


Abstract
MutS homologues are highly conserved enzymes engaged in DNA mismatch repair (MMR), meiotic recombination and other DNA modifications. Genome sequencing projects have revealed that bacteria and plants possess a MutS homologue, MutS2. MutS2 lacks the mismatch-recognition domain of MutS, but contains an extra C-terminal region called the small MutS-related (Smr) domain. Sequences homologous to the Smr domain are annotated as ‘proteins of unknown function’ in various organisms ranging from bacteria to human. Although recent in vivo studies indicate that MutS2 plays an important role in recombinational events, there had been only limited characterization of the biochemical function of MutS2 and the Smr domain. We previously established that Thermus thermophilus MutS2 (ttMutS2) possesses endonuclease activity. In this study, we report that a Smr-deleted ttMutS2 mutant retains the dimerization, ATPase and DNA-binding activities, but has no endonuclease activity. Furthermore, the Smr domain alone was stable and functional in binding and incising DNA. It is noteworthy that an endonuclease activity is associated with a MutS homologue, which is generally thought to recognize specific DNA structures.
INTRODUCTION
Escherichia coli MutS is known to be a key enzyme of DNA mismatch repair (MMR), which corrects mismatched bases produced during DNA replication (1,2). MutS homologues are widely conserved in almost all organisms, ranging from bacteria to human, and are known to play multiple roles in DNA repair or recombination. These DNA transactions are essential for retaining the fidelity of the genetic information and increasing genetic diversity. Eukaryotes have several MutS homologues (MSHs; MSH1-7) in the genome, whilst the majority of bacteria possess two MutS homologues (MutS and MutS2). Bacterial MutS homodimer and eukaryotic MSH2–MSH6 and MSH2–MSH3 heterodimers recognize mismatched bases and stimulate downstream repair reactions (3–5). However, eukaryotic MSH4–MSH5 heterodimer is not involved in MMR but participates in meiotic recombination (6,7). These characteristics are attributed to the lack of the mismatch-recognition motif in MSH4 and MSH5 (8).
Bacterial MutS2, which lacks the mismatch-recognition domain (Figure 1B), has an unknown function. Although, disruption of the mutS2 gene in Deinococcus radiodurans (9) and Bacillus subtilis (10) does not appear to affect the phenotype, recent in vivo studies showed that the disruption of Helicobacter pylori mutS2 resulted in an increase in the rate of homologous recombination (11,12). These results suggest that H.pylori MutS2 is involved in the inhibition of homologous recombination. More recently, Wang et al. observed that the disruption of H.pylori mutS2 also caused increased sensitivity to oxidative stress induced by agents, such as hydrogen peroxide or oxygen (13). Their data showed that H.pylori MutS2 is involved not only in the inhibition of homologous recombination but also in repair of oxidative DNA lesions.
Figure 1.

Limited proteolysis of ttMutS2. (A) An aliquot of 4 μM ttMutS2 was reacted with thermolysin or trypsin as described in Materials and Methods. The concentrations of protease are indicated at the top of the figure. For the thermolysin digestion, the reaction mixtures contained 10 mM CaCl2. The digests were separated by 7.5% SDS–PAGE. Arrows indicate the bands derived from digestions. (B) The same samples were also separated by 12.5% SDS–PAGE. (C) Schematic representation of ttMutS, ttMutS2 and the fragmentation of ttMutS2. Regions represented as mismatch-recognition, dsDNA-binding and dimerization domains in ttMutS are the counterparts of T.aquaticus MutS whose crystal structure has been solved (24). The dsDNA-binding and ATPase domains of ttMutS2 show >30% identity in the respective domains of ttMutS. Three bands indicated by asterisks in A and B were manually excised and subjected to PMF analysis. Arrows indicate the fragmentation of ttMutS2, and the white regions were detected by PMF analysis.
MutS2 proteins from bacteria and plants have a well-conserved C-terminal sequence called the small MutS-related (Smr) domain (14,15). Eukaryotic proteins also contain a sequence homologous to the bacterial Smr domain (14). Furthermore, Smr-like sequences also exist as stand-alone proteins (14). This wide range of distribution implies that the Smr domain is engaged in an important role within the cell. Although it was reported that the Smr-like domain of human BCL-3-binding protein, which is not a member of MutS family, exhibits endonuclease activity (16), there has been no experimental evidence to establish the biochemical function of the Smr domain in MutS2.
In this study, we investigate the biochemical function of MutS2 from an extremely thermophilic bacterium, Thermus thermophilus HB8. T.thermophilus is a Gram-negative eubacterium that can grow at temperatures greater than 75°C (17). In general, proteins isolated from T.thermophilus are heat-stable and ideally suited to physicochemical examination. Moreover, this bacterium retains the fundamental set of genes essential for the living cell (18). Our laboratory has studied the functions and characteristics of T.thermophilus MutS (19–22) and ttMutS2 (23). We previously reported that ttMutS2 possesses ATPase activity and has the ability to dimerize and bind double-stranded DNA (dsDNA). The ATPase activity was promoted by the addition of dsDNA. These characteristics are similar to those of bacterial MutS proteins. The crystal structures of E.coli and Thermus aquaticus MutSs showed that these proteins bind to dsDNA in a dimeric form, and that dimerization is necessary to form a nucleotide-binding site (24,25). However, ttMutS2 did not display specificity for a mismatched dsDNA. In addition, ttMutS2 showed an endonuclease activity which MutS does not possess. Here we describe the detailed biochemical characteristics of ttMutS2 and in particular that of the Smr domain. Smr-deleted ttMutS2 (N60-ttMutS2) and the N-terminal 60 kDa-deleted mutant (ttSmr) were generated and analyzed biochemically. N60-ttMutS2 retained the dimerization, ATPase and dsDNA-binding abilities but lost endonuclease activity. In contrast, ttSmr displayed endonuclease activity, suggesting that this activity is confined to the ttSmr domain of ttMutS2.
MATERIALS AND METHODS
Bacterial strains, media and plasmids
E.coli strains DH5α, BL21(DE3) and Rosetta-gami(DE3) (Novagen, Madison, WI) were cultured in Luria–Bertani (LB) broth at 37°C. Plasmid pET-11a/ttmutS2, derived from pET-11a (Novagen), contains the complete ttmutS2 gene under control of a T7 promoter (23).
Construction of smr-deleted ttmutS2 and ttsmr genes
DNA fragments expressing Smr-deleted ttMutS2 (N60-ttMutS2) and Smr domain of ttMutS2 (ttSmr) were generated by PCR using pET-11a/ttmutS2 plasmid as template. The following pairs of primers were used for amplification of each fragment: 5′-ATATCATATGATGCGTGACGTCCTCGAGGTCCTGGAGTTC-3′ and 5′-ATATAGATCTTTATTACTCAAAGCGGGCCTCCCGCTCGGCCAGGGC-3′ (N60-ttmutS2) and 5′-ATATCATATGGTGGTGGAGCTCCGTGGGGAGGAGGTCCTGGTCCA-3′ and 5′-ATATAGATCTTTATTATCAAGGCCGAAGCGCCACCACGGTAACCCC-3′ (ttsmr). Forward and reverse primers contained NdeI and BglII restriction sites, respectively (underlined). The amplified fragments were ligated into the pT7Blue vector (Novagen). Sequence analysis revealed that the constructions were error-free. The genes were then digested with the restriction enzymes NdeI and BglII and ligated into the compatible sites of the expression vector pET-11a, creating the plasmids pET-11a/N60-ttmutS2 and pET-11a/ttsmr under control of a T7 promoter.
Overexpression and purification of ttMutS2, N60-MutS2 and ttSmr
ttMutS2 was overexpressed and purified as described previously (23). N60-ttMutS2 was overexpressed and purified by the same method as ttMutS2. E.coli Rosetta-gami(DE3) was transformed with pET-15b/ttsmr and grown at 37°C in 1.5 l YT medium containing 50 μg/ml ampicillin. When the density of cultures reached 4 × 108 cells/ml, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to 0.4 μM. Cells were grown at 37°C for 6 h after induction and harvested by centrifugation. Cells were lysed by sonication in buffer I [20 mM Tris–HCl (pH 7.9), 500 mM NaCl and 25% sucrose] and then heated to 70°C for 10 min. After centrifugation at 48 000 g for 60 min, the supernatant was loaded onto a His-bind resin (Novagen) column (bed volume 10 ml) equilibrated with buffer II [20 mM Tris–HCl (pH 7.9) and 500 mM NaCl]. The column was washed with 100 ml of buffer II and eluted with a 100 ml gradient of 0–1000 mM imidazole in buffer II. The fractions containing ttSmr were detected by SDS–PAGE and concentrated using a Vivaspin (5000 cut-off) concentrator. The concentrated solution was applied to a Superdex 75 HR 10/30 column (bed volume 24 ml) previously equilibrated with buffer III [50 mM Tris–HCl (pH 7.5) and 100 mM KCl] and eluted with the same buffer using an ÄKTA explorer (GE Healthcare Biosciences, Uppsala, Sweden). N-terminal sequencing of the purified proteins gave the expected sequence for N60-ttMutS2 and ttSmr. Protein solutions were stored at 4°C.
Size exclusion chromatography
Size exclusion chromatography was performed at 25°C on a Superdex 200 HR and a Superdex 75 HR column (1 × 30 cm; GE Healthcare Biosciences) using an ÄKTA system. Samples of ttMutS2 (4 μM), N60-ttMutS2 (4 μM) or ttSmr (20 μM) were eluted at a flow rate of 0.1 ml/min in 50 mM Tris–HCl (pH 7.5) and 300 mM KCl. The elution profiles were monitored by recording the absorbance at 280 and 220 nm. Superdex 200 HR column was calibrated using apoferritin (443 000 Da), β-amylase (200 000 Da), alcohol dehydrogenase (150 000 Da), thyroglobulin (66 900 Da), albumin (66 000 Da), carbonic anhydrase (29 000 Da) and cytochrome c (12 400 Da). Superdex 75 HR column was calibrated using alcohol dehydrogenase (150 000 Da), albumine (66 000 Da), ovalbumin (45 000 Da), carbonic anhydrase (29 000 Da), cytochrome c (12 400 Da) and aprotinin (6500 Da).
Protein crosslinking
ttMutS2 (2 μM), N60-ttMutS2 (2 μM), ttSmr (10 μM) or BSA (2 μM) were incubated with 0.1, 0.3, 1 or 2 μM 4,4′-diisothiocyanatostilbene-2,2′disulfonic acid (DIDS) in 20 mM HEPES (pH 7.5), 100 mM KCl, 5 mM MgCl2, 5 mM ADP and 1 mM DTT, for 30 min at 37°C. Reaction products were separated on 12.5% and 7.5% SDS–polyacrylamide gels and stained with CBB.
ATPase assay
Hydrolysis of ATP was measured at 25°C by an enzyme-coupled spectrophotometric assay (26). The assay was performed at 25°C because the enzymes used to couple the reactions were not thermostable. The change of absorbance at 340 nm was measured with a Hitachi spectrophotometer model U-3000 (Hitachi, Tokyo, Japan). ATP was reacted with 100 nM protein in 50 mM Tris–HCl (pH 7.5), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 2 mM phosphoenolpyruvate, 0.32 mM NADH, 25 U/ml pyruvate kinase (Sigma-Aldrich, St Louis, MO) and 25 U/ml lactate dehydrogenase (TOYOBO, Osaka, Japan). The kinetic parameters were determined using the Michaelis–Menten equation.
Gel shift assay
DNA substrates were made by annealing combinations of oligonucleotides. Chemically synthesized 20mer single-stranded DNA (ssDNA), 5′-ATGTGAATCAGTATGGTTTC-3′, was radiolabeled at the 5′-end with [γ-32P]ATP using polynucleotide kinase. The 5′ 32P-labeled ssDNA was annealed to the complementary ssDNA (5′-GAAACCATACTGATTCACAT-3′) to obtain dsDNA. The 32P-labeled ssDNA (20 nM) or dsDNA (20 nM) were incubated with ttMutS2 or N60-ttMutS2 (protein concentrations are given in the figure legends) in 50 mM Tris–HCl (pH 7.5), 100 mM KCl, 1 mM DTT and 1 mM EDTA for 30 min at 37°C. The mixture (10 μl) was loaded on to a 7.5% polyacrylamide gel and electrophoresed in 1× TBE buffer (89 mM Tris–borate and 2 mM EDTA). The gel was dried and placed in contact with an imaging plate. The bands were visualized and analyzed using a BAS2500 image analyzer (Fuji Film).
Nuclease assay using plasmid DNA
A total of 50 ng/μl supercoiled plasmid DNA (pT7Blue) was incubated with or without ttMutS2, N60-ttMutS2 and ttSmr (protein concentrations are given in the figure legends) in 50 mM Tris–HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2 and 1 mM DTT at 37°C. The reactions were stopped by adding 5× loading buffer (5 mM EDTA, 1% SDS, 50% glycerol and 0.05% bromophenol blue). Reaction solutions were then loaded on to a 1.0% agarose S (Takara, Kyoto, Japan) gel containing 0.5× TBE buffer and 10 μg/ml ethidium bromide and electrophoresed. DNA fragments were stained with ethidium bromide and detected under ultraviolet (UV) light at 254 nm. In the examination of the pH dependence of activity, the following buffers were used: 50 mM sodium acetate (pH 4.5), 50 mM potassium phosphate (pH 5.5–6.0), 50 mM Tris–HCl (pH 7.0–9.0), 50 mM glycine-NaOH (pH 9.5–11.3) and 50 mM sodium phosphate (pH 11.7), each of which contained 100 mM KCl, 5 mM MgCl2 and 1 mM DTT.
Nuclease assay using oligonucleotides
The 32P-labeled 20 bp dsDNA were incubated with various concentrations of ttMutS2 or ttSmr in a buffer containing 50 mM Tris–HCl (pH 7.5), 100 mM KCl, 1 mM DTT and 2 mM MgCl2. Reactions were performed at 37°C and stopped by addition of an equal volume of sample buffer [5 mM EDTA, 80% deionized formamide, 10 mM NaOH, 0.1% bromophenol blue and 0.1% xylene cyanol]. The reaction mixtures were loaded on to 25% acrylamide gels (8 M urea and 1× TBE buffer) and electrophoresed with 1× TBE buffer. The gels were analyzed as described above. The initial rate (v0) was calculated by quantifying the proportion of products to unreacted substrate, and then kinetic parameters were determined from the following equation using the software Igor 4.03 (WaveMatrics, Lake Oswego, OR).
| (1) |
Fitting plot of pH-dependent assay
In the examination of the pH dependence of activity, the following buffers were used: 50 mM sodium acetate (pH 4.5), 50 mM potassium phosphate (pH 5.0–6.5), 50 mM Tris–HCl (pH 7.0–8.5), each of which contained 100 mM KCl, 5 mM MgCl2 and 1 mM DTT. Assuming Scheme 1 for the ttMutS2 and ttSmr nuclease reactions, the following equations were generated. Since the substrates were mixed with a large excess of enzyme, the following postulations were applied.
Scheme 1.
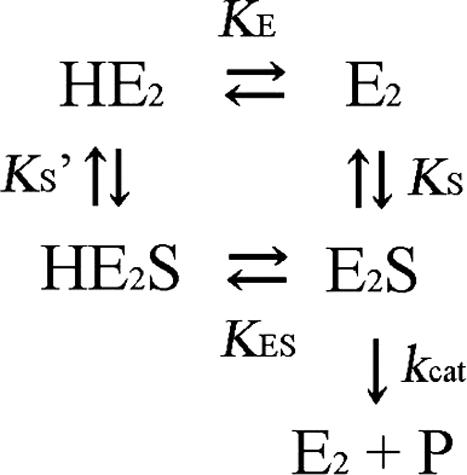
The ionization equilibrium of free enzyme and complex.
E2 represents the dimeric form of the enzyme. The initial rate at each pH is defined by
| (2) |
The ionization and dissociation constants were defined by
By substitution of these equations to Equation 2, the pH dependence of initial rate was obtained.
| (3) |
The pH dependence of kcat was derived from Equation 3.
| (4) |
pKES value was calculated by fitting the data to Equation 4 using Igor 4.03.
Site-directed mutagenesis
The pT7Blue plasmid containing the ttmutS2 gene was used as a template to generate H701A using Quick Change Site-Directed Mutagenesis (Stratagene) with primers 5′-CCCTAGGCCTTTCCACCCTCCGCCTCCTCGCCGGCAAGG-3′ and 5′-CCTTGCCGGCGAGGAGGCGGAGGGTGGAAAGGCCTAGGG-3′. Expression and purification of H701A ttMutS2 was done according to the protocol for wild-type ttMutS2 (23).
CD spectrometry
Circular dichroism (CD) measurements were carried out with a Jasco spectropolarimeter, model J-720W (Jasco, Tokyo, Japan). All measurements were performed using a 0.1 cm cell. The residue molar ellipticity [θ] was defined as 100 θobs (lc)−1, where θobs was the observed ellipticity; l was the length of the light path in centimeters, and c was the residue molar concentration of each protein. The stability of 2 μM ttMutS2 to pH was examined by the pH-dependent changes in CD values at 222 nm. The measurements were performed after incubation at 25°C for 24 h under each pH condition. The buffers used were: 50 mM sodium acetate (pH 4.5), 50 mM potassium phosphate (pH 5.5–6.5), 50 mM Tris–HCl (pH 7.0–9.0), 50 mM glycine-NaOH (pH 10.0–11.3) and 50 mM sodium phosphate (pH 11.7). All samples contained 100 mM KCl.
RESULTS
Limited proteolysis of ttMutS2
To reveal the organization of the structural domains of ttMutS2 (82.5 kDa), we analyzed the limited proteolysis profile of the purified protein using two endoproteases of different substrate specificities, trypsin and thermolysin. Under mild conditions, endoproteases are expected to preferentially hydrolyze a protein at the sites exposed to the solvent, which are often within interdomain linker regions. As shown in Figure 1A, treatment of ttMutS2 with trypsin produced two fragments of 60 and 20 kDa even at a low protease concentration. Similarly, treatment with thermolysin generated two fragments of 60 and 10 kDa. These fragments were relatively resistant to further digestion, indicating the presence of stable domains. Peptide mass fingerprinting (PMF) revealed that the 60 kDa fragment generated by thermolysin digestion included the putative dsDNA-binding and the ATPase (dimerization) domains, while 20 and 10 kDa fragments contained the putative Smr domain (Figure 1B). Amino acid sequence comparison between bacterial MutS2 and MutS indicates that ttMutS2 comprises the regions corresponding to the dsDNA-binding and dimerization domains of MutS (Figure 1B). ttMutS2 also has an additional C-terminal extension, called the Smr domain, which is absent from MutS (23). The region between the Smr domain and the N-terminal portion of ttMutS2 is poorly conserved and presumably constitutes an interdomain linker (23).
Knowledge of the likely domain boundaries within ttMutS2, based on limited proteolysis and amino acid sequence analyses, allowed us to design truncated versions of the protein with a degree of confidence. The 1–544 fragment of ttMutS2, N60-ttMutS2 and the 621–744 fragment of ttMutS2, ttSmr, were successfully expressed and purified to homogeneity (Figure 2A). The far-ultraviolet (UV) CD spectra of intact ttMutS2, N60-ttMutS2 and ttSmr showed negative double maxima at 209 and 220 nm, characteristic of an α-helical structure (data not shown), indicating that all three recombinant proteins were folded. The thermostability of each protein was examined based on the mean residue ellipticity at 222 nm. As a result, intact ttMutS2, N60-ttMutS2 and ttSmr were all stable up to 80°C (data not shown). These results indicate that the ttSmr domain and the N-terminal region retain stability even when expressed as individual domains.
Figure 2.

Size exclusion chromatography. (A) Recombinant proteins were purified as described in Materials and Methods and subjected to 12% SDS–PAGE. The calculated molecular mass of ttMutS2, N60-ttMutS2 and ttSmr were 82.5, 60 and 15 kDa, respectively. (B) The elution profiles of 4 μM ttMutS2 (light gray line) and 4 μM N60-ttMutS2 (black line). (C) The elution profile of 20 μM ttSmr. The elution positions for some of the size markers are shown at the top of the figure. Vo indicates the void volume of the column. The apparent molecular mass of peaks a, b, c, d and e were 410, 170, 290, 120 and 31 kDa, respectively.
Self-association
Size exclusion chromatography was performed to investigate the self-association ability of ttMutS2, N60-ttMutS2 and ttSmr (Figure 2B and C). The elution profile of ttMutS2 (calculated mass of 82.5 kDa) consisted of two peaks of 410 and 170 kDa, corresponding to oligomeric and dimeric forms of the protein, respectively. Likewise, the profile of N60-ttMutS2 (calculated mass of 60 kDa) consisted of two peaks of 290 and 120 kDa, indicating that N60-ttMutS2 also exists in an oligomeric and dimeric state in solution. These results strongly suggest that not only ttMutS2 but also N60-ttMutS2 retain the ability to dimerize. Indeed, this is consistent with the fact that ttMutS2 and N60-ttMutS2 both contain the region corresponding to the dimerization domain of ttMutS. The profile of ttSmr (calculated mass of 16.2 kDa) comprised a single peak corresponding to an apparent molecular mass of 31 kDa, indicating that ttSmr exists in solution as a dimeric form only.
The ability of ttMutS2 and the truncated proteins to dimerize was also supported by crosslinking experiments using the homo-bifunctional crosslinker, DIDS, which crosslinks two amino groups that are in close proximity to each other. The crosslinked species became detectable as the concentration of crosslinker was increased (Figure 3A–C). In each case, the apparent size of the crosslinked complex by SDS–PAGE suggests the formation of a homodimer. BSA was used as a negative control (Figure 3D). There was no significant difference in the oligomerization abilities between intact ttMutS2 and N60-ttMutS2. Therefore, the N-terminal 60 kDa region of ttMutS2 and the Smr domain both appear to undergo independent dimerization.
Figure 3.

Protein crosslinking by DIDS. (A) N60-ttMutS2. (B) ttMutS2. (C) ttSmr. (D) BSA. N60-ttMutS2 (2 μM), ttMutS2 (2 μM), ttSmr (10 μM) and BSA (2 μM) were incubated with homo-bifunctional crosslinker DIDS at 37°C for 30 min. The concentration of DIDS is indicated at the top of figure. Reaction mixtures were subjected to 7.5 or 12% SDS–PAGE and stained with Coomassie blue. Arrows indicate crosslinked dimeric proteins. The calculated molecular mass of dimeric N60-ttMutS2, ttMutS2 and ttSmr were 120, 165 and 30 kDa, respectively.
Previous studies have revealed that bacterial MutS binds to substrate DNA in a homodimeric form. The crystal structures of dimeric E.coli and T.aquaticus MutS showed that the C-terminal region of bacterial MutS forms the dimerizing interface (24,25). This region corresponds to residues 320–520 in ttMutS2, which is also present in N60-ttMutS2 (Figure 1C).
ATPase activity
To establish whether N60-ttMutS2 or ttSmr retain ATPase activity, we performed an enzymatic coupling assay using pyruvate kinase and lactate dehydrogenase (see Materials and Methods). As shown in Figure 4, N60-ttMutS2 exhibited ATPase activity while ttSmr did not. The kinetic constants were determined by assuming a Michaelis–Menten type reaction. The catalytic constants (kcat) of ttMutS2 and N60-ttMutS2 were 2.1 and 2.0 min−1, respectively. The Km values of ttMutS2 and N60-ttMutS2 were 48 and 40 μM, respectively. The kinetic parameters for N60-ttMutS2 were nearly identical to those of ttMutS2, showing that the Smr domain is not involved in the ATPase activity of ttMutS2. Furthermore, the kinetic parameters are also similar to those of T.thermophilus MutS (data not shown) and E.coli MutS (27). Thin layer chromatography (23) showed that the ATPase activity was also retained at an incubation temperature of 70°C (data not shown). We previously revealed that intact ttMutS2 has an ATPase activity (23). ttMutS2 includes the region corresponding to the ATPase domain of ttMutS, which contains the Walker's A type ATPase motif. This region in MutS is also within the dimerization domain, and it is known that dimerization is necessary to form the nucleotide-binding site (24,25).
Figure 4.

ATP hydrolysis in the presence of ttMutS2 (circles), N60-ttMutS2 (triangles) or ttSmr (square). ATPase activity was assayed at 25°C using an enzyme-coupled assay.
DNA-binding activity
Gel shift assays were performed to examine the DNA-binding activity of ttMutS2 and N60-ttMutS2 using 20 bp dsDNA as substrate in the absence of divalent cations. We confirmed that divalent cations are required for ttMutS2 to cleave DNA (data not shown). As shown in Figure 5A, complexes of both proteins with substrate DNA were detected. Plots of the percentage of shifted band versus protein concentration were almost identical for ttMutS2 and N60-ttMutS2 (Figure 5B), indicating that the dsDNA-binding domain in N60-ttMutS2 and ttMutS2 are equally functional.
Figure 5.

Gel shift assay. (A) ttMutS2 (left), N60-ttMutS2 (center) and ttSmr (right) were incubated with 20 nM 32P-labeled 20 bp dsDNA at 37°C for 30 min in the solution containing 50 mM Tris–HCl (pH 7.5), 100 mM KCl, 1 mM DTT and 1 mM EDTA. The concentrations of the proteins are indicated at the top of figure. (B) The percentage of complexed substrate to all substrate was quantified and plotted against protein concentration. Circles: N60-ttMutS2; triangles: ttMutS2. (C) The binding of ttSmr to 32P-labeled ssDNA and dsDNA was analyzed as above. Circles: dsDNA; triangles: ssDNA.
We examined the DNA-binding ability of ttSmr using the same substrate. As shown in Figure 5A, ttSmr alone bound to dsDNA, suggesting that the Smr sequence in isolation forms a functional domain. It should be mentioned that the DNA-binding activity of ttSmr for dsDNA is significantly weaker than seen for the full-length and N60-ttMutS2, suggesting that the N-terminal 60 kDa region rather than the Smr domain is primarily responsible for binding to dsDNA. Higher concentrations of ttSmr bound not only to dsDNA but also to ssDNA (Figure 5C). In contrast, binding of intact ttMutS2 or N60-ttMutS2 to ssDNA was never detected (data not shown), although studies at high protein concentration were not possible due to solubility limitations.
Nuclease activity
We examined the nicking endonuclease activity of N60-ttMutS2 and ttSmr using supercoiled plasmid DNA as substrate. The DNA was degraded by ttSmr to produce an open circular form of the plasmid DNA and linear DNA (Figure 6A). An identical degradation pattern was generated by intact ttMutS2. In contrast, N60-ttMutS2 did not degrade the plasmid DNA even at higher concentrations of protein (∼5 μM) (data not shown). Thus, the endonuclease activity of ttMutS2 can be ascribed to the Smr domain.
Figure 6.

Nuclease activity. (A) Nuclease activities of the proteins were examined using plasmid DNA as substrate. Reaction mixtures were subjected to 0.7% agarose gel electrophoresis and stained with ethidium bromide. Arrows CCC, OC and Linear indicate the closed circular form, open circular form of the plasmid and linear dsDNA, respectively. (B) Nuclease activities of the proteins were examined using an oligonucleotide. The sizes of the digested DNA molecules were estimated referring to our previous result (23) and shown at the right of the panel. (C) The substrates were incubated with various concentrations of ttMutS2 or ttSmr under the same condition as (B) for 20 min. Initial rates (vo) were calculated on the basis of the proportion of all products to unreacted substrates, and plotted against protein concentration. The theoretical curve was drawn by fitting the data to Equation 1 (See Materials and Methods). Circles: ttMutS2; triangles: ttSmr.
Next, the nuclease activity for linear DNA was examined using 20 bp dsDNA as substrate. As shown in Figure 6B, ttSmr digested the linear dsDNA whereas N60-ttMutS2 did not. The reaction with ttSmr produced several discrete bands of DNA, which was identical to the pattern obtained using intact ttMutS2. We have determined the three major bands to be 7, 11 and 14mer oligonucleotides (23). These results are entirely consistent with the proposal that the nuclease activity of ttMutS2 is confined to the Smr domain.
A plot of the initial rate (v0) for ttMutS2 nuclease activity versus ttMutS2 concentration gave a sigmoidal curve with a Hill constant of approximately 2 (Figure 6C). This result suggests that ttMutS2 dimerization is required for nuclease activity. Indeed, ttMutS2 dimerization may be required for DNA-binding, as is the case with MutS from E.coli and T.aquaticus (24,25). The kcat and Km values for ttMutS2 were 0.012 min−1 and 0.26 μM, respectively, whereas the kcat and Km values for ttSmr were 0.041 min−1 and 0.29 μM, respectively. Interestingly, the activity of ttSmr was much greater than that of intact ttMutS2: the kcat value of ttSmr being ∼4 times that of ttMutS2. This suggests that the N-terminal 60 kDa region of ttMutS2 may regulate the nuclease activity of ttMutS2 or the configuration of the domains may define the substrate specificity.
The pH dependence of nuclease activity was examined to obtain information concerning the catalytic residues of ttMutS2 and ttSmr. The pH-dependent changes to the CD values showed that both ttMutS2 and ttSmr retain the proper secondary structure across the pH range used for the nuclease activity studies (Figure 7A). Stability of the substrate plasmid DNA was confirmed by incubation in the buffers at each pH level in the absence of protein (Figure 7B). There was no apparent difference between the digestion patterns obtained under the various pH conditions (Figure 7C). However, the apparent velocity of the ttMutS2 nuclease activity drastically changed in the range pH 5 to 6. The same pH dependence was observed when plasmid DNA was incubated with ttSmr (data not shown). The activity at each pH value was quantified using 20 bp dsDNA as substrate (Figure 7D). Because of the instability of short dsDNA at pH values greater than 10, reactions were carried out at pH 4.5–8.5. The logarithmic plots of kcat against pH are shown in Figure 7E. By fitting the data to Equation 4, pKES values for ttMutS2 and ttSmr nuclease reactions were determined as 5.6 and 5.4, respectively. Because the substrate DNA molecule has no dissociation group with a pKa value in the pH range used in these experiments, we propose that the rate depends on the basic form of the catalytic residues of pKa 5.6 and 5.4 in ttMutS2 and ttSmr, respectively. The good agreement between the pKES values for ttMutS2 and ttSmr further supports the idea that the nuclease activity of ttMutS2 is confined to the Smr domain. In addition, sequence analysis revealed that bacterial Smr domain contains a highly conserved histidine residue (His-701 in ttMutS2). To test the involvement of this residue in catalysis, we generated H701A ttMutS2 mutant. The CD spectrum suggested that H701A was properly folded (data not shown). As shown in Figure 8, the activity of H701A to incise plasmid DNA and oligonucleotide was greatly decreased compared with that of wild-type ttMutS2. The kcat and Km values for H701A were 8.6 × 10−5 min−1and 0.39 μM, respectively. Though there was no great effect on Km, a 140-fold decrease in kcat was observed. This result strongly suggests that His-701 is involved in catalysis. However, this decrease seems to be too small to support the notion that His-701 acts as a general base in catalysis. The unessential role of His-701 as a catalyst is also supported by the fact that the pKES value of H701A was almost the same as that of wild-type (data not shown).
Figure 7.

The pH dependence of ttMutS2 nuclease activity. (A) The pH-dependent change of CD value at 222 nm. CD spectra of 2 μM ttMutS2 were analyzed under various pH conditions after incubation at 25°C for 24 h under each pH condition. CD values at 222 nm were plotted relative to pH. Triangles and circles are ttSmr and ttMutS2, respectively. (B) Substrate plasmid DNA was incubated without ttMutS2 under various pH conditions at 37°C for 120 min. (C) Substrate plasmid DNA was incubated with 200 nM ttMutS2 under various pH conditions at 37°C for 120 min. Reaction mixtures were subjected to 0.7% agarose gel electrophoresis and stained with ethidium bromide. Cont. represents substrate without incubation. (D) 32P-labeled 20 bp dsDNA (20 nM) was incubated with the indicated concentration of ttMutS2 at 37°C for 20 min under various pH conditions. Initial rates (vo) were calculated on the basis of the proportion of all products to unreacted substrates and then plotted against the protein concentration. (E) kcat values at each pH were determined by fitting the data in (C) to Equation 1, and then plotted relative to pH. This plot was fitted to Equation 4 (see Materials and Methods), and the theoretical curve was drawn. Triangles and circles are ttSmr and ttMutS2, respectively.
Figure 8.

The effect of substitution of His-701 with an alanine residue. (A) pT7Blue plasmid DNA was incubated with the indicated concentration of wild-type ttMutS2 (WT ttMutS2) or H701A at 37°C for 20 min. (B) 32P-labeled 20 bp dsDNA was incubated with the indicated concentration of ttMutS2 at 37°C for 120 min. Initial rates (vo) were calculated based on the proportion of all products to unreacted substrates and then plotted against the protein concentration. Triangles and circles are wild-type ttMutS2 and H701A, respectively.
DISCUSSION
The results obtained in this study indicate that the putative Smr domain exists as a stable protein and possesses endonuclease activity. Although, the ttSmr domain can dimerize (Figures 2 and 3), no higher oligomeric state could be detected even at extremely high protein concentrations. This result contrasts with intact ttMutS2 and N60-ttMutS2, which readily form oligomeric forms. Indeed, the absence of oligomeric states of ttSmr is presumably related to the higher solubility of this domain compared to intact ttMutS2 and N60-ttMutS2. We also revealed that ttSmr can bind DNA (Figure 5), although the lack of the Smr domain did not influence the DNA-binding activity of N60-ttMutS2. Therefore, the N-terminal 60 kDa region and the Smr domain of ttMutS2 are able to bind DNA independently. We have also demonstrated that the ttSmr possesses endonuclease activity (Figure 6). Indeed, this is the first report to reveal the contribution of the Smr domain to the endonuclease activity of MutS2.
The ttSmr domain used in this study was composed of 124 amino acid residues. Many endonucleases possess a similar number of residues: e.g. E.coli MutH and T7 endonuclease I contain 100 and 149 amino acid residues, respectively (28,29). Hence, we believe that ttSmr contains a sufficient number of residues to constitute an endonuclease. In addition, dimerization is also a common feature among many endonucleases, such as restriction enzymes and Holliday junction resolvases (30,31). Sequence analysis of the Smr domain shows that the protein lacks a known nuclease motif. The pH dependence of ttSmr nuclease activity implies that amino acid residues with pKa values of 5–6 are critical for catalysis (Figure 7). Sequence alignments show that bacterial Smr sequences include some highly conserved acidic and basic amino acid residues, including a histidine residue (His-701 in ttMutS2). Substitution of His-701 with an alanine residue caused a 140-fold decrease in the activity; supporting the notion, that His-701 plays a significant role in catalysis. However, it should be mentioned that this mutant ttMutS2 retained residual activity to cleave substrate DNA. The residual activity of H701A may be explained by the fact that many metal-dependent nucleases have a catalytic mechanism in which multiple amino acid residues, including histidine, concertedly act as ligands to a metal ion (32,33).
ttSmr displayed much greater endonuclease activity than intact ttMutS2 (Figure 6). This result indicates that the apparent non-specific endonuclease activity of the Smr domain may be regulated by the N-terminal 60 kDa region in ttMutS2. We suspect the regulation of the endonuclease activity is related to the recognition of a particular DNA structure and/or sequence of nucleotides. The apparent non-specific nicking endonuclease activity was also observed for the junction-resolving enzyme T7 endonuclease I (34). The heterodimeric protein containing a single catalytic domain of T7 endonuclease I act as a non-specific endonuclease, which degrades supercoiled plasmid DNA to short fragments of linear DNA (35). A similar result was obtained in this study using ttSmr. This common feature between the two enzymes implies that an appropriate configuration of dsDNA-binding domain and catalytic domain is required for discrimination of specific DNA substrates.
The experiments described in this paper confirm the endonuclease activity of ttMutS2, a MutS homologue. This result is perhaps surprising given that many MutS homologues recognize specific DNA structures, such as mismatched bases, bulge loops, oxidatively-damaged bases or Holliday junctions. Endonuclease activity is generally required to repair or process those DNA structures. However, the structure of the natural DNA substrate of ttMutS2 is unknown. Recent studies indicate that H.pylori MutS2 might be involved in the inhibition of homologous recombination (11,12) or oxidative DNA damage (13). Therefore, intermediates of recombination or oxidatively-damaged DNA might be good candidate substrates for ttMutS2. As Kang et al. assumed (12), it is possible that bacterial MutS2 incises a primary intermediate in recombination to limit intergenomic recombination.
Interestingly, smr-like sequences are widely distributed in almost all organisms, not only as a part of the mutS2 gene but also as a stand-alone gene (e.g. E.coli ydaL and Saccharomyces cerevisiae YPL199c) (14). In this study, we engineered the ttSmr domain of ttMutS2 for heterologous expression and have analyzed the biochemical properties of the recombinant protein. Thus, our results are relevant to the function of stand-alone Smr proteins from a wide range of organisms, including eukaryotes.
Acknowledgments
The authors thank H. Omori for his help in sequence analyses. This work was supported in part by Grants-in-aid for Scientific Research to S.K. and R.M. from the Ministry of Education, Science, Sports and Culture of Japan. K.F. is the recipient of a Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists (no. 9716). Funding to pay the Open Access publication charges for this article was provided by Research fellowship of JSPS for Young Scientists (no. 9716).
Conflict of interest statement. None declared.
REFERENCES
- 1.Friedberg E.C., Walker G.C., Siede W. DNA Repair and Mutagenesis. Washington, D.C.: ASM Press; 1995. p. 1. [Google Scholar]
- 2.Worth L., Clark S., Radman M., Modrich P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc. Natl Acad. Sci. USA. 1994;91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Modrich P. Methyl-directed DNA mismatch correction. J. Biol. Chem. 1989;264:6597–6600. [PubMed] [Google Scholar]
- 4.Jiricny J. Eukaryotic mismatch repair: an update. Mutat. Res. 1998;409:107–121. doi: 10.1016/s0921-8777(98)00056-1. [DOI] [PubMed] [Google Scholar]
- 5.Buermeyer A.B., Deschenes S.M., Baker S.M., Liskay R.M. Mammalian DNA mismatch repair. Annu. Rev. Genet. 1999;33:533–564. doi: 10.1146/annurev.genet.33.1.533. [DOI] [PubMed] [Google Scholar]
- 6.Ross-Macdonald P., Roeder G.S. Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell. 1994;79:1069–1080. doi: 10.1016/0092-8674(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 7.Hollingsworth N.M., Ponte L., Halsey C. MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes Dev. 1995;9:1728–1739. doi: 10.1101/gad.9.14.1728. [DOI] [PubMed] [Google Scholar]
- 8.Eisen J.A. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 1998;26:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mennecier S., Coste G., Servant P., Bailone A., Sommer S. Mismatch repair ensures fidelity of replication and recombination in the radioresistant organism Deinococcus radiodurans. Mol. Gen. Genomics. 2004;272:460–469. doi: 10.1007/s00438-004-1077-6. [DOI] [PubMed] [Google Scholar]
- 10.Rossolillo P., Albertini A.M. Functional analysis of the Bacillus subtilis yshD gene, a mutS paralogue. Mol. Gen. Genet. 2001;264:809–818. doi: 10.1007/s004380000370. [DOI] [PubMed] [Google Scholar]
- 11.Pinto A.V., Mathieu A., Marsin S., Veaute X., Ielpi L., Labigne A., Radicella J.P. Suppression of homologous and homeologous recombination by the bacterial MutS2 protein. Mol. Cell. 2005;17:113–120. doi: 10.1016/j.molcel.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 12.Kang J., Huang S., Blaser M.J. Structural and functional divergence of MutS2 from Bacterial MutS1 and eukaryotic MSH4-MSH5 homologs. J. Bacteriol. 2005;187:3528–3537. doi: 10.1128/JB.187.10.3528-3537.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang G., Alamuri P., Humayun M.Z., Taylor D.E., Maier J. The Helicobacter pylori MutS protein confers protection from oxidative DNA damage. Mol. Microbiol. 2005;58:166–176. doi: 10.1111/j.1365-2958.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 14.Moreira D., Philippe H. Smr: a bacterial and eukaryotic homologue of the C-terminal region of the MutS2 family. Trends Biochem. Sci. 1999;24:298–300. doi: 10.1016/s0968-0004(99)01419-x. [DOI] [PubMed] [Google Scholar]
- 15.Malik H.S., Henikoff S. Dual recognition-incision enzymes might be involved in mismatch repair and meiosis. Trends Biochem. Sci. 2000;25:414–418. doi: 10.1016/s0968-0004(00)01623-6. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe N., Wachi S., Fujita T. Identification and characterization of BCL-3-binding protein: implications for transcription and DNA repair or recombination. J. Biol. Chem. 2003;278:26102–26110. doi: 10.1074/jbc.M303518200. [DOI] [PubMed] [Google Scholar]
- 17.Oshima T., Imahori K. Description of Thermus thermophilus (Yoshima and Oshima) comb. nov., a nonsporulating thermophilic bacterium from a Japanese thermal spa. Int. J. Syst. Bacteriol. 1974;24:102–112. [Google Scholar]
- 18.Yokoyama S., Hirota H., Kigawa T., Yabuki T., Shirouzu M., Terada T., Ito Y., Matsuo Y., Kuroda Y., Nishimura Y., et al. Structural genomics in Japan. Nature Struct. Biol. 2000;7:943–945. doi: 10.1038/80712. [DOI] [PubMed] [Google Scholar]
- 19.Kato R., Kataoka M., Kamikubo H., Kuramitsu S. Direct observation of three conformations of MutS protein regulated by adenine nucleotides. J. Mol. Biol. 2001;309:227–238. doi: 10.1006/jmbi.2001.4752. [DOI] [PubMed] [Google Scholar]
- 20.Tachiki H., Kato R., Kuramitsu S. DNA binding and protein–protein interaction sites in MutS, a mismatched DNA recognition protein from Thermus thermophilus HB8. J. Biol. Chem. 2000;275:40703–40709. doi: 10.1074/jbc.M007124200. [DOI] [PubMed] [Google Scholar]
- 21.Tachiki H., Kato R., Masui R., Hasegawa K., Itakura H., Fukuyama K., Kuramitsu S. Domain organization and functional analysis of Thermus thermophilus MutS protein. Nucleic Acids Res. 1998;26:4153–4159. doi: 10.1093/nar/26.18.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takamatsu S., Kato R., Kuramitsu S. Mismatch DNA recognition protein from an extremely thermophilic bacterium, Thermus thermophilus HB8. Nucleic Acids Res. 1996;24:640–647. doi: 10.1093/nar/24.4.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukui K., Masui R., Kuramitsu S. Thermus thermophilus MutS2, a MutS paralogue, possesses an endonuclease activity promoted by MutL. J. Biochem. (Tokyo) 2004;135:375–384. doi: 10.1093/jb/mvh045. [DOI] [PubMed] [Google Scholar]
- 24.Obmolova G., Ban C., Hsieh P., Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 25.Lamers M.H., Perrakis A., Enzlin H.J., Winterwerp H.H.K., Wind N., Sixma T.K. The crystal structure of DNA mismatch repair protein MutS binding to a G·T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 26.Pugh B.F., Cox M.M. High salt activation of recA protein ATPasein the absence of DNA. J. Biol. Chem. 1988;263:76–83. [PubMed] [Google Scholar]
- 27.Acharya S., Foster P.L., Brooks P., Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 28.Ban C., Yang W. Structural basis for MutH activation in E. coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J. 1998;17:1526–1534. doi: 10.1093/emboj/17.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parkinson M.J., Lilley D.M.J. The junction-resolving enzyme T7 endonuclease I: quaternary structure and interaction with DNA. J. Mol. Biol. 1997;270:169–178. doi: 10.1006/jmbi.1997.1128. [DOI] [PubMed] [Google Scholar]
- 30.Pingoud A., Fuxreiter M., Pingoud V., Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lilley D.M., White M.F. Resolving the relationships of resolving enzymes. Proc. Natl Acad. Sci. USA. 2000;97:9351–9353. doi: 10.1073/pnas.97.17.9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pingoud A., Fuxreiter M., Pingoud V., Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Lite Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamagata A., Kakuta Y., Masui R., Fukuyama K. The crystal structure of exonuclease RecJ bound to Mn2+ ion suggests how its characteristic motifs are involved in exonuclease activity. Proc. Natl Acad. Sci. USA. 2002;99:5908–5912. doi: 10.1073/pnas.092547099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mashal R.D., Koontz J., Sklar J. Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nature Genet. 1995;9:177–183. doi: 10.1038/ng0295-177. [DOI] [PubMed] [Google Scholar]
- 35.Guan C., Kumar S. A single catalytic domain of the junction-resolving enzyme T7 endonuclease I is a non-specific nicking endonuclease. Nucleic Acids Res. 2005;33:6225–6234. doi: 10.1093/nar/gki921. [DOI] [PMC free article] [PubMed] [Google Scholar]