

Abstract
Friedreich ataxia (FRDA), the most common of the inherited ataxias, is an autosomal recessive degenerative disorder, characterized clinically by onset before the age of 25 of progressive gait and limb ataxia, absence of deep tendon reflexes, extensor plantar responses, and loss of position and vibration sense in the lower limbs. FRDA is caused by a GAA triplet expansion in the first intron of the FRDA gene on chromosome 9q13 in 97% of patients. The FRDA gene encodes a widely expressed 210-aa protein, frataxin, which is located in mitochondria and is severely reduced in FRDA patients. Frataxin function is still unknown but the knockout of the yeast frataxin homologue gene (YFH1) showed a severe defect of mitochondrial respiration and loss of mtDNA associated with elevated intramitochondrial iron. Here we report in vivo evidence of impaired mitochondrial respiration in skeletal muscle of FRDA patients. Using phosphorus magnetic resonance spectroscopy we demonstrated a maximum rate of muscle mitochondrial ATP production (Vmax) below the normal range in all 12 FRDA patients and a strong negative correlation between mitochondrial Vmax and the number of GAA repeats in the smaller allele. Our results show that FRDA is a nuclear-encoded mitochondrial disorder affecting oxidative phosphorylation and give a rationale for treatments aimed to improve mitochondrial function in this condition.
Friedreich ataxia (FRDA) is the most common form of inherited ataxia with a frequency of 1 in 50,000 live births. FRDA is an autosomal recessive degenerative disorder, characterized clinically by onset before the age of 25 of progressive gait and limb ataxia, absence of deep tendon reflexes, extensor plantar responses, and loss of position and vibration sense in the lower limbs (1). Cardiomyopathy as defined by echocardiography is present in more than 60% of FRDA patients (2). The cause of FRDA is a GAA triplet expansion in the first intron of the FRDA gene on chromosome 9q13 (3). Ninety-seven percent of FRDA patients are homozygous for the GAA expansion, the remainder carrying a repeat expansion in one FRDA allele and a point mutation in the other (2, 3).
The FRDA gene encodes a widely expressed 210-aa protein, frataxin, which is located in mitochondria (4–6) and is severely reduced in FRDA patients (4). Although frataxin function is still unknown, yeast strains carrying a disruption in the frataxin homologue gene (YFH1) showed a severe defect of mitochondrial respiration (5–8) and loss of mtDNA (7, 8) associated with elevated intramitochondrial iron (5, 8).
In view of the mitochondrial localization of frataxin (4–6), the evidence from the YFH1 knockout model for mitochondrial dysfunction (5–8), and the similarities of the cardinal clinical features present in FRDA with primary mitochondrial diseases (9), we used in vivo 31phosphorus magnetic resonance spectroscopy (31P-MRS) to test for the presence of mitochondrial dysfunction in skeletal muscle of 12 FRDA patients. Skeletal muscle is an ideal tissue in which to assess in vivo mitochondrial ATP production rate by 31P-MRS as it can be studied conveniently at rest, during exercise, and during the subsequent aerobic recovery phase (10, 11). Our results show a deficit of the maximum rate of muscle mitochondrial ATP production, Vmax (12), in all FRDA patients and a strong negative correlation between mitochondrial Vmax and the number of GAA repeats in the smaller allele, indicating that FRDA is a nuclear-encoded mitochondrial disorder that affects oxidative phosphoryation.
METHODS
Subjects.
Twelve FRDA patients (seven males; age range 16 to 54 years; 30 ± 10 years, mean ± SD), 12 disease controls (seven with Becker muscular dystrophy, one with limb girdle muscular dystrophy, and four with inclusion body myositis) (11 males, age range 15 to 56; 31 ± 14, mean ± SD), and 18 control subjects (10 males; age range 18 to 54; 30 ± 10, mean ± SD) were studied. Among the FRDA patients four could walk without support and three with support, three could stand with support, and two were wheelchair bound whereas among the disease controls three could walk without support and four with support, three could stand with support, and two were wheelchair bound. The four patients who could not stand had been wheelchair bound for 2–4 years before the 31P-MRS exam.
Informed consent was obtained from each patient, and normal volunteer and studies were carried out with approval of the local hospital ethics committees.
DNA Analysis.
DNA was extracted from the patients’ blood by using the Nucleon I DNA Isolation kit (Scotlab, Strathclyde, U.K.). A portion of the FRDA gene was amplified by using the Expand Long Template PCR System (Boehringer) using the GAAF and GAAR primers described (3) and the PCR protocol of 94°C for 3 min, followed by 94°C for 10 sec, 63°C for 30 sec, and 68°C for 3 min for 30 cycles, with the addition of 20 sec per cycle for the elongation step for the last 20 cycles and a final elongation of 72°C for 10 min. The GAA repeat length was calculated according to the size of the PCR product (457 + 3n bp, n = number of GAA triplets).
31P-MRS.
The study used a 2.0-T superconducting magnet (Oxford Magnet Technology, Oxford, U.K.) interfaced to a Bruker spectrometer (Bruker, Coventry, U.K.). Subjects lay supine with a 6-cm diameter surface coil centered on the maximal circumference of the right calf muscle. Spectra were acquired by using a 2-sec interpulse delay at rest (64 scans) and during exercise (32 scans) and recovery. As soon as the last 32-scan exercise spectrum was collected, an additional eight-scan spectrum was recorded, to be considered zero time of recovery, and the exercise stopped immediately afterward. Data were collected for 10.7 min during recovery (four eight-scan spectra followed by four of 16 scans, three of 32 scans, and two of 64 scans). The muscle was exercised by plantar flexion at 0.5 Hz, lifting a weight of 10% of lean body mass (calculated from body weight and skin fold thickness) (13) through a distance of 7 cm. After four spectra the weight was incremented by 2% of lean body mass for each subsequent spectral acquisition.
Relative concentrations of inorganic phosphate (Pi), phosphocreatine (PCr), and ATP were obtained by a time-domain fitting program (varpro/mrui) and were corrected for magnetic saturation. Absolute concentrations were obtained by assuming that the concentration of ATP in normal muscle is 8.2 mM (i.e., mmol/liter of intracellular water) (14). Intracellular pH was calculated from the chemical shift of the Pi peak relative to PCr (δPi, measured in ppm), as
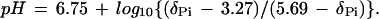 |
Free cytosolic [ADP] was calculated from pH and [PCr] by using a creatine kinase equilibrium constant of 1.66 × 109 M−1 (15) and assuming a normal total creatine content (TCr) of 42.5 mM (i.e., mmol/liter of intracellular water) (14) as
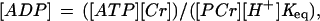 |
where [Cr] is the concentration of creatine calculated as [TCr] − [PCr].
PCr recovery half-times were calculated from the slope of semilogarithmic plots (12) by using the end-exercise and the first three recovery spectra. Initial rates of PCr resynthesis after exercise, V (mM/min), were calculated from the exponential rate constant of PCr recovery (k = 0.693/t1/2) and the total fall in [PCr] during exercise (Δ[PCr]) as V = k.Δ[PCr] (12). Using the hyperbolic ADP control model for mitochondrial respiration (16, 17) and a normal Km for ADP of 30 μM (12, 18), we calculated the maximum rate of mitochondrial ATP synthesis (Vmax) from the initial rate of PCr postexercise resynthesis (V) and the end-exercise [ADP] ([ADP]end):
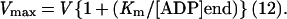 |
Statistical significance, determined by Student’s unpaired t test, was taken as P < 0.05. Linear regression analysis was used to calculate correlation coefficients.
RESULTS
All 12 FRDA patients were homozygous for the GAA triplet repeat expansion with a range of 290 to 900 (Table 1). MRS revealed abnormalities of skeletal muscle in FRDA even at rest. Although the mean PCr concentration was not different in the patient and normal control groups (33.3 ± 2.2 and 33.8 ± 2.2, respectively, P = 0.5), the mean Pi concentration was significantly elevated (4.5 ± 0.7 vs. 3.9 ± 0.7, P = 0.01) in FRDA (Table 1). Consequently the PCr/Pi ratio was also lower in FRDA than in control group (7.5 ± 1.3 vs. 9.0 ± 1.6, P = 0.01). This ratio showed an apparent bimodal distribution (data not shown); the reason for this result is not clear because there was no correlation between this ratio and the specific indices of mitochondrial function, discussed below, or the clinical indices of the disease.
Table 1.
31P-MRS data from calf muscle of FRDA patients, controls, and disease
| Case no. | n of GAA repeats in the smaller allele | Rest, [Pi] (mM) | End-exercise, [ADP] (μM) | Recovery
|
|
|---|---|---|---|---|---|
| V (mM/min) | Vmax (mM/min) | ||||
| 1 | 900 | 5.5 | 46 | 6 | 10 |
| 2 | 590 | 5.1 | 98 | 18 | 24 |
| 3 | 890 | 4.1 | 25 | 5 | 11 |
| 4 | 540 | 3.4 | 86 | 12 | 16 |
| 5 | 460 | 5.2 | 29 | 10 | 20 |
| 6 | 650 | 4.6 | 72 | 11 | 16 |
| 7 | 290 | 5.1 | 54 | 16 | 25 |
| 8 | 450 | 4.1 | 47 | 12 | 19 |
| 9 | 630 | 4.0 | 51 | 11 | 18 |
| 10 | 780 | 3.5 | 69 | 11 | 16 |
| 11 | 620 | 5.1 | 59 | 7 | 11 |
| 12 | 400 | 4.5 | 58 | 11 | 17 |
| FRDA patients (n = 12) | 4.5 ± 0.7 | 58 ± 21 | 11 ± 4 | 17 ± 5 | |
| Controls | 3.9 ± 0.7 | 58 ± 17 | 33 ± 12 | 51 ± 16 | |
| (n = 18) | (2.4–4.9) | (25–94) | (19–60) | (27–82) | |
| (FRDA vs controls) | P, 0.01 | P, 0.3 | P, 0.0001 | P, 0.0001 | |
| Disease controls (DC) (n = 12) | 4.7 ± 1.6 | 75 ± 24 | 33 ± 13 | 48 ± 18 | |
| (FRDA vs DC) | P, 0.7 | P, 0.07 | P, 0.0001 | P, 0.0001 | |
ADP, free cytosolic; V, initial rate of PCr postexercise resynthesis; Vmax, maximum rate of mitochondrial ATP production. Data are expressed as mean ± SD (range).
An increase in skeletal muscle resting [Pi] and, sometimes, a reduction in [PCr] are present in patients with mitochondrial encephalomyopathies caused by mtDNA mutations (19, 20) and have been interpreted as indices of altered state 4 mitochondrial respiration (19). However, a rise in intracellular [Pi] also is found in other neuromuscular disorders where a defect of mitochondrial respiration cannot be demonstrated (19, 21–23), which is true for the disease control group reported in Table 1. Therefore, the available evidence suggests that the increased Pi concentration is a nonspecific indicator of abnormal skeletal muscle metabolism. Oxidative metabolism is better evaluated from data collected after the end of exercise. The rate of muscle oxidative metabolism is very low at rest, and a better assessment of mitochondrial function can be obtained by stimulating mitochondrial respiration. With an in-magnet aerobic exercise protocol (see Methods) it is possible to assess the transition from the resting state 4 to the active state 3 of mitochondrial respiration. During incremental exercise the intracellular concentration of free ADP, which exerts the primary control on mitochondrial respiration (12, 17) increases (Table 1), stimulating oxidative phosphorylation. In these experimental conditions the rate of oxidative ATP production can be precisely assessed from the postexercise PCr recovery rate, which is entirely oxidative (14). The initial rate of PCr recovery, V, was significantly slower in FRDA patients (P = 0.0001) (Fig. 1, Table 1). Skeletal muscle maximum rate of mitochondrial ATP synthesis (Vmax), calculated from the initial rate of PCr recovery (V) and the end-exercise [ADP] (12), was severely reduced in FRDA patients compared with controls (P = 0.0001). In all 12 patients mitochondrial Vmax was below the normal range (Table 1, Fig. 2). Mitochondrial V and Vmax in FRDA patients were also significantly lower than in a group of disease controls (Table 1, Fig. 2) with similar maximal motor ability (see Methods), indicating that disability per se does not account for the reduced mitochondrial Vmax in FRDA patients. In FRDA patients muscle Vmax strongly depended on the size of the GAA repeats in the smaller allele (r = −0.75; P = 0.004) (Fig. 3). No correlation was found between resting concentration of Pi and PCr and the size of the GAA repeats in the smaller allele (data not shown).
Figure 1.

Calf muscle 31P-MRS spectra. (Upper) Spectra (eight free induction decays) from a normal subject (A) and patient 2 (B) at the end of in-magnet work. (Lower) Spectra (eight free induction decays) collected between 16 and 32 sec of recovery from the same subjects (C, normal subject; D, patient 2). During incremental exercise there is a progressive reduction of the PCr peak, hydrolyzed via the creatine kinase reaction to buffer ATP concentration, and an increase in the Pi peak produced by the ATP hydrolysis during muscle contraction (A and B). As soon the exercise is stopped PCr and Pi concentrations begin to return to their pre-exercise values: PCr is resynthesized from ATP, via the creatine-kinase reaction, and Pi is used for ATP synthesis (C and D). The ATP production during recovery from exercise is entirely caused by oxidative phosphorylation (10), thus the PCr resynthesis rate reflects precisely the mitochondrial rate of ATP production. At the same recovery time point, the PCr peak is smaller in patient 2 (D) than in the control subject (C), indicating a reduced rate of muscle mitochondrial ATP production in the FRDA patient. The abscissa reports the chemical shift in ppm and ordinate the relative signal intensity in arbitrary units.
Figure 2.

Deficit of mitochondrial ATP production in skeletal muscle of patients with FRDA. Maximum rate of mitochondrial ATP production (Vmax) in 12 FRDA patients compared with 18 controls and 12 diseased controls (DC).
Figure 3.

Correlation between skeletal muscle maximum rate of mitochondrial ATP production (Vmax) and the number of GAA repeats in the smaller allele in the 12 FRDA patients.
DISCUSSION
The expansion of the GAA repeat in intron 1 of the FRDA gene results in a reduction of frataxin expression. Although skeletal muscle involvement is not a prominent clinical feature of patients with FRDA, it has been shown that in skeletal muscle, which normally expresses intermediate levels of frataxin mRNA (3), frataxin is absent or severely reduced in FRDA patients (4). Elevated plasma lactate levels in FRDA patients support their compromised oxidative phosphorylation capacity (24).
Our findings demonstrate that the GAA triplet repeat expansion in FRDA is associated with a profound deficit of skeletal muscle mitochondrial ATP production and strongly support mitochondrial involvement in the pathogenesis of FRDA. In FRDA patients homozygous for the GAA triplet repeat expansion there is a correlation between age at onset, rate of disease progression and cardiomyopathy, and the GAA repeat number on the smaller allele (2). The strong negative correlation we found in our FRDA patients between the number of GAA repeats on the smaller allele and skeletal muscle mitochondrial Vmax is evidence that the expansion is the cause of the mitochondrial deficit and may suggest a link between degree of mitochondrial respiration deficit and clinical expression of the disease in other tissues. In lymphoblastoid cell lines of FRDA patients the length of the GAA expansion has been shown to determine the amount of frataxin expressed (4). Therefore, frataxin expression probably determines the reduced skeletal muscle mitochondrial ATP production rate we detected in vivo. Our results contribute to the growing body of evidence that, in certain diseases, biochemical abnormalities as detected by in vivo MRS can reflect the magnitude of a genetic abnormality (25, 26).
The yeast YFH1 knockout model exhibits defective mitochondrial respiration, decreased mtDNA levels, and iron accumulation. In a separate study (J.L.B., J.M.C., S. Chamberlain, and A.H.V.S., unpublished observations) we have identified severe reductions in the activities of the Fe-S containing respiratory chain enzyme complexes II/III and aconitase in FRDA cardiac and skeletal muscle, and an additional complex I defect in cardiac muscle. There was iron accumulation in FRDA heart but not in skeletal muscle, which may reflect different iron handling mechanisms in these tissues. There are therefore strong biochemical parallels between the yeast model and FRDA. The abnormalities identified may result from abnormal intramitochondrial iron handling giving rise to altered Fe-S assembly, the defective respiratory chain function, increased oxidative stress, and a loss of mtDNA (5, 8, 27, 28).
The involvement of oxidative stress and damage in FRDA is supported further by the fact that a FRDA-like phenotype is induced by vitamin E deficiency (29, 30). Vitamin E is a lipid-soluble antioxidant that localizes in membranes, including mitochondrial membranes (31), and protects against both lipid peroxidation (31, 32) and mtDNA damage (32). The connection with mitochondrial respiratory chain dysfunction is highlighted further by the fact that vitamin E deficiency can lead to mitochondrial damage as illustrated in a report of a deficit of brain and skeletal muscle respiration in a family with vitamin E deficit associated with hypobetalipoproteinemia (33) and in a rat model of vitamin E deficiency (34).
In conclusion, the present study shows that GAA repeat expansions within the FRDA nuclear gene are associated with a profound in vivo deficit of mitochondrial respiration in FRDA patients, which is inversely correlated with the GAA expansion length. Based on parallels with the YFH1 knockout model, this deficit may result from abnormal mitochondrial iron handling, which could cause an abnormality of iron sulfur assembly or function that would induce a deficiency of iron sulfur containing mitochondrial enzymes, and enhanced oxidative stress and damage. Despite the oxidative phosphorylation deficit skeletal muscle dysfunction is not a prominent clinical feature of FRDA. It is possible that signs of muscle weakness and fatigue may not be evident at the activity level of most FRDA patients and also may be masked by the more prominent features associated with ataxia.
In contrast to other neurodegenerative disorders FRDA can be diagnosed by genetic analysis either presymptomatically or in the early stage of disease, before central nervous system and cardiac damage become established. Our findings provide a very strong rationale for treatments aimed to enhance mitochondrial function and reduce toxic radical production in these patients.
Acknowledgments
We are indebted to all of the patients and their families for participating in the study. We would like to thank Dr. B. Rajagopalan for his help with some of the 31P-MRS studies and Mrs. E. Gower for helping with their organization. This work was supported by the Medical Research Council and the Friedreich’s Ataxia Group (United Kingdom). R.L. is a Junior Research Fellow at Wolfson College, Oxford, United Kingdom and is supported by an European Economic Community grant in the framework of the BIOMED program (Contract BMH4CT965017).
ABBREVIATIONS
- FRDA
Friedreich ataxia
- 31P-MRS
31phosphorus magnetic resonance spectroscopy
- PCr
phosphocreatine
Footnotes
A Commentary on this article begins on page 10948.
References
- 1.Harding A E. Brain. 1981;104:598–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- 2.Durr A, Cossee M, Agid Y, Capuzano V, Mignard C, Penet C, Mandel J L, Brice A, Koenig M. N Engl J Med. 1996;335:1169–1175. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- 3.Campuzano V, Montermini L, Moltò M D, Pianese L, Cassee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, et al. Science. 1996;271:1423–1426. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 4.Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish S J, Faucheux B, Trouillas P, et al. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 5.Babcock M, DeSilva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 6.Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M. Nat Genet. 1997;16:345–351. doi: 10.1038/ng0897-345. [DOI] [PubMed] [Google Scholar]
- 7.Wilson R B, Roof D M. Nat Genet. 1997;16:352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- 8.Foury F, Cazzalini O. FEBS Lett. 1997;411:373–377. doi: 10.1016/s0014-5793(97)00734-5. [DOI] [PubMed] [Google Scholar]
- 9.Johns D R. N Engl J Med. 1995;333:638–644. doi: 10.1056/NEJM199509073331007. [DOI] [PubMed] [Google Scholar]
- 10.Radda G K. Diabetes. 1996;45, Suppl. 1:88–92. doi: 10.2337/diab.45.1.s88. [DOI] [PubMed] [Google Scholar]
- 11.Argov Z, Bank W. Ann Neurol. 1991;30:90–97. doi: 10.1002/ana.410300116. [DOI] [PubMed] [Google Scholar]
- 12.Kemp G J, Taylor D J, Radda G K. NMR Biomed. 1993;6:66–72. doi: 10.1002/nbm.1940060111. [DOI] [PubMed] [Google Scholar]
- 13.Durnin J V G A, Womersley J. Brit J Nutr. 1974;32:77–97. doi: 10.1079/bjn19740060. [DOI] [PubMed] [Google Scholar]
- 14.Arnold D L, Matthews P M, Radda G K. Magn Reson Med. 1984;1:307–315. doi: 10.1002/mrm.1910010303. [DOI] [PubMed] [Google Scholar]
- 15.Veech R L, Lawson J W R, Cornell N W, Krebs H A. J Biol Chem. 1979;254:6538–6547. [PubMed] [Google Scholar]
- 16.Chance B, Williams G R. J Biol Chem. 1955;217:383–393. [PubMed] [Google Scholar]
- 17.Chance B, Leigh J, Jr, Kent J, McCully K, Nioka S, Clark B, Maris J, Graham T. Proc Natl Acad Sci USA. 1986;83:9458–9462. doi: 10.1073/pnas.83.24.9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nioka S, Argov Z, Dobson G P, Forster R E, Subramanian H V, Veech R L, Chance B. J Appl Physiol. 1992;72:521–528. doi: 10.1152/jappl.1992.72.2.521. [DOI] [PubMed] [Google Scholar]
- 19.Matthews P M, Allaire C, Shoubridge E A, Karpati G, Carpenter S, Arnold D L. Neurology. 1991;41:114–120. doi: 10.1212/wnl.41.1.114. [DOI] [PubMed] [Google Scholar]
- 20.Taylor D J, Kemp G J, Radda G K. J Neurol Sci. 1994;127:198–206. doi: 10.1016/0022-510x(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 21.Kemp G J, Taylor D J, Dunn J F, Frostick S P, Radda G K. J Neurol Sci. 1993;116:201–206. doi: 10.1016/0022-510x(93)90326-t. [DOI] [PubMed] [Google Scholar]
- 22.Lodi R, Taylor D J, Tabrizi S J, Hilton-Jones D, Squier M V, Seller A, Styles P, Schapira A H V. Brain. 1998;121:2119–2126. doi: 10.1093/brain/121.11.2119. [DOI] [PubMed] [Google Scholar]
- 23.Argov Z, Taivassalo T, DeStefano N, Genge A, Karpati G, Arnold D L. Muscle Nerve. 1998;21:1523–1525. doi: 10.1002/(sici)1097-4598(199811)21:11<1523::aid-mus22>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 24.Finocchiaro G, Baio G, Micossi P, DiDonato S. Neurology. 1988;38:1292–1296. doi: 10.1212/wnl.38.8.1292. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins B G, Rosas H D, Chen Y C I, Makabe T, Myers R, MacDonald M, Rosen B R, Beal M F, Koroshetz W J. Neurology. 1998;50:1357–1365. doi: 10.1212/wnl.50.5.1357. [DOI] [PubMed] [Google Scholar]
- 26.Chang L, Ernst T, Osborn D, Seltzer W, Leonide-Yee M, Poland R E. Arch Neurol. 1998;55:305–311. doi: 10.1001/archneur.55.3.305. [DOI] [PubMed] [Google Scholar]
- 27.Rotig A, Lonlay P D, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 28.Aust S D, Svingen B A. In: Free Radicals in Biology. Pryor W A, editor. New York: Academic; 1982. pp. 1–28. [Google Scholar]
- 29.Kayden H J. Neurology. 1993;43:2167–2169. doi: 10.1212/wnl.43.11.2167. [DOI] [PubMed] [Google Scholar]
- 30.Ouahchi K, Arita M, Kayden H, Hentati F, BenAmida M, Sokol R, Arai H, Inoue K, Mandel J L, Koenig M. Nat Genet. 1995;9:141–145. doi: 10.1038/ng0295-141. [DOI] [PubMed] [Google Scholar]
- 31.Thomas S M, Gebicki J M, Dean R T. Biochim Biophys Acta. 1989;1002:189–197. doi: 10.1016/0005-2760(89)90286-5. [DOI] [PubMed] [Google Scholar]
- 32.Hruszkewycz A M. Biochem Biophys Res Commun. 1988;153:191–197. doi: 10.1016/s0006-291x(88)81207-5. [DOI] [PubMed] [Google Scholar]
- 33.Lodi R, Rinaldi R, Gaddi A, Iotti S, D’Alessandro R, Scoz N, Battino M, Carelli V, Azzimondi G, Zaniol P, et al. J Neurol Neurosurg Psychiatry. 1997;62:574–580. doi: 10.1136/jnnp.62.6.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas P K, Cooper J M, King R H M, Workman J M, Schapira A H V, Goss-Sampson M A. J Anat. 1993;183:451–461. [PMC free article] [PubMed] [Google Scholar]