus basalis of Meynert are a major source of cholinergic projections for the cerebral cortex, it is unclear whether cortical neurons exhibit intrinsic CHT1 activity that is altered in AD. We now report that primary cortical neurons express intrinsic and biologically active CHT1, and that, in these neurons, CHT1-mediated choline uptake activity is significantly reduced in PS-1 M146V mutant knock-in mice. Further kinetic studies using HC-3 binding and cell surface biotinylation assays showed that the PS-1 mutation inhibits CHT1 mediated choline uptake by reducing the ligand binding affinity of CHT1 without significantly altering levels of CHT1 expression in the plasma membrane. Since human neocortex has recently been shown to possess intrinsic cholinergic innervation, our results indicate that alterations in CHT1-mediated high affinity choline uptake in cortical neurons may contribute to Alzheimer’s dementia.
Keywords: choline, high affinity choline transporter-1, presenilin-1, Alzheimer’s disease, hemicholinium-3, cerebral cortex
1. INTRODUCTION
Cholinergic neurotransmission is involved in regulation of many complex motor, autonomic, behavioral and cognitive functions (Dickinson-Anson et al., 2003; Warburton et al., 2003; Winkler et al., 1995). Profound loss of cholinergic function has been implicated in several neurodegenerative diseases, including Alzheimer’s disease (AD) (Auld et al., 2002; Coyle et al., 1983; Fine et al., 1985; Fischer et al., 1987; Gibbs and Aggarwal, 1998; Gottfries et al., 1983; Price et al., 1985; Price et al., 1993; Whitehouse et al., 1982). High affinity choline uptake, which is mediated by the activity of the high-affinity, Na+-dependent, hemicholinium-3 (HC-3) sensitive choline transporter-1 (CHT1), is generally believed to be the rate-limiting step in acetylcholine synthesis, and is essential for cholinergic transmission (Breer and Knipper, 1990; Guo et al., 1999a; Okuda and Haga, 2000; Okuda et al., 2000; Pascual et al., 1991; Zapata et al., 2000). Thus, understanding of the regulatory machinery of CHT1-mediated choline uptake should have significant implications in the understanding of normal cholinergic neurotransmission and pathogenesis of cholinergic hypoactivity in AD.
Mutations in presenilin-1 (PS-1) are responsible for majority of early onset familial AD, and have been shown to be involved in regulation of neuronal apoptosis and aberrant production and aggregation of amyloid β-peptide (Annaert et al., 1999; Borchelt et al., 1997; Cai et al., 2006; Capell et al., 2000; Chui et al., 1999; Duff et al., 1996; Guo et al., 1996; Guo et al., 1997; Guo et al., 1998; Guo et al., 1999c; Mattson et al., 1998; Scheuner et al., 1996). Mutations in presenilins have also been shown to regulate cholinergic signaling, although the precise roles of these mutations in in vitro and in vivo models of AD are still unsettled (De Sarno et al., 2001; Hartmann et al., 2004; Hernandez et al., 2001; Mattson et al., 1998; Pedersen et al., 1997; Vaucher et al., 2002). Using gene targeting and expression protocols, we previously generated and characterized presenilin-1 mutant M146V knock-in (PS-1 M146V KI) mice and found an increased vulnerability of neurons from these mice to insults relevant to the pathogenesis of AD (Guo et al., 1999a; Guo et al., 1999b; Guo et al., 1999c). Since the PS-1 mutation was targeted to the endogenous PS-1 locus and the mutant PS-1 protein was expressed at normal physiological levels, these M146V KI mice provided a physiologically relevant and pathologically meaningful model of familial AD. However, the alterations in cholinergic signaling in these animals have not been fully characterized.
We showed previously that expression of the presenilin-1 L286V mutation caused a nerve growth factor-independent reduction of ChAT activity in PC12 cells (Pedersen et al., 1997). Another study also demonstrated a prominent diminution in the density of cholinergic synapses in the frontal cortex and a reduction in the size of these synapses in the frontal cortex and hippocampus in mice transgenic for APP K670N/M671L and PS-1 M146L mutations (Wong et al., 1999). These results suggest that the cholinergic signaling can be affected by mutations in presenilin-1. Of importance, we recently found that Par-4, a pro-apoptotic protein that mediates the adverse effect of the mutant presenilin-1, may directly alter CHT1 mediated choline uptake (Xie et al., 2001; Xie and Guo, 2004). These results raised the possibility that choline uptake might be altered in the knockin mouse expressing the AD-linked PS-1 mutation.
Extensive neuronal cell death was found in the cerebral cortex in AD. Although cholinergic neurons associated with the nucleus basalis of Meynert are a major source of cholinergic projections for the entire cerebral cortex, several studies identified significant amount of choline acetyltransferase (ChAT) positive cell bodies in the cerebral cortex of various species of mammals and thus indicated that there was an intrinsic source of cholinergic innervation of the cerebral cortex, in addition to extrinsic sources (Eckenstein and Baughman, 1984; Houser et al., 1985; Johnston et al., 1981; Kristt, 1979). Although there was a perception that cholinergic innervation in the human cerebral cortex is exclusively of subcortical origin, recent studies that employ highly specific monoclonal antibody against choline acetyltransferase (ChAT) have shown that the human neocortex, like that in the mouse and the rat, also contain intrinsic cholinergic neurons (Benagiano et al., 2003). In surgical samples of human parietal association neocortex, a region severely affected in AD brain, specific ChAT immunoreactivity in the cytoplasmic compartments, the perikarya and the neuronal processes was detected in subpopulations of pyramidal neurons located in layers II and III (Benagiano et al., 2003). Because Na+-dependent, HC-3 sensitive high affinity choline uptake mediated by CHT1 is essential for cholinergic transmission, we examined if CHT1 is expressed in primary cortical neurons, and if CHT1-mediated choline uptake is altered in primary cortical neurons from PS-1 M146V KI mice. Our data showed that primary cortical neurons express intrinsic and biologically active CHT1 activity, and that, in these neurons, CHT1-mediated choline uptake activity is significantly reduced by the Alzheimer’s PS-1 M146V mutation, through a mechanism involving altered ligand binding affinity of CHT1.
2. RESULTS
CHT1 colocalizes with ChAT in primary cortical neurons and levels of CHT1 expression in the cell surface is not altered by PS-1 M146V mutation
We asked the question of whether primary cortical neurons express functional CHT1 protein, and if so, whether levels of CHT1 expression and/or its choline uptake activity in these neurons can be altered by the PS-1 M146V mutation linked to the pathogenesis of familial AD. Primary cultures of mouse cortical neurons were established (Fig. 1a) and levels of CHT1 were measured in 7-day old cultures by immunocytochemistry. As shown in Fig. 1(c), in both wild-type and PS-1 M146V mutant knockin mice, CHT1 immunoreactivity was observed in some but not all neurons, and was predominantly intracellular (left panel). No specific staining was observed when the immunostaining procedure was carried out without the primary CHT1 antibody (Fig. 1b, right panel). These results demonstrate the existence of CHT1 in the cell bodies of cortical neurons. We further counted the CHT1 positive neurons in 7-day old cultures of primary cortical neurons, and found that about 30% of cortical neurons from both wild-type and PS-1 M146V mutant mice were clearly immunostained with CHT1 (Fig. 1c). To further demonstrate that CHT1 is also expressed on the cell surface of these neurons, we measured CHT1 immunoreacted in 7-day old cultures using cell surface biotinylation assay and Western blot analysis. Biotinylated proteins were considered to be mostly expressed on the cell surface because they were greatly depleted of the intracellular marker actin (Fig. 1d, bottom panel) (Xie and Guo, 2004). As shown in Figure 1d (top panel) and Fig. 1(e), significant amount of CHT1 was found in biotinylated proteins in primary mouse cortical neurons from wild-type as well as PS-1 M146V mutant mice. Given that cholinergic hypoactivity is a prominent feature of AD, we predicted that the expression of CHT1 would be decreased in primary cortical neurons from PS-1 M146V KI mice. Contrary to our expectations, cell surface levels of CHT1 proteins in primary neurons from PS-1 M146V KI mice were not significantly different compared with those in wild-type animals (Fig. 1d and 1e). Using double-labeling immunocytochemistry, we further found that the CHT1 positive cortical neurons also expressed the classical cholinergic marker ChAT (Fig. 1f), indicating that the CHT1 expression was located in intrinsic cholinergic neurons in the cerebral cortex. These data suggest that cortical neurons have intrinsic CHT1 activity, and that the PS-1 M146V mutation is not likely to be involved in altering the levels of CHT1 expression on the cell surface.
Figure 1. CHT1 is present in primary cortical neurons and cell surface expression of CHT1 is not altered by PS-1 M146V mutation.
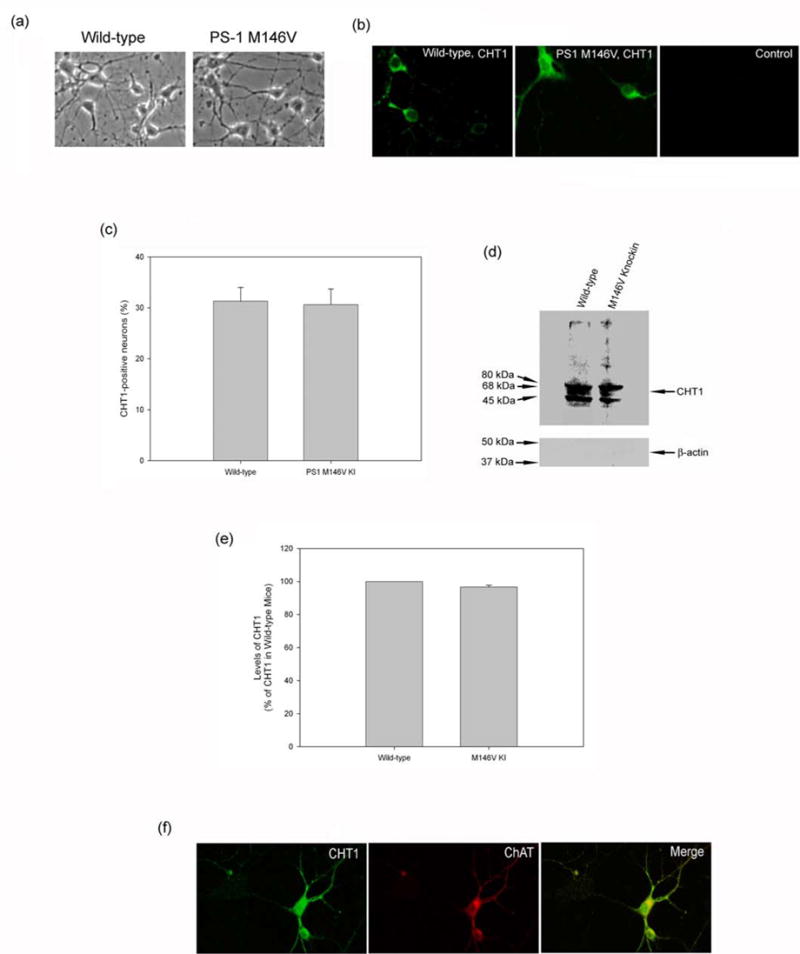
(a) Representative phase contrast microscope images of primary cultures of cortical neurons established from postnatal day 1 mouse pups of wild-type and PS-1 M146V mutant knockin mice. (b) Representative confocal laser scanning microscope images of CHT1 immunoreactivity in 7-day old primary cortical neurons from control wild-type and PS-1 M146V mutant knockin mice. CHT1 immunoreactivity was observed in some but not all neurons, and was predominantly intracellular (left panel). To control for non-specific staining, additional sections were subjected to the immunostaining procedure without the primary CHT1 antibody (right panel). (c) The percentage of CHT1 positive cells in 7-day old cultures of primary cortical neurons. Values are the mean and SE of determinations made in 6 separate cultures, and at least 120 neurons were examined per culture dish. Similar amounts of CHT1 positive neurons were observed in neuronal cultures from the wild-type and PS-1 M146V mutant knockin mice. (d) Representative Western blot analysis of biotinylated proteins showing CHT1 is also present in cell surface of primary mouse cortical neurons cultured for 7 days. Biotinylated proteins were considered to be mostly expressed on the cell surface because they were greatly depleted of the intracellular marker actin (bottom panel). (e) Statistical analysis of relative levels of cell surface CHT1 on Western blots in primary neurons from wild-type and PS-1 M146V mutant knock-in mice. Values are the mean and S.E. of determinations made in six separate Western blots. No significant difference was observed in the levels of cell surface CHT1 in neurons from wild-type and those from the PS-1 M146V KI mice. (f) CHT1 colocalizes with ChAT in cortical neurons. CHT1 and ChAT immunoreactivity was indicated in green and red fluorescence, respectively. Merged image shows the areas of colocalization of CHT1 and ChAT, as indicated in yellow fluorescence.
CHT1-mediated Na+-dependent and HC-3 sensitive choline uptake was significantly reduced in primary cortical neurons from PS-1 M146V KI mice
If CHT1 is expressed in cortical neurons, the next question is whether CHT1 is physiologically functional and mediates high-affinity choline uptake in those neurons. Since CHT1 is a Na+-dependent, HC-3 sensitive choline transporter, we examined whether Na+-dependent, HC-3 sensitive choline transporter activity is present in primary cultures of cortical neurons. As shown in Fig. 2, normal choline uptake (in the presence of Na+ but in the absence of HC-3) was clearly observed in these neurons. The sensitivity of the [3H] choline uptake to HC-3 was assessed by measuring [3H] choline uptake in the presence of 1 μM HC-3 in the culture media. Addition of HC-3 significantly reduced choline uptake by up to 64% (Fig. 2). The Na+ dependence of [3H] choline uptake was assessed using isotonic replacement of NaCl with LiCl in the culture media. Depletion of Na+ significantly reduced choline uptake by up to 76% (Fig.2). These data suggest that primary cortical neurons express significant amount of Na+-dependent, HC-3 sensitive choline uptake activity mediated by CHT1.
Figure 2. Na+-dependent and HC-3 sensitive choline uptake in mouse primary cortical neurons.
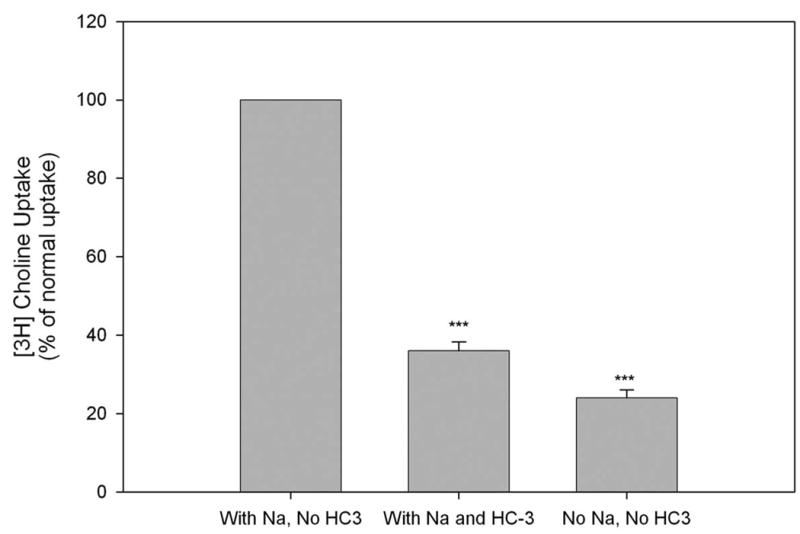
Primary cultures of cortical neurons from the wild-type mice were established and [3H] choline uptake was measured with 1 μM of [3H] choline in 7-day old cultures, and presented as percentage of normal uptake in the presence of Na+ but in the absence of HC-3. The sensitivity of [3H] choline uptake to HC-3 was assessed by measuring [3H] choline uptake in the presence of 1 μM HC-3. The Na+ dependence of [3H] choline uptake was assessed using isotonic replacement of NaCl with LiCl. Values are the mean and S.E. of determinations made in six separate cultures. ***P<0.001 compared with the value of normal choline uptake in the presence of Na+ but in the absence of HC-3. ANOVA with Scheffe’s post-hoc tests.
Next, we examined if HC-3 sensitive choline uptake was altered in neurons from PS-1 M146V KI mice. Surprisingly, although cell surface levels of CHT1 were not significantly altered by PS-1 M146V mutation (Fig. 1d), there was a significant and age-dependent decrease in HC-3 sensitive choline uptake in cortical neurons from PS-1 M146V mutant mice compared with that in neurons from the wild-type animals (Fig 3a). Further kinetic studies with increasing concentrations of [3H] choline showed that there was a significant decrease in specific [3H] choline uptake in primary cortical neurons from PS-1 M146V mutant mice, which resulted in a significant increase in Km in the mutant animals compared with those in wild-type mice (Fig 3b, Table 1). The change in Vmax values in PS-1 M146V mutant mice was not significantly different from that in wild-type mice (Fig 3b, Table 1). These results indicate that primary cortical neurons exhibit biologically active CHT1 choline transport activity that is significantly reduced by the PS-1 M146V mutation, possibly by a mechanism involving a decreased ligand binding affinity of CHT1.
Figure 3. Altered HC-3 sensitive choline uptake in primary neurons from PS-1 M146V knock-in mice.
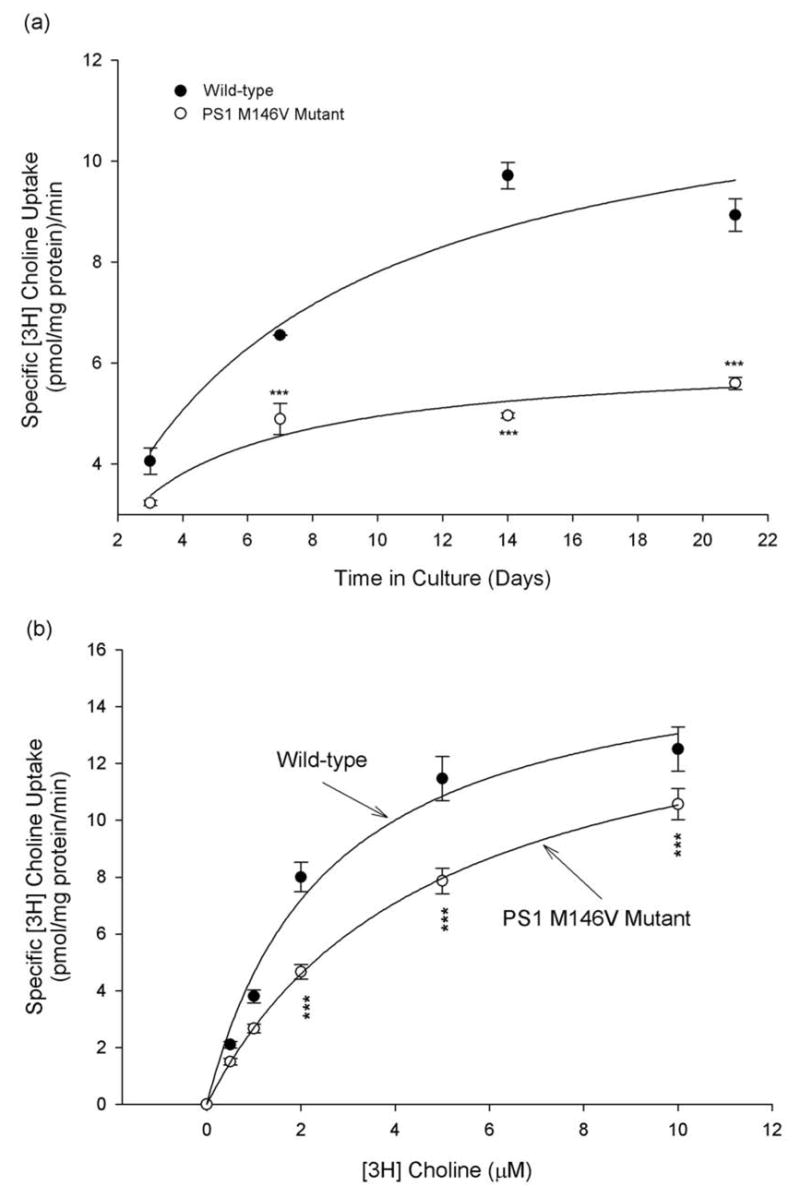
(a) Primary cultures of cortical neurons from wild-type and PS-1 M146V mutant mice were established and maintained for different time periods, and specific [3H] choline uptake was measured with 1 μM of [3H] choline, and quantified by subtracting non-specific uptake (as determined by the choline uptake in the presence of 1 μM HC-3) from the total uptake. Significant decline in specific choline uptake in cortical neurons from PS-1 M146V mutant mice was clearly observed. (b) [3H] choline uptake was analyzed in 4-day old cultures of primary cortical neurons from wild-type and PS-1 M146V mutant mice with increasing concentrations of [3H] choline, and specific [3H] choline uptake was quantified by subtracting non-specific uptake (as determined by the choline uptake in the presence of 1 μM HC-3) from the total uptake. ***P<0.001 compared with corresponding values in cells from the wild-type mice. ANOVA with Scheffe’s post-hoc tests.
Table 1. PS-1 M146V mutation alters functional properties of CHT1 in primary neurons.
Primary cultures of cortical neurons from wild-type and PS-1 M146V mutant mice were established and maintained for 4 days, and specific [3H] choline uptake was measured with increasing concentrations (0.5, 1.0, 2.0, 5.0 and 10 μM) of [3H] choline, and quantified by subtracting non-specific uptake (as determined by the choline uptake in the presence of 1 μM HC-3) from the total uptake. While no significant difference was observed in values of Vmax in neurons from wild type and PS-1 M146V mutant mice, a significant decrease in Km was observed in neurons from the PS-1 M146 mutant mice. Ligand binding affinity of CHT1 (Kd) and levels of CHT1 in the plasma membrane (Bmax) in primary neurons from the wild-type and PS-1 M146V mutant mice were measured using HC-3 binding assays. Expression of the PS-1 M146V mutation resulted in a significant increase in Kd compared with that in neurons from the wild-type mice. The difference in Bmax between wild-type and PS-1 M146V mutant neurons was not statistically significant. These results indicate that PS-1 M146V mutation may reduce the choline uptake by altering CHT1’s ligand binding affinity without significantly altering cell surface expression of CHT1.
| Neuronal Genotype | Choline Uptake | [3H] HC-3 binding | ||
|---|---|---|---|---|
| Vmax(pmol/mg protein/min) | Km(μM) | Bmax(fmol/mg protein) | Kd(nM) | |
| Wild-type | 16.4 ± 1.1 | 2.5 ± 0.4 | 17.7 ± 2.6 | 6.1 ± 2.1 |
| PS-1M146V mutant | 15.6 ± 2.5 | 4.8 ± 0.6*** | 15.4 ± 2.1 | 9.1 ± 2.5 *** |
P<0.001 compared with corresponding values in wild-type mice. ANOVA with Scheffe’s post-hoc tests.
PS-1 M146V mutation reduces the choline uptake in cortical neurons by altering the ligand binding affinity of CHT1
To further confirm that the PS-1 M146V mutation decreases CHT1-mediated choline uptake by reducing the ligand binding affinity of CHT1, we measured ligand binding affinity of CHT1 (Kd) using HC-3 binding assays (see Materials and Methods). Kinetic analysis of the HC-3 binding assays in primary cortical neurons from the wild-type and PS-1 M146V mutant mice demonstrated that expression of the PS-1 M146V mutation resulted in a significant increase in Kd, with no apparent alterations in Bmax, compared with those in neurons from the wild-type mice (Fig. 4, see also Table 1). The increase in Kd clearly suggest a significant decrease in CHT1’s affinity to its binding ligand. Because HC-3 is hydrophilic and impermeable to cell membrane, the HC-3 binding activity represents the expression level of the transporter in the plasma membrane (Xie and Guo, 2004). The observation that there was no significant difference in Bmax values among neurons from the wild-type mice and those from the PS-1 M146V mutant animals suggests that levels of CHT1 protein in the plasma membrane was not significantly altered by the PS-1 mutation (Fig. 4, and Table 1). These data were certainly consistent with those obtained from cell surface biotinylation/Western blot analyses shown in Fig. 1.
Figure 4. PS-1 M146V mutation reduces the choline uptake by altering CHT1’s ligand binding affinity without affecting levels of CHT1 on the cell surface.
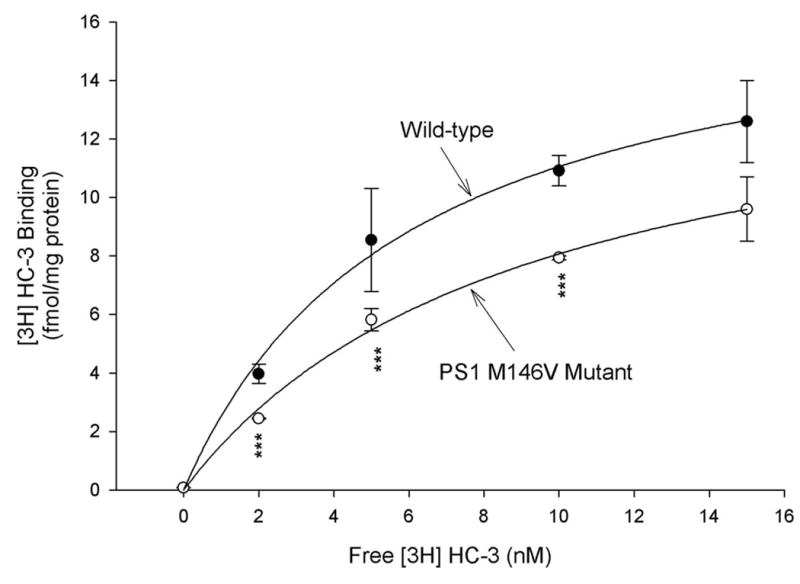
Primary cultures of cortical neurons from the wild-type and PS-1 m146V mutant mice were established. Ligand binding affinity of CHT1 (Kd) and levels of CHT1 in the plasma membrane (Bmax) were then measured using HC-3 binding assays described in Materials and Methods. Kinetic analysis indicated that expression of the PS-1 M146V mutation resulted in a significant increase in Kd, with no apparent alterations in Bmax in HC-3 binding assays (see also Table 1). ***P<0.001 compared with corresponding value in cells from the wild-type mice. ANOVA with Scheffe’s post-hoc tests.
3. DISCUSSION
Previous reports have showed a significant amount of choline acetyltransferase (ChAT) positive cell bodies in the cerebral cortex of mammals (Eckenstein and Baughman, 1984; Houser et al., 1985). Although whether human cerebral cortex contains intrinsic cholinergic neurons was a matter of debate, recent studies that employ highly specific monoclonal antibody against choline acetyltransferase (ChAT) confirmed that, in surgical samples of human parietal association neocortex (a region severely affected in AD brain), specific ChAT immunoreactivity was detected in the cell bodies in subpopulations of pyramidal neurons located in layers II and III (Benagiano et al., 2003). In this study, the expression of CHT1 on the cell surface of cortical neurons was confirmed using cell surface biotinylation assay and Western blot analysis. Furthermore, the CHT1 positive cortical neurons also expressed the classical cholinergic marker ChAT, indicating that CHT1 activity was located in cortical cholinergic neurons. These data provides additional supporting evidence that there is indeed an intrinsic source of cholinergic innervation of the cerebral cortex.
Neuronal dysfunction and cell death in the cerebral cortex is a hallmark of AD (Bennett et al., 2005; Bonthius et al., 2005; Delatour et al., 2004; Geula and Mesulam, 1995; Minger et al., 2001; Rosenberg et al., 2000; Samuel et al., 1994). The present study shows that primary cortical neurons express intrinsic and biologically active CHT1 activity, and that, in these neurons, CHT1-mediated choline uptake activity is significantly reduced by the Alzheimer’s PS-1 M146V mutation. Several potential mechanisms may be considered for the observed decrease in CHT1-mediated choline uptake in cortical neurons from PS-1 M146V mutant mice. Although CHT1 is predominantly intracellular, CHT1 shuttles constitutively between the plasma membrane and endocytic compartments, and synaptic vesicles may function as a reservoir to deliver CHT1 to plasma membrane following neuronal excitation (Ribeiro et al., 2003; Ribeiro et al., 2005). One potential mechanism by which the PS-1 mutation may decrease the CHT1-mediated choline uptake is by decreasing the expression and/or incorporation of CHT1 in cell surface (plasma membrane). However, kinetic analyses and data from [3H] HC-3 binding studies showed no significant difference in Bmax values among cortical neurons from the wild-type mice and those from the PS-1 M146V mutant animals, indicating that the PS-1 mutation did not significantly alter the incorporation of CHT1 to plasma membrane. (Fig. 4, and Table 1). This conclusion was further supported by data from Western blotting analysis of the biotinylated proteins (which are mostly cell surface proteins) showing that cell surface levels of CHT1 proteins in primary neurons from PS-1 M146V KI mice were not significantly different from those in wild-type animals (Fig. 1). The lack of a significant change in cell surface expression of CHT1 in cortical neurons from PS-1 M146V mutant animals suggests that the PS-1mutation is unlikely to be involved in the regulation of expression and/or incorporation of CHT1 protein in the plasma membrane.
Another potential mechanism by which the PS-1 mutation may decrease the CHT1-mediated choline uptake is by reducing the ligand binding affinity of CHT1 and/or the substrate transport activity of CHT1. Our observations that expression of PS-1 M146V mutation in primary cortical neurons resulted in a significant increase in Km in [3H] choline uptake assays and in Kd in [3H] HC-3 binding assays were consistent with this hypothesis, and demonstrated that the binding affinity of CHT1 to its substrate was significantly decreased in the PS-1 mutant animals. It is still unclear how mutations in PS-1 alter the ligand binding affinity of CHT1. Mounting evidence suggests that aberrant production and aggregation of the neurotoxic amyloid β-peptide 1–42 (Aβ42) play a key role in the pathogenesis of Alzheimer’s disease. Mutations in presenilins have been shown to significantly increase the production of Aβ42 (Annaert et al., 1999; Borchelt et al., 1997; Cai et al., 2006; Capell et al., 2000; Chui et al., 1999; Duff et al., 1996; Guo et al., 1996; Guo et al., 1997; Guo et al., 1998; Guo et al., 1999c; Mattson et al., 1998; Scheuner et al., 1996). Of importance, a recent report found that, in freely moving PDAPP mice (a well-characterized transgenic mouse model of AD), Aβ42 formed a complex with CHT1 in co-immunoprecipitation assays, which may impair steady-state and on-demand ACh release (Bales et al., 2006). Treatment of PDAPP mice with the anti-Aβ antibody could rapidly and completely restore hippocampal ACh release and high-affinity choline uptake while greatly reducing impaired habituation learning in these mice (Bales et al., 2006). These observations raised the intriguing possibility that the significant reduction in choline uptake activity of CHT1 observed in this study in PS-1 M146V mutant mice might be mediated by an aberrant production of Aβ42 associated with the PS-1 mutation.
The PS-1 mutant knockin mouse strain used in this study was originated on a B6;129 background, and has been backcrossed to C57BL/6 for multiple generations. We have reported that the homozygous PS1M146VKI mice produced only the mutant gene product. However, during the transfer of the knock-in to the B6 background, the flanking regions of the PS-1 knock-in that are of 129 origin might have also been transferred. It is therefore possible that there might be genes in this flanking region that are causing the observed changes in CHT1-mediated choline uptake. Although there is no clear evidence for the existence of such passenger genes of 129 origin in our PS-1 M146 knockin strain, this possibility can be a potential concern in early generations of our PS-1 knockin mice in which there might have been a significant contribution from the 129 strain. However, many of ours PS-1 knockin animals have been backcrossed to C57BL/6 for over 20 generations now. Moreover, we found that wild-type 129S1/svImJ mice backcrossed to C57BL/6 for at least 10 generations showed no significant difference in specific choline uptake in cortical neurons when compared with specific choline uptake in cortical neurons from C57BL/6 inbred animals (data not shown). These data suggest that the effect of PS-1 mutation on CHT1- mediated choline uptake observed in this study was unlikely due to the contribution from the 129 mouse strain.
The experimental design of the present study was to focus on intrinsic CHT1 activity in cortical neurons rather than that in basal forebrain neurons. This design was important for several reasons: (1) the existence of the intrinsic cholinergic neurons in the cerebral cortex has been reported in rodents as well as in humans; (2) although the precise molecular mechanisms by which PS-1 M146V mutation alters CHT1 activity still need to be elucidated, this report provides, to our knowledge, the first experimental evidence for an adverse action of an Alzheimer’s PS-1 mutation in altering the intrinsic CHT1 activity in cortical neurons. Since massive degeneration of cortical neurons has been known to be one of the hallmark features of AD pathology, and since cholinergic neurotransmission is critically involved in cognitive functions, our data suggest that alterations in CHT1-mediated high affinity choline uptake in cortical neurons may contribute to the pathogenesis of Alzheimer’s dementia. The adverse effects of presenilin mutations on CHT1 activity in basal forebrain neurons are now being investigated in a eparate study in our lab.
4. EXPERIMENTAL PROCEDURES, ACKNOWLEDGEMENTS, REFRENCES
Generation and Characterization of PS-1 M146V Mutant Knock-in Mice
The targeting strategy for generating PS-1 M146V KI mice is described in detailed elsewhere (Guo et al., 1999a). The targeted allele (PS-1M146VKI) causes a mutation of the mouse presenilin-1 gene that results in expression of a presenilin-1 protein with the human familial Alzheimer's disease-linked mutation PS-1M146V. A targeting vector containing neomycin resistance and herpes simplex virus thymidine kinase genes was utilized in constructing this mutant. A mutagenized DNA sequence of exon 5 of the mouse presenilin-1 gene was targeted for the presenilin-1 allele. The construct was electroporated into 129X1/SvJ x 129S1/Sv-derived R1 embryonic stem (ES) cells. Correctly targeted ES cells were injected into recipient blastocysts. The resulting chimeric male animals were bred to C57BL/6 females to produce mice heterozygous for the mutation (Guo et al., 1999a). These mice were then bred to a CMV-cre transgenic strain to delete the neo cassette from the targeted allele. We have reported that the homozygous PS-1M146VKI mice produce only the mutant gene product at normal physiological levels (Guo et al., 1999a). The wild-type mouse was used as the control. The PS-1 M146V KI mice and the wild-type mice were bred in house and were maintained on the same genetic background (BL6/129Sv) in the animal facility at the University of Oklahoma health Science Center. In vivo and in vitro studies also showed that hippocampal and cortical neurons from PS-1 M146V KI mice showed increased vulnerability to excitotoxicity and apoptosis induced by amyloid beta-peptide (Aβ) (Guo et al., 1999a; Guo et al., 1999c). Assessments of expression from the targeted allele and genotype assignments have also been described in detail in our previous studies (Guo et al., 1999a; Guo et al., 1999c). The animal protocols used in this study are consistent with regulations for experimental animal care and use, and have been approved by the Institutional Animal Care and Use committee (IACUC) at the University of Oklahoma Health Sciences Center.
Primary Neuronal Cultures
Dissociated cortical neuronal cultures were prepared from postnatal day 1 mouse pups using methods similar to those described previously (Guo et al., 1999a). Briefly, cerebral cortical tissues from PS-1 M146V KI or wild-type control mice were removed and incubated for 15 min in Ca2+- and Mg2+-free Hank's Balanced Saline Solution (Gibco BRL) containing 0.2% papain. Cells were dissociated by trituration and plated into polyethyleneimine-coated plastic or glass-bottom culture dishes containing Minimum Essential Medium with Earle's salts supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 1 mM pyruvate, 20 mM KCl, 10 mM sodium bicarbonate and 1 mM Hepes (pH 7.2). Following cell attachment (3–6 hr post-plating), the culture medium was replaced with Neurobasal Medium with B27 supplements (Gibco BRL).
[3H] Choline Uptake Assay
[3H] Choline uptake assays were performed essentially as previously described (Okuda et al., 2002; Xie and Guo, 2004). In brief, cells were washed twice with Krebs-Ringer’s–HEPES buffer (NaCl 130 mM, KCl 1.3 mM, MgSO4, 1.2 mM, CaCl2 2.2 mM, KH2PO4 1.2 mM, glucose 10 mM, HEPES 10 mM, pH 7.4) and incubated at 37°C for 2 hour , followed by an additional incubation in Krebs-Ringer’s–HEPES buffer containing 1 μM [methyl-3H] choline chloride (Amersham Biosciences; 83 Ci/mmol) for 10 min. Choline uptake was terminated by washing cells three times with ice-cold Krebs-Ringer’s-HEPES buffer. The level of accumulated [3H] choline was determined by solubilizing cells in 1% SDS, 0.2 N NaOH and then radioactivity was measured with a Liquid Scintillation Counter (Beckman Coulter LS 6500). The sensitivity of [3H] choline uptake to HC-3 was assessed by measuring [3H] choline uptake in the presence of 1 μM HC-3. Specific [3H] choline uptake was determined by subtracting non-specific uptake (as determined by the choline uptake in the presence of 1 μM HC-3) from the total uptake. The Na+ dependence of [3H] choline uptake was assessed in buffer A containing 140 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 5 mM Tris, 10 mM glucose (pH 7.4) using isotonic replacement of NaCl with LiCl, as previously described (Xie and Guo, 2004).
Cell Surface Biotinylation Assay and Western Blot Analysis
These assays were used to analyze the cell surface expression of CHT1 because biotinylated proteins were considered to be mostly expressed on the cell surface and because they were greatly depleted of the intracellular marker actin (Okuda et al., 2002; Xie and Guo, 2004) (see also Fig. 1d). The methods were essentially the same as previously described (Okuda et al., 2002; Xie and Guo, 2004). In brief, cells were treated with Sulfo-NHS-SS-Biotin (1 mg/ml, Pierce) in PBS/calcium-magnesium buffer (138 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 9.6 mM Na2HPO4, 0.1 mM CaCl2, 1 mM MgCl2, pH 7.4) at 4°C for 30 min followed by washing with 100 mM glycine in PBS/calcium-magnesium. Cells were solubilized with the assay buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton-X-100, 1% sodium deoxycholate, 0.1% SDS) and then centrifuged at 17,000 X g for 30 min at 4°C. Biotinylated proteins were separated from nonbiotinylated proteins by incubation with UltraLinkTM Plus Immobilized Streptavidin Gel (Pierce) for 1 h at room temperature. Beads were washed 5 times with 1 ml of the assay buffer, and adsorbed proteins were eluted with SDS sample buffer (50 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 6% β-mercaptoethanol, and 0.001% bromphenol blue) for 30 min at room temperature. 100 μg of biotinylated proteins were separated by electrophoresis on a 4–12% gradient SDS-polyacrylamide gel and then transferred to a nitrocellulose sheet. Western blot analysis with an affinity purified rabbit polyclonal anti-CHT1 antibody was then performed as previously described (Xie and Guo, 2004). The lack of the intracellular marker actin in biotinylated proteins was confirmed using a monoclonal anti-β-actin antibody (Sigma Aldrich, St Louis, MO, USA). The CHT1 antibody was raised against an oligopeptide encoding the final 16 amino acid residues of CHT1 conserved at the carboxyl-terminus in mouse, rat and human. The antibody recognized 3 major bands of about 80 kDa, 68 kDa and 45kDa. The 68 kDa band corresponds closely to the molecular weight predicted from mouse CHT1 primary structure, while various other bands may represent oligomers and/or heterogeneous glycosylation of the CHT1 protein. Immunoblot images were acquired and quantified using Kodak Image Station 2000R and Kodak Digital Science 1D 3.6 software (Eastman Kodak Company, New Haven, CT, USA).
Immunocytochemistry and Confocal Laser Scanning Microscopy
These methods have been described in our previous studies (Guo et al., 1998; Xie and Guo, 2004; Xie et al., 2005). In brief, for immunocytochemical analysis of CHT1 expression, the cultured cells were fixed for 30 min in 4% paraformaldehyde/PBS, and membranes were permeabilized by incubation in 0.2% Triton-X100 in PBS. Cells were incubated for 1 h in blocking serum (5% normal goat serum in PBS). Cells were then exposed to 1:1000 dilution of the rabbit anti-CHT1 polyclonal antibody overnight at 4 °C, followed by incubation for 1 h with fluorescein-labeled anti-rabbit secondary antibody (Vector Laboratories, Burlingame, California). For immunocytochemical analysis of colocalization of CHT1 and choline acetyltransferase (ChAT) in primary neurons, the cultured cells were exposed to 1:1000 dilution of rabbit anti-CHT1 polyclonal antibody and 1:250 dilution of mouse anti-ChAT monoclonal antibody (Chemicon) overnight at 4 °C, followed by incubation for 1 h with a mixture of Texas Red-labeled anti-mouse and fluorescein-labeled anti-rabbit secondary antibodies (Vector Laboratories, Burlingame, California). Images of CHT1 and ChAT immunofluorescence were acquired using a Zeiss LSM 510 confocal laser scanning microscope (dual wavelength scan) with a 60X oil immersion objective. All images were acquired using the same laser intensity and photodector gain, to allow quantitative comparisons of relative levels of fluorescence in the cells. The average pixel intensity per cell and sites of colocalization of immunoreactivities were determined using the LSM 510 image analysis software. The percentage of CHT1 positive neurons may vary from one microscopic field to another, sometimes significantly. Therefore, in order to obtain more reliable and statistically meaningful data, random fields were selected to count the CHT1 positive neurons and the total number of neurons separately. This provides a reliable estimation of the actual average percentage of CHT1 positive neurons across the areas of the entire neuronal culture.
[3H] Hemicholinium-3 Binding Assay
[3H]HC-3 (PerkinElmer Life Sciences, 128 Ci/mmol) binding experiments were performed as previously described (Okuda et al., 2002; Xie and Guo, 2004). In brief, cells were washed with Krebs-Ringer’s-HEPES buffer and preincubated for 1–2 h at 37°C for equilibrium. Binding assays were performed at 4 °C for 1–2 h and terminated by three washes with ice-cold Krebs-Ringer’s-HEPES buffer. Saturation binding was determined using 0.1–10 nM [3H] HC-3. Because HC-3 is hydrophilic and impermeable to cell membrane, the HC-3 binding activity (Bmax) represents the expression level of the transporter in the plasma membrane (Xie and Guo, 2004). Kd for HC-3, which represents ligand binding affinity of CHT1, was calculated by nonlinear least-square fits using the Hill equation (SigmaPlot 8.0, SSPS, Inc.).
Acknowledgments
We thank Dr. Takashi Okuda (University College London, UK) for providing specific polyclonal antibody against CHT1 and other related materials. This work was supported by grants to Q.G. from the National Institute of Neurological Disorders and Stroke of NIH (R01NS043296), The Alzheimer’s Association, and American Federation for Aging Research (AFAR).
Abbreviations
- CHT1
high affinity choline transporter-1
- PS-1
presenilin-1
- HC-3
hemicholinium-3
- ChAT
choline acetyltransferase
- AD
Alzheimer’s disease
- S.E
standard error
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–94. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol. 2002;68:209–45. doi: 10.1016/s0301-0082(02)00079-5. [DOI] [PubMed] [Google Scholar]
- Bales KR, Tzavara ET, Wu S, Wade MR, Bymaster FP, Paul SM, Nomikos GG. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-Abeta antibody. J Clin Invest. 2006;116:825–32. doi: 10.1172/JCI27120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benagiano V, Virgintino D, Flace P, Girolamo F, Errede M, Roncali L, Ambrosi G. Choline acetyltransferase-containing neurons in the human parietal neocortex. Eur J Histochem. 2003;47:253–6. doi: 10.4081/835. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–41. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Solodkin A, Van Hoesen GW. Pathology of the insular cortex in Alzheimer disease depends on cortical architecture. J Neuropathol Exp Neurol. 2005;64:910–22. doi: 10.1097/01.jnen.0000182983.87106.d1. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Breer H, Knipper M. Regulation of high affinity choline uptake. J Neurobiol. 1990;21:269–75. doi: 10.1002/neu.480210202. [DOI] [PubMed] [Google Scholar]
- Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, Foster DA, Sisodia SS, Xu H, Gorelick FS, Greengard P. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci U S A. 2006;103:1941–6. doi: 10.1073/pnas.0510708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A, Steiner H, Romig H, Keck S, Baader M, Grim MG, Baumeister R, Haass C. Presenilin-1 differentially facilitates endoproteolysis of the beta-amyloid precursor protein and Notch. Nat Cell Biol. 2000;2:205–11. doi: 10.1038/35008626. [DOI] [PubMed] [Google Scholar]
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–4. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–90. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Lesort M, Bijur GN, Johnson GV, Jope RS. Cholinergic- and stress-induced signaling activities in cells overexpressing wild-type and mutant presenilin-1. Brain Res. 2001;903:226–30. doi: 10.1016/s0006-8993(01)02428-3. [DOI] [PubMed] [Google Scholar]
- Delatour B, Blanchard V, Pradier L, Duyckaerts C. Alzheimer pathology disorganizes cortico-cortical circuitry: direct evidence from a transgenic animal model. Neurobiol Dis. 2004;16:41–7. doi: 10.1016/j.nbd.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Dickinson-Anson H, Winkler J, Fisher LJ, Song HJ, Poo M, Gage FH. Acetylcholine-secreting cells improve age-induced memory deficits. Mol Ther. 2003;8:51–61. doi: 10.1016/s1525-0016(03)00145-x. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Pereztur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–3. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Eckenstein F, Baughman RW. Two types of cholinergic innervation in cortex, one co-localized with vasoactive intestinal polypeptide. Nature. 1984;309:153–5. doi: 10.1038/309153a0. [DOI] [PubMed] [Google Scholar]
- Fine A, Dunnett SB, Bjorklund A, Iversen SD. Cholinergic ventral forebrain grafts into the neocortex improve passive avoidance memory in a rat model of Alzheimer disease. Proc Natl Acad Sci U S A. 1985;82:5227–30. doi: 10.1073/pnas.82.15.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Wictorin K, Bjorklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–8. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- Geula C, Mesulam MM. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis Assoc Disord. 1995;9(Suppl 2):23–8. doi: 10.1097/00002093-199501002-00005. [DOI] [PubMed] [Google Scholar]
- Gibbs RB, Aggarwal P. Estrogen and basal forebrain cholinergic neurons: implications for brain aging and Alzheimer's disease-related cognitive decline. Horm Behav. 1998;34:98–111. doi: 10.1006/hbeh.1998.1451. [DOI] [PubMed] [Google Scholar]
- Gottfries CG, Adolfsson R, Aquilonius SM, Carlsson A, Eckernas SA, Nordberg A, Oreland L, Svennerholm L, Wiberg A, Winblad B. Biochemical changes in dementia disorders of Alzheimer type (AD/SDAT) Neurobiol Aging. 1983;4:261–71. doi: 10.1016/0197-4580(83)90002-7. [DOI] [PubMed] [Google Scholar]
- Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. Neuroreport. 1996;8:379–83. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–22. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP. Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer disease. Nat Med. 1998;4:957–62. doi: 10.1038/nm0898-957. [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999a;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sebastian L, Sopher BL, Miller MW, Glazner GW, Ware CB, Martin GM, Mattson MP. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a PS1 mutation. Proc Natl Acad Sci U S A. 1999b;96:4125–30. doi: 10.1073/pnas.96.7.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. J Neurochem. 1999c;72:1019–29. doi: 10.1046/j.1471-4159.1999.0721019.x. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Erb C, Ebert U, Baumann KH, Popp A, Konig G, Klein J. Central cholinergic functions in human amyloid precursor protein knock-in/presenilin-1 transgenic mice. Neuroscience. 2004;125:1009–17. doi: 10.1016/j.neuroscience.2004.02.038. [DOI] [PubMed] [Google Scholar]
- Hernandez D, Sugaya K, Qu T, McGowan E, Duff K, McKinney M. Survival and plasticity of basal forebrain cholinergic systems in mice transgenic for presenilin-1 and amyloid precursor protein mutant genes. Neuroreport. 2001;12:1377–84. doi: 10.1097/00001756-200105250-00018. [DOI] [PubMed] [Google Scholar]
- Houser CR, Crawford GD, Salvaterra PM, Vaughn JE. Immunocytochemical localization of choline acetyltransferase in rat cerebral cortex: a study of cholinergic neurons and synapses. J Comp Neurol. 1985;234:17–34. doi: 10.1002/cne.902340103. [DOI] [PubMed] [Google Scholar]
- Johnston MV, McKinney M, Coyle JT. Neocortical cholinergic innervation: a description of extrinsic and intrinsic components in the rat. Exp Brain Res. 1981;43:159–72. doi: 10.1007/BF00237760. [DOI] [PubMed] [Google Scholar]
- Kristt DA. Acetylcholinesterase-containing neurons of layer VIb in immature neocortex: possible component of an early formed intrinsic cortical circuit. Anat Embryol (Berl) 1979;157:217–26. doi: 10.1007/BF00305161. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Guo Q, Furukawa K, Pedersen WA. Presenilins, the endoplasmic reticulum, and neuronal apoptosis in Alzheimer's disease. J Neurochem. 1998;70:1–14. doi: 10.1046/j.1471-4159.1998.70010001.x. [DOI] [PubMed] [Google Scholar]
- Minger SL, Honer WG, Esiri MM, McDonald B, Keene J, Nicoll JA, Carter J, Hope T, Francis PT. Synaptic pathology in prefrontal cortex is present only with severe dementia in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:929–36. doi: 10.1093/jnen/60.10.929. [DOI] [PubMed] [Google Scholar]
- Okuda T, Haga T. Functional characterization of the human high-affinity choline transporter. FEBS Lett. 2000;484:92–7. doi: 10.1016/s0014-5793(00)02134-7. [DOI] [PubMed] [Google Scholar]
- Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I. Identification and characterization of the high-affinity choline transporter. Nat Neurosci. 2000;3:120–5. doi: 10.1038/72059. [DOI] [PubMed] [Google Scholar]
- Okuda T, Okamura M, Kaitsuka C, Haga T, Gurwitz D. Single nucleotide polymorphism of the human high affinity choline transporter alters transport rate. J Biol Chem. 2002;277:45315–22. doi: 10.1074/jbc.M207742200. [DOI] [PubMed] [Google Scholar]
- Pascual J, Fontan A, Zarranz JJ, Berciano J, Florez J, Pazos A. High-affinity choline uptake carrier in Alzheimer's disease: implications for the cholinergic hypothesis of dementia. Brain Res. 1991;552:170–4. doi: 10.1016/0006-8993(91)90676-m. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Guo Q, Hartman BK, Mattson MP. Nerve growth factor-independent reduction in choline acetyltransferase activity in PC12 cells expressing mutant presenilin-1. J Biol Chem. 1997;272:22397–400. doi: 10.1074/jbc.272.36.22397. [DOI] [PubMed] [Google Scholar]
- Price DL, Kitt CA, Struble RG, Whitehouse PJ, Cork LC, Walker LC. Neurobiological studies of transmitter systems in aging and in Alzheimer-type dementia. Ann N Y Acad Sci. 1985;457:35–51. doi: 10.1111/j.1749-6632.1985.tb20798.x. [DOI] [PubMed] [Google Scholar]
- Price DL, Koliatsos VE, Clatterbuck RC. Cholinergic systems: human diseases, animal models, and prospects for therapy. Prog Brain Res. 1993;98:51–60. doi: 10.1016/s0079-6123(08)62380-8. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Alves-Silva J, Volknandt W, Martins-Silva C, Mahmud H, Wilhelm A, Gomez MV, Rylett RJ, Ferguson SS, Prado VF, Prado MA. The hemicholinium-3 sensitive high affinity choline transporter is internalized by clathrin-mediated endocytosis and is present in endosomes and synaptic vesicles. J Neurochem. 2003;87:136–146. doi: 10.1046/j.1471-4159.2003.01974.x. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Black SA, Cregan SP, Prado VF, Prado MA, Rylett RJ, Ferguson SS. Constitutive high-affinity choline transporter endocytosis is determined by a carboxyl-terminal tail dileucine motif. J Neurochem. 2005;94:86–96. doi: 10.1111/j.1471-4159.2005.03171.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg CK, Pericak-Vance MA, Saunders AM, Gilbert JR, Gaskell PC, Hulette CM. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol (Berl) 2000;100:145–52. doi: 10.1007/s004019900155. [DOI] [PubMed] [Google Scholar]
- Samuel W, Terry RD, DeTeresa R, Butters N, Masliah E. Clinical correlates of cortical and nucleus basalis pathology in Alzheimer dementia. Arch Neurol. 1994;51:772–8. doi: 10.1001/archneur.1994.00540200048015. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–70. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Vaucher E, Fluit P, Chishti MA, Westaway D, Mount HT, Kar S. Object recognition memory and cholinergic parameters in mice expressing human presenilin 1 transgenes. Exp Neurol. 2002;175:398–406. doi: 10.1006/exnr.2002.7915. [DOI] [PubMed] [Google Scholar]
- Warburton EC, Koder T, Cho K, Massey PV, Duguid G, Barker GR, Aggleton JP, Bashir ZI, Brown MW. Cholinergic neurotransmission is essential for perirhinal cortical plasticity and recognition memory. Neuron. 2003;38:987–96. doi: 10.1016/s0896-6273(03)00358-1. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–9. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Winkler J, Suhr ST, Gage FH, Thal LJ, Fisher LJ. Essential role of neocortical acetylcholine in spatial memory. Nature. 1995;375:484–7. doi: 10.1038/375484a0. [DOI] [PubMed] [Google Scholar]
- Wong TP, Debeir T, Duff K, Cuello AC. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J Neurosci. 1999;19:2706–16. doi: 10.1523/JNEUROSCI.19-07-02706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Chang X, Zhang X, Guo Q. Aberrant induction of Par-4 is involved in apoptosis of hippocampal neurons in presenilin-1 M146V mutant knock-in mice. Brain Res. 2001;915:1–10. doi: 10.1016/s0006-8993(01)02803-7. [DOI] [PubMed] [Google Scholar]
- Xie J, Guo Q. Par-4 inhibits choline uptake by interacting with CHT1 and reducing its incorporation on the plasma membrane. J Biol Chem. 2004;279:28266–75. doi: 10.1074/jbc.M401495200. [DOI] [PubMed] [Google Scholar]
- Xie J, Awad KS, Guo Q. RNAi knockdown of Par-4 inhibits neurosynaptic degeneration in ALS-linked mice. J Neurochem. 2005;92:59–71. doi: 10.1111/j.1471-4159.2004.02834.x. [DOI] [PubMed] [Google Scholar]
- Zapata A, Capdevila JL, Trullas R. Role of high-affinity choline uptake on extracellular choline and acetylcholine evoked by NMDA. Synapse. 2000;35:272–80. doi: 10.1002/(SICI)1098-2396(20000315)35:4<272::AID-SYN5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]