

Abstract
Background
Somatic genetic CDKN2A, TP53, and DNA content abnormalities are common in many human cancers and their precursors, including esophageal adenocarcinoma (EA) and Barrett's esophagus (BE), conditions for which aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) have been proposed as possible chemopreventive agents; however, little is known about the ability of a biomarker panel to predict progression to cancer nor how NSAID use may modulate progression. We aimed to evaluate somatic genetic abnormalities with NSAIDs as predictors of EA in a prospective cohort study of patients with BE.
Methods and Findings
Esophageal biopsies from 243 patients with BE were evaluated at baseline for TP53 and CDKN2A (p16) alterations, tetraploidy, and aneuploidy using sequencing; loss of heterozygosity (LOH); methylation-specific PCR; and flow cytometry. At 10 y, all abnormalities, except CDKN2A mutation and methylation, contributed to EA risk significantly by univariate analysis, ranging from 17p LOH (relative risk [RR] = 10.6; 95% confidence interval [CI] 5.2–21.3, p < 0.001) to 9p LOH (RR = 2.6; 95% CI 1.1–6.0, p = 0.03). A panel of abnormalities including 17p LOH, DNA content tetraploidy and aneuploidy, and 9p LOH was the best predictor of EA (RR = 38.7; 95% CI 10.8–138.5, p < 0.001). Patients with no baseline abnormality had a 12% 10-y cumulative EA incidence, whereas patients with 17p LOH, DNA content abnormalities, and 9p LOH had at least a 79.1% 10-y EA incidence. In patients with zero, one, two, or three baseline panel abnormalities, there was a significant trend toward EA risk reduction among NSAID users compared to nonusers (p = 0.01). The strongest protective effect was seen in participants with multiple genetic abnormalities, with NSAID nonusers having an observed 10-y EA risk of 79%, compared to 30% for NSAID users (p < 0.001).
Conclusions
A combination of 17p LOH, 9p LOH, and DNA content abnormalities provided better EA risk prediction than any single TP53, CDKN2A, or DNA content lesion alone. NSAIDs are associated with reduced EA risk, especially in patients with multiple high-risk molecular abnormalities.
In a ten-year study of people with Barrett's esophagus, nonsteroidal anti-inflamatory drugs were associated with reduced risk of esophageal adenocarcinoma, especially in patients with multiple high-risk molecular abnormalities.
Editors' Summary
Background.
Normally, the cells in the human body divide only when extra cells are needed, after an injury, for example. Sometimes, however, cells accumulate genetic changes (mutations) that allow them to divide uncontrollably to form a disorganized mass or tumor. If these altered cells also acquire mutations that allow them to spread around the body, a malignant tumor or cancer results. Scientists have identified numerous genetic changes that occur in tumors and are now investigating whether these molecular abnormalities can be used as “biomarkers” to choose the best treatments for patients, to identify who will benefit from cancer-prevention strategies, to detect cancer early, and to predict which cancers are most likely to become life-threatening. This last application is particularly important for cancers with a well-defined premalignant stage. Because the cells in premalignant tissues have acquired some of the genetic changes required for cancer development, they are more likely to become malignant than normal cells. Barrett's esophagus, for example, is a premalignant disorder of the muscular tube that takes food from the mouth to the stomach. People with Barrett's esophagus are much more likely to develop esophageal cancer than the general population.
Why Was This Study Done?
Esophageal cancer is often incurable by the time it is detected, so it would be helpful to know which people with Barrett's esophagus are most likely to develop esophageal cancer—only 1 in 200 of them develop cancer each year. In this study, the researchers evaluated whether a panel of genetic alterations could identify this subset of patients. They also investigated whether the regular use of aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) affects the risk of developing esophageal cancer in people with Barrett's esophagus—other evidence suggests that NSAIDs may help to prevent several types of cancer, including esophageal cancer.
What Did the Researchers Do and Find?
The researchers took esophageal tissue samples from patients with Barrett's esophagus and looked for alterations in the genes encoding the tumor-suppressor proteins TP53 and CDKN2A. These proteins normally stop cells dividing but are often inactivated in cancer cells by mutation of one of the two gene copies that encode each of them and also loss of the other copy (so-called “loss of heterozygosity” or LOH). The researchers also looked for changes in the cellular DNA content of the samples (tumor cells often contain unusual amounts of DNA) and asked the study participants about their NSAID use before waiting to see which participants developed esophageal cancer. After 10 y, the participants whose tissue samples had LOH of the short arms (p) of Chromosome 17 or 9 (the sites of the genes encoding TP53 and CDKN2A, respectively), or an altered DNA content, were more likely to have developed esophageal cancer than those without these abnormalities; those whose samples contained all three abnormalities had the highest risk of developing esophageal cancer. Overall, just 12% of patients with no abnormalities but nearly 80% of patients with three abnormalities developed esophageal cancer. NSAID use reduced the risk of cancer development in all the participants, but its effect was greatest in those with three genetic abnormalities.
What Do These Findings Mean?
These findings suggest that the combined measurement of 17pLOH, 9pLOH, and cellular DNA content might be a powerful way to identify those patients with Barrett's esophagus who are most likely to develop esophageal cancer. They also suggest that NSAID use is associated with a reduced risk of esophageal cancer, particularly in patients with multiple genetic abnormalities. Because very few participants developed cancer during the study, these results need confirming in more patients. Also, the ability of NSAIDs to prevent the progression of Barrett's esophagus to esophageal cancer needs testing in multicenter randomized trials; the use of the panel of abnormalities described here to identify the people with Barrett's esophagus most at risk of developing esophageal cancer should facilitate such studies.
Additional Information.
Please access these Web sites via the online version of this summary at http://dx.doi.org/10.1371/journal.pmed.0040067.
CancerQuest information from Emory University (Atlanta, Georgia, United States) on cancer biology, including the role of tumor suppressor proteins
US National Cancer Institute patient and physician information on esophageal cancer and its prevention
MedlinePlus encyclopedia pages on Barrett's esophagus and esophageal cancer
Cancerbackup (UK charity) patient information on esophageal cancer and Barrett's esophagus
Introduction
Rapid advances in understanding the molecular pathogenesis of neoplasia have raised the possibility that molecular abnormalities may be used as “biomarkers” for cancer risk stratification and early detection as well as possible entry criteria for cancer-prevention trials [1–4]. Identification of inherited, highly penetrant mutations in some cancer-susceptibility genes is being incorporated into clinical practice as well as cancer-prevention strategies for patients with many familial cancer syndromes, including inherited breast cancer, hereditary nonpolyposis colon cancer, and adenomatous polyposis coli [5–10]. Although progress in developing predictive biomarkers from common somatic genetic abnormalities in at-risk tissues has been less striking, there is some evidence that this approach may be successful. Based on a genetic progression model for head and neck cancer [11,12], a series of retrospective, longitudinal studies have been performed on patients with the premalignant condition oral leukoplakia, resulting in potential biomarker panels for risk stratification [13–16]. Some of these biomarkers have been proposed as entry criteria for a randomized cancer-prevention trial using cyclo-oxygenase-2 and epidermal growth factor receptor inhibitors [4]. Understanding how modifiable exposures interact with the somatic genetic composition of a neoplasm will be important for individualized interventions and cancer prevention.
Barrett's esophagus (BE) is a premalignant condition that predisposes to esophageal adenocarcinoma (EA). The incidence of this cancer has risen dramatically in the United States, Western Europe, Australia, and other developed countries over the past three decades, with little sign of abating [17,18]. Unfortunately, EA is typically lethal unless detected early, having an overall survival rate of only 13.7% [19]. Endoscopic surveillance is recommended for early detection in BE [20], and there has been recent interest in the potential for cancer prevention using nonsteroidal anti-inflammatory drugs (NSAIDs) based on data from case control and cohort studies [21–25], as well as preclinical models [26,27]. Analysis of genetic progression of BE has identified abnormalities in the tumor-suppressor genes TP53 and CDKN2A, as well as DNA content abnormalities (tetraploidy and aneuploidy) as critical events in the evolution of EA [28–40]. Several prospective studies have suggested that individual somatic genetic abnormalities derived from this progression model may identify those patients with BE who are at increased risk for progression to EA, but no study has evaluated the combined contributions of genetic abnormalities for EA risk prediction [34,41–45].
Using data collected prospectively over a 10-y period from a long-standing cohort study of patients with BE, we investigated the role of host factors in combination with somatic genetic abnormalities in the development of EA. The aims of the study were to determine whether somatic genetic abnormalities involving TP53, CDKN2A, and DNA content were predictors of progression to EA and whether regular NSAID use modulates risk of these abnormalities for future EA.
Methods
Study Design
This was a prospective, longitudinal study of the Seattle Barrett's Esophagus Study cohort. It constituted a phase 4 study according to the National Cancer Institute's Early Detection Research Network classification [3]. The Seattle Barrett's Esophagus Study was approved by the Human Subjects Division of the University of Washington in 1983 and was renewed annually thereafter with reciprocity from the Institutional Review Board of the Fred Hutchinson Cancer Research Center from 1993 to 2001. Since 2001, the study has been approved annually by the Institutional Review Board of the Fred Hutchinson Cancer Research Center with reciprocity from the Human Subjects Division of the University of Washington.
Participants
Two hundred and seventy-four participants were eligible as defined by a diagnosis of metaplastic columnar epithelium with intestinal metaplasia in esophageal biopsies, the absence of esophageal malignancy at or prior to baseline endoscopy, and having had at least one follow-up endoscopy. Two hundred and forty-three participants had sufficient tissue for flow cytometry, mutation, and LOH analysis in the same DNA samples and represented the final study cohort. Study participants entered surveillance from 1983 to 1999 (Table 1). The baseline endoscopy was defined as the first endoscopy from 5 January 1995 to 2 December 1999. Since this is a long-standing cohort, we assessed the enrollment-time effect by performing statistical analyses with all patients, and also with the subset of 211 patients, excluding the 32 patients who entered the cohort before 1990, and found no change in the conclusions of the study. This study was conducted at a specialty research and referral center, and thus our cohort is considered a high-risk patient population. We included all cancers that developed subsequent to the baseline evaluation so that accurate risk-stratification models could be developed based on findings at a single baseline endoscopy [44].
Table 1.
Cohort Characteristics
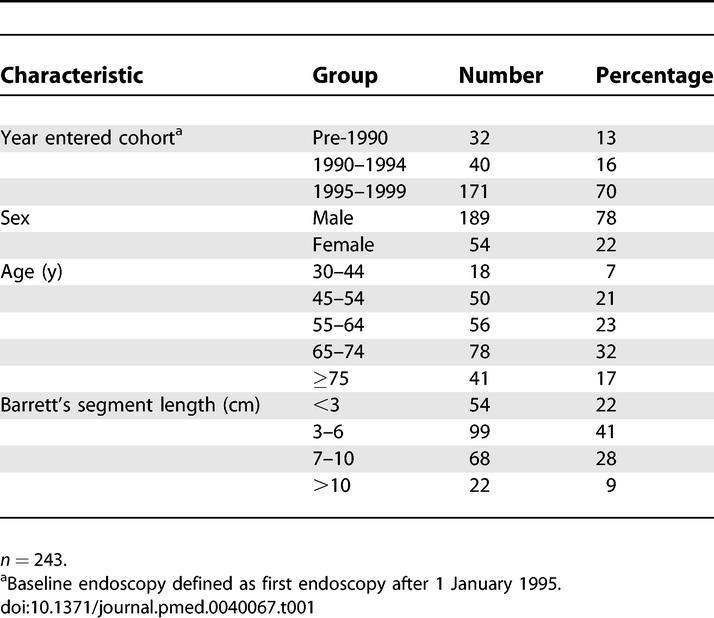
All research participants were counseled concerning the risks and benefits of endsocopic biopsy surveillance for BE. Patients with high-grade dysplasia (HGD) were also counseled concerning the risks and benefits of esophagectomy and endoscopic therapies. Endoscopic biopsy protocols used in the Seattle Barrett's Esophagus Study have been published previously [46]. Briefly, all patients had one biopsy evaluated for biomarkers every 2 cm in the Barrett's segment regardless of histologic diagnosis. In patients without HGD, four-quadrant biopsies were obtained for histologic evaluation every 2 cm in the Barrett's segment. If, after fully informed consent, participants with HGD opted for endoscopic biopsy surveillance reserving intervention for cancer if detected, they were evaluated by an intensive protocol of four-quadrant biopsies every 1 cm in the Barrett's epithelium at closely timed intervals for 4 mo, after which time endoscopies were typically repeated approximately every 6 mo [47]. Exclusion of patients who were diagnosed with EA that developed within 4 mo of their baseline procedure (n = 4) did not alter the conclusions of the study. Participants with maximum baseline diagnosis of HGD had an average of 7.3 endoscopies (median = 6.5, range 2–18), compared to those with less HGD at baseline, who had an average of 4.8 endoscopies (median = 4, range 2–20).
The cohort is typical for gender, age, and Barrett's segment length compared to other specialty research centers (Table 1) [48–51]. The cohort included 189 males and 54 females with a mean age of 62 y (median = 64 y, range 30–87 y) at entry into the study. This cohort was comprised of participants with maximum baseline histologic diagnoses including negative for dysplasia (n = 67), indefinite (n = 78), low-grade dysplasia (n = 48), and HGD (n = 50) using previously published pathology criteria [43,52]. The mean Barrett's segment length, defined by total centimeters between the ora serrata and the distal end of the tubular esophagus proximal to the flattening of the gastric folds, was 5.6 cm (median = 5 cm, range 0–20 cm). Participants were followed for a total of 17,139 patient-months with a mean of 71 mo (median = 80.5 mo, range 2.3–130.8 mo).
Endoscopy and Biopsy
Endoscopy and biopsy were performed using a standard protocol [43]. Participants with a history of HGD (n = 50) had histologic evaluation of four-quadrant biopsies at 1-cm intervals in the Barrett's segment, whereas those without a history of HGD (n = 193) had histologic analysis of four-quadrant biopsies at 2-cm intervals. Histologic analyses were evaluated in biopsies obtained at the baseline evaluation and at all follow-up endoscopies, whereas TP53, CDKN2A, and DNA content analyses were evaluated at a single baseline endoscopy. For each baseline endoscopy, samples were characterized for 17p LOH spanning TP53, TP53 mutations, DNA content abnormalities including tetraploidy and aneuploidy, 9p LOH spanning the CDKN2A (p16) locus, CDKN2A promoter methylation, and CDKN2A mutation in one biopsy every 2 cm in the Barrett's segment (average = 3.2, range 1–11 biopsies analyzed per study participant). For a participant to be counted as having a molecular abnormality at baseline, the abnormality must have been detected in more than one flow-purified DNA fraction or in a single sample that has been confirmed in a second, independent reaction.
Flow Cytometry
Biopsies were processed by DNA content (DAPI) and Ki67/DNA content flow cytometry to determine 4N fraction and ploidy, and to purify proliferating cells using previously validated techniques [43,45,53]. Flow-purified fractions are defined as Ki67-positive (proliferating) 2N, 4N, increased 4N/tetraploid (cells with DNA content between 3.85 and 4.10N that comprise >6% of the total cells), and aneuploid populations (distinct DNA content peak representing >2.5% of cells). Cell-cycle analysis was performed on 1,569 flow-purified fractions with a median of 6.5 (range 1–29) cell-cycle fractions depending on Barrett's segment length.
DNA Extraction, Whole-Genome Amplification and LOH Analysis
DNA was extracted from flow-purified cell populations using either standard phenol/chloroform or the Puregene DNA Isolation Kit as recommended by the manufacturer (Gentra Systems, Minneapolis, Minnesota, United States). Whole-genome amplification using primer extension preamplification was performed with each sorted fraction and three constitutive controls per participant [53]. LOH data was obtained from 1,331 and 1,284 flow-purified fractions at 17p and 9p loci, respectively, as detailed previously [34,44,53]. Thirteen microsatellite loci were evaluated, including the 17p loci D17S1298 (3.87 Mbp), D17S1537 (6.10 Mbp), TP53-ALU (AAAAT)n in intron 1 (7.77 Mbp), TP53 (CA)n (7.77 Mbp), D17S786 (9.01 Mbp), D17S974 (10.72 Mbp), D17S1303 (11.06 Mbp), and Chromosome 9p loci D9S2169 (5.19 Mbp), D9S935 (5.19 Mbp), D9S925 (18.28 Mbp), D9S932 (24.43 Mbp), D9S1121 (25.39 Mbp), and D9S1118 (31.92 Mbp). Physical map locations were determined from the University of California, Santa Cruz (Santa Cruz, California, United States) version hg16 July 2003 assembly (http://genome.cse.ucsc.edu). LOH was defined as QLOH ≤ 0.4 or ≥ 2.5 for loss which spanned TP53 or CDKN2A [34,44,53]. For convenience, we used the nomenclature of 17p LOH and 9p LOH to describe LOH events spanning these genes. LOH was not scored for rare telomeric or centomeric loss events that did not span TP53 or CDKN2A. 17p LOH for all participants and 9p LOH in HGD patients have been previously published [34,44].
CDKN2A Promoter Methylation Analysis
Genomic DNA from flow-purified Barrett's epithelium was evaluated for CDKN2A promoter methylation in 175 flow-purified fractions from 121 participants. Only a subset (n = 121) of the patients had CDKN2A methylation tested because the bisulfite treatment assays used required large amounts of DNA, and the DNA from our flow-purified fractions was insufficient for all molecular assays to be performed. This patient subset was representative of the entire 243-patient cohort with no statistically significant difference in follow-up time, sex, age, segment length, and cancer outcome between the methylation-assayed group and the non-assayed group. DNA was bisulfite treated and methylation-specific PCR was performed with modifications as detailed previously [31,54,55]. Human genomic DNA treated in vitro with Sss I methyltransferase (New England Biolabs, Beverly, Massachusetts, United States) was used as the methylated control. In a subset of cases, promoter methylation was determined and/or verified by directly sequencing PCR products of bisulfite-treated genomic DNA using published primers and methods [31]. Methylation data from a subset of patients has been previously reported [31].
DNA Sequencing
Genomic or primer extension preamplification DNA was sequenced using either BigDye or BigDyeV3 Terminator cycle sequencing (Applied Biosystems, Foster City, California, United States) on either an ABI 377, ABI 3730, or ABI 3700 DNA sequencer. Wild-type sequences for each participant were confirmed using constitutive samples. All mutations were confirmed by at least two independent PCR and sequencing reactions and, in cases of ambiguity, by direct sequencing of genomic DNA. Evaluation of mutation of exons 5–9 of the TP53 gene was performed on 1,118 flow-purified fractions under conditions described previously [56]. TP53 mutations from patients with HGD were reported by Prevo et al. [56], and additional new TP53 mutations not previously reported are available from the corresponding author (PCG). Mutation analysis of exon 2 of the CDKN2A gene was performed in 1,109 flow-purified fractions, detailed by Paulson 2006 (unpublished data) and with methods described previously [31].
Use of Aspirin and Other NSAIDs
Questionnaires and methods for determining host variables were as described previously [25] and were available for 241 participants. Use of aspirin and other NSAIDs in the current study was defined in the same way as NSAID use, including NSAID use in follow-up, as described in Vaughan et al. [25]. Briefly, duration and frequency of regular aspirin and NSAID use were assessed, with “regular” defined by use at least once per week for ≥6 mo. The NSAID variable took into account changes in aspirin and NSAID medication use during the follow-up period. It has been shown previously that there are no significant differences between the protective associations of aspirin and other NSAIDs in this cohort [25]. Patients were classified as being users (including patients who were regular aspirin or other NSAID users within 1 y of the baseline interview or at any time during follow-up, n = 157), or nonusers (former users or those who had never used NSAIDs, n = 84).
Statistical Analysis
We sought to evaluate the extent to which specific tissue-based, mechanistically derived molecular markers, both alone and in combination, predict risk of progression to EA in patients with BE. The analytic approach is summarized as follows. (1) Univariate Cox proportional hazard analyses (not adjusted for host variables or other markers) were used to determine the EA risk of patients with each marker, measured at baseline at 2-, 6-, and 10-y follow-up time points (Table 2). (2) Stepwise multivariate Cox regression was used to select, from among all of the available molecular markers, a subset which independently contributes to EA risk prediction (Table 3). (3) Cumulative EA incidence and relative risk (RR) of progression to EA at 10 y were calculated for patients with different numbers of the selected markers at baseline relative to patients with no abnormalities (Table 4). To determine the contribution of known or suspected nongenetic host factors to EA risk prediction, we incorporated these host factors together with all molecular markers in a multivariate Cox model. Only NSAID use showed significant independent prediction in combination with molecular markers in this multivariate model. The predictive ability of the selected molecular markers was evaluated among NSAID users and nonusers (Figures 1 and 2).
Table 2.
Univariate Analysis of RR for EA during Follow-up

Table 3.
Stepwise Selection for Molecular Markers and RR for EA of Final Selected Markers
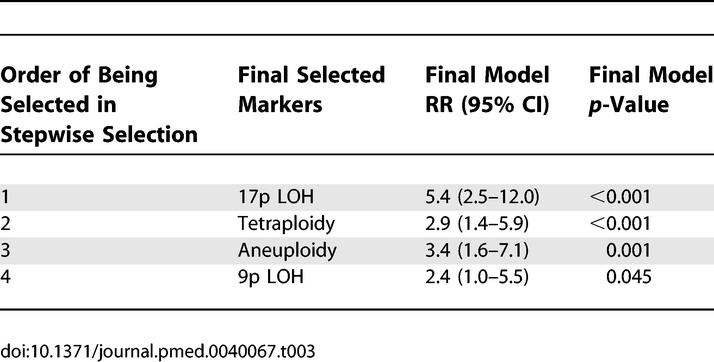
Table 4.
Cumulative EA Incidence and RR of Different Baseline Abnormality Combinations

Figure 1. Modulation of EA Risk by NSAIDs in Participants with Different Baseline Abnormalities.

Two hundred and forty-one patients are classified according to whether they have (A) baseline 17p LOH (n = 46), (B) baseline DNA content abnormalities (aneuploidy and/or tetraploidy) (n = 41), (C) baseline 9p LOH (n = 144), or (D) more than one baseline abnormality (top two curves) or one or less abnormality (lower two curves). Shown are Kaplan-Meier curves of cancer incidence rates in patients who are NSAID nonusers (former or never users, red curves) or NSAID users (current or user during follow-up, black curves).
Figure 2. Cumulative EA Incidence with Combinations of Abnormalities (17p LOH, DNA Content Abnormality, 9p LOH) in NSAID Nonusers and NSAID Users.

Cancer incidence rates are shown for participants with no selected abnormalities (17p LOH, DNA content abnormalities [aneuploidy and/or tetraploidy], or 9p LOH) at baseline (red), any one abnormality (green), any combination of two abnormalities (blue), or all three abnormalities (black). (A) All participants. When comparing NSAID nonusers (B) and NSAID users (C) there is a strong significant trend toward EA risk reduction in the NSAID users group for all abnormality combinations (Mantel-Haenszel test p = 0.01).
Two hundred and forty-three patients had informative data for all of the molecular markers (17p LOH, TP53 mutation, tetraploidy, aneuploidy, 9p LOH, and CDKN2A mutation). Except for CDKN2A methylation, all markers in the analysis were binary variables coded as 0 (no), 1 (yes) for each participant. CDKN2A methylation status was coded as yes, no, or no data in the statistical model because only 121 patients had sufficient quantity of DNA to be evaluated for this marker. In the Cox regression models for molecular marker selection, the statistical significance levels used in the initial process of stepwise selection for variable entry and removing were 0.25 and 0.15, respectively. Based on initial selection results, p = 0.05 was used as the significance threshold for the final selection (second Cox model). Association relationship among binary markers was also assessed using coefficient Φ. The Kaplan-Meier method was used to estimate the probability of EA in different stratified groups. The Gehan-Wilcoxon test was used to test differences in the cancer incidence curves. The Grambsch-Therneau test was used to examine the significance of a change in RR ratio over time for all markers. We adjusted p-values for multiple comparisons in Figure 2 using the Benjamini and Hochberg method [57]. All of the analyses were performed with Statistical Analysis System (SAS) software Version 9.0; (SAS Institute, http://www.sas.com).
Results
Univariate Analysis of LOH, Mutation, Methylation, Tetraploidy, and Aneuploidy for EA Risk Prediction
The cohort characteristics for participants used for molecular and DNA content analysis are shown in Table 1. During the follow-up period of this study, 34 participants developed EA. The number of participants with each genetic abnormality at baseline and the cumulative number who developed cancer at 2, 6, and 10 y are shown in Table 2. The risk of EA during follow-up in patients with each molecular and DNA content abnormality was assessed with univariate analysis using Cox regression models (2, 6, and 10 y presented to show general trend, Table 2). The RRs presented in Table 2 were not adjusted for host variables, but the significance of the results does not change when such adjustments are performed. At 10 y, each molecular and DNA content abnormality, when analyzed alone in a patient at baseline, made a significant contribution to prediction of EA risk, with the exception of CDKN2A mutation (10-y RR = 1.8; 95% CI 0.8–4.1, p = 0.13) and CDKN2A methylation (RR = 2.1; 95% CI 0.8–4.1, p = 0.09). For example, the univariate RRs ranged from a high for 17p LOH (10-y RR = 10.6; 95% CI 5.2–21.3, p < 0.001) to that for 9p LOH (10-y RR = 2.6; 95% CI 1.1–6.0, p = 0.03). Although RRs at intermediate time points may be unstable owing to small numbers, 9p LOH had a higher RR at early follow-up intervals. The RR in univariate analysis for 9p LOH was 8.9 (95% CI 1.2–67.9, p = 0.04) at 2-y follow-up, but decreased to RR = 2.6 by 10 y. With the current dataset, 9p LOH was the only abnormality that showed a statistically significant change in RR over time (nonproportional hazard over time, p = 0.004, Grambsch-Therneau test for global trend at 10 y).
Comprehensive Analysis of Relationships among the Multiple Molecular Abnormalities for EA Risk Prediction
To determine a set of biomarkers that are independent predictors of EA risk, baseline molecular markers were evaluated using multivariate Cox models with backward and forward stepwise selection. Starting with all molecular abnormalities for selection, 17p LOH was the most significant abnormality selected by the statistical model, and remained a strong predictor when accounting for TP53 mutation status in a step-wise analysis process. Although TP53 mutation was highly significant in univariate analysis, it became nonsignificant for EA risk prediction when 17p LOH was included in the model. Tetraploidy was the next selected marker, followed by aneuploidy. Finally, 9p LOH improved the model for risk prediction significantly in combination with 17p LOH and DNA content abnormalities (likelihood ratio test p = 0.03). CDKN2A methylation provided marginal additional risk but was not statistically significant in the multivariate model (p = 0.28). CDKN2A mutation did not provide significant additional contribution to EA risk prediction. The final selected molecular markers that independently contributed to EA risk prediction included 17p LOH, tetraploidy, aneuploidy, and 9p LOH (Table 3). To further evaluate the associations among the markers, we calculated the association coefficient (Φ) for all binary markers. TP53 mutation showed strong association with 17p LOH (Φ = 0.63) and aneuploidy (Φ = 0.56). 17p LOH is also strongly associated with aneuploidy (Φ = 0.56). No other strong associations were observed by this method. Although TP53 mutation was significantly related to EA risk in univariate analysis, it was strongly associated with 17p LOH and aneuploidy, which may explain why it was not retained in the model selection. The adjusted RRs for future EA at 10 y, using the final selected model with 17p LOH, tetraploidy, aneuploidy, and 9p LOH, were RR = 5.4 (95% CI 2.5–12.0), RR = 2.9 (95% CI 1.4–5.9), RR = 3.4 (95% CI 1.6–7.1), and RR = 2.4 (95% CI 1.0–5.5), respectively (Table 3).
Effect of Combinations of Selected Panel Abnormalities on Cumulative EA Incidence
Each study participant had the potential to have any combination of selected panel abnormalities at baseline. Stratification by all possible combinations of the selected abnormalities was limited by an inadequate number of patients for analysis in some strata, and we therefore grouped participants based on the number of abnormalities at baseline. Table 4 lists the cumulative EA incidence at 2, 6, and 10 y, and the RR at maximum follow-up (10 y) for future development of EA for participants with no panel abnormalities, a single abnormality (either 17p LOH, any DNA content abnormality, or 9p LOH), any two selected abnormalities, or all three selected abnormalities at baseline. Tetraploidy and aneuploidy are two measures of DNA content and were treated as a single variable in this section (Figures 1 and 2; Table 4). Study participants with no baseline abnormalities (85/243) remained cancer free to almost 8 y (95 mo) and had a relatively low 10-y cumulative cancer incidence of 12%. In participants with only a single baseline abnormality (104/243), the 6-y EA incidence was 5.65%, with an overall 10-y EA incidence of 19.88%. It is of note that 9p LOH was the single abnormality in 98/104 of those participants, indicating that it was rare for 17p LOH or a DNA content abnormality to be detected alone. Participants with two abnormalities detected at baseline (32/243) had increasing cumulative EA incidence of 16.83%, 28.4%, and 35.56% at 2, 6, and 10 y, respectively, showing a significantly higher EA risk than for patients with no abnormalities at baseline (RR = 9.0; 95% CI 2.4–33.3, p < 0.001). Participants (22/243) with all three abnormalities (17p LOH, DNA content abnormality, and 9p LOH) had EA incidence rates of 40.2% and 79.12% at 2 and 6 y, respectively. Owing to sample size, the EA incidence rate at the 10-y time point was not estimated. These patients had the highest risk of cancer relative to those with a no baseline abnormalities (RR = 38.7; 95% CI 10.8–135.5, p < 0.001). Of these cancer cases, 80% occurred within 36 mo, and all cancers that developed in patients with three baseline abnormalities did so within a 55-mo follow-up period.
Association of NSAID Use with Reduction of EA Risk in Participants with 17p LOH, DNA Content Tetraploidy and Aneuploidy, and 9p LOH
Multiple nongenetic host factors and demographic variables have been previously suggested as potential modifiers of EA progression including age, gender, waist-to-hip ratio, smoking status, segment length, and NSAID use [24,58–67]. To determine the host variables that provide independent prediction for EA when combined with molecular markers, we incorporated these nongenetic host factors with all molecular markers into a new Cox model for variable selection. The selected independent variables were 17p LOH, DNA content abnormalities (tetraploidy and aneuploidy), 9p LOH, and NSAID use. In combination with the selected molecular abnormalities, NSAID use was associated with a statistically significant reduction of EA risk (p < 0.001). Thus, NSAID use was evaluated as a potential modulator of cancer risk in combination with selected genetic abnormalities. For a given selected molecular marker, Kaplan-Meier analysis showed that NSAID use during follow-up provided a significant association with protection against development of EA, relative to participants who were NSAID nonusers (former or never) (Figure 1). This protective effect was significant in patients with baseline 17p LOH (p = 0.004, Gehan-Wilcoxon test), DNA content abnormalities (tetraploidy and/or aneuploidy) (p = 0.01), and 9p LOH (p <0.001) (Figure 1A–C). For patients with more than one panel abnormality at baseline, NSAID use was associated with a significant reduction in EA incidence (p = 0.001): NSAID nonusers had an observed cumulative EA risk of 68%, compared to 30% for NSAID users at 6 y, and 79% compared to 30% for NSAID users at 10 y (Figure 1D). In participants with either no abnormalities or one abnormality at baseline (predominantly 9p LOH, Table 4), cancer risk was low, and the protective effect of NSAIDs for future EA was not significant (p = 0.28 at 10-y follow-up).
EA Incidence in NSAID Nonusers and NSAID Users Stratified by Combinations of Baseline Abnormalities
Cancer incidence of all participants based on the overall number of select panel abnormalities at baseline is shown in Kaplan-Meier curves (Figure 2A). When comparing NSAID nonusers (Figure 2B, n = 84) and NSAID users (Figure 2C, n = 157) within all categories (zero, one, two, or three abnormalities at baseline), there was a strong significant trend toward EA risk reduction in the NSAID user's group (Mantel-Haenszel test p = 0.01). NSAID nonusers with two or three abnormalities at baseline had a highly significant increased EA incidence compared to NSAID nonusers with zero or one lesion (two versus zero p < 0.001; two versus one p <0.001; three versus zero p < 0.001; three versus one p < 0.001; adjusted for multiple comparisons with Benjamini and Hochberg method [57] [Figure 2B]). All other comparisons for NSAID nonusers, for example between participants with two versus three abnormalities, were nonsignificant. Participants who used NSAIDs regularly (Figure 2C) and had all three abnormalities at baseline had a significantly increased cumulative EA incidence compared to NSAID users with zero, one, and two abnormalities (p < 0.001, p < 0.001, and p = 0.045, respectively). NSAID users with two baseline abnormalities had a significant or marginally significant increased cancer incidence relative to NSAID users with zero or one abnormality (p = 0.013 and p = 0.07, respectively).
Discussion
This investigation reports the results of a prospective cohort study of mechanistic-based genetic abnormalities evaluated as predictors of EA and demonstrates the modulating effect of NSAIDs on EA risk in patients with BE. We hypothesized that a panel of somatic genetic abnormalities involving TP53, CDKN2A, and DNA content could improve prediction of progression to EA and that NSAID use may modulate EA risk. In this longitudinal study spanning more than a decade, we showed that a combination of 17p LOH, 9p LOH, and DNA content tetraploidy and aneuploidy provide significant, independent EA risk prediction. NSAID use is associated with reduction of EA risk, and the protective effect was highly significant for patients who have multiple high-risk molecular abnormalities at baseline. These analyses include 34 EA endpoints, which is second only to our previous 15-y report of histology and flow cytometry (42 EAs) and substantially larger than most other longitudinal studies of biomarkers in BE from other centers, which have typically reported 12 or fewer incident cancers [41,42,68,69]. This prospective study has been conducted in a single center with a high-risk cohort; studies in other centers will be required to determine whether our results can be generalized to other patient populations and to validate the results for clinical application. Our results are consistent, however, with previous longitudinal studies of single biomarkers from other centers, including TP53 abnormalities and flow cytometry [41,42,70]. To our knowledge, no previous studies in patients with BE or any other human premalignant condition have prospectively evaluated the contributions of TP53 and CDKN2A gene inactivation (methylation, mutation, and LOH) and DNA content abnormalities in combination with candidate interventions to assess their potential utility as biomarkers for future cancer risk and cancer prevention.
The mechanisms by which TP53 and CDKN2A regulate the cell cycle under normal and abnormal conditions have been investigated extensively in elegant molecular studies in vitro and in model organisms. Perturbations of these genes and the pathways in which they act have profound, mechanistic associations with human cancer based upon evidence accumulated in numerous laboratories [1,71–75]. In BE, neoplastic progression is characterized by clonal evolution in which genetic instability generates variants on which natural selection acts, resulting in waves of clonal expansion, generation of new variants, and further selection [76,77]. TP53 abnormalities typically arise in clones with CDKN2A abnormalities [78], creating a condition permissive for clonal variants, including tetraploid and aneuploid populations, to survive and expand [79]. Thus, the abnormalities in this biomarker panel assess viable clones that undergo expansion (CDKN2A) and survive chromosomal instability (TP53, DNA content). Assessment of multiple stages of clonal evolution may be the basis for improved risk stratification compared to single biomarkers.
We simultaneously measured DNA content abnormalities (tetraploidy and aneuploidy), inactivation of TP53 (mutation and LOH), and inactivation of CDKN2A (mutation, methylation, and LOH), all of which have been shown to be mechanistically related to neoplastic progression in BE [28,29,34,80–82]. TP53 abnormalities have been shown in numerous studies to be predictive of EA [41,70,83–85]. In the present study, TP53 mutations were strongly associated with 17p LOH and aneuploidy and were not selected in the multivariate analysis. Selection of LOH over methylation or mutation in the Cox model could occur because LOH is a common manifestation of the chromosomal instability that is characteristic of neoplastic progression in BE [29]. LOH could also be selected as the “second hit” for inactivating TP53 and CDKN2A. Alternatively, LOH events often span large chromosomal regions that could include other genes such as HIC1 (17p13.3) [86] and p14ARF (9p21) [72,87] that may confer additional selective advantages over mutational or methylation events that affect only TP53 and CDKN2A. Recently, Maley et al. reported that clonal diversity measures derived from evolutionary biology retained significant independent EA risk prediction with 17p (TP53) LOH and abnormal ploidy, but 9p LOH became nonsignificant when incorporating evolutionary variables [88]. The nonproportional hazard varying with time that we found for 9p LOH in the present study may reflect the genetic background of the CDKN2A clone. For example, expansion of a CDKN2A abnormal clone that is otherwise genetically stable may homogenize the neoplasm, minimizing diversity on which natural selection might act to promote progression [76,77]. In contrast, expansion of a CDKN2A abnormal clone predisposed to genetic instability through either environmental or somatic genetic factors would result in increased diversity that could promote progression.
Although this study had a large number of EA endpoints for published studies of BE, the number of cancers was relatively small compared to studies in breast and colon, for example. Therefore, estimates of RR over time showing the 9p LOH nonproportional hazard over time need to be further investigated in future studies. In addition, RR for EA presented in Table 2 at intermediate follow-up times should be treated with caution. We do not recommend that clinicians manage patients on these data alone. Within the BE research field, there is a paucity of data concerning progression to EA in patients with different molecular abnormalities. We present the intermediate time points to inform design of future intervention and multicenter studies, while maintaining statistical rigor in our analyses.
Identification of host genetic factors, including inherited, highly penetrant mutations in cancer susceptibility for hereditary breast cancer (BRCA1, BRCA2), familial polyposis coli (APC), and those predisposing to hereditary non-polyposis colon cancer, among others, in combination with knowledge of environmental factors, have the potential to reduce cancer morbidity and mortality by early detection and prevention [5,6,9,10,89–91]. In contrast to inherited mutations in relatively uncommon susceptibility genes, less is known concerning temporal progression of somatic genetic abnormalities in more prevalent sporadic premalignant conditions. Genetic progression models have been proposed for many types of cancers based largely on cross-sectional data [11,13,14,92–104]. TP53, CDKN2A, and DNA content (tetraploidy and aneuploidy) abnormalities are among the most common abnormalities in cancers and premalignant conditions affecting multiple organs, including head and neck, lung, breast, bladder, and pancreas, among others [1,71–73]. Few data exist as to their ability to predict future cancer or how these lesions can be modulated by chemoprevention efforts. Advances have been made in cancer risk prediction for patients with oral premalignant lesions in multiple retrospective, longitudinal studies [13–16]. Lee et al. combined multiple biomarkers and patient characteristics in Cox regression analysis and found that chromosome polyploidy, together with high p53 expression, LOH, and histology was the best predictor of cancer risk in a 10-y study of 70 patients with 22 cancer outcomes [13]. Thus, genetic progression models may be a rich source of hypotheses for retrospective longitudinal and prospective biomarker validation studies.
In experimental model systems and observational studies, aspirin and other NSAIDs have been reported to inhibit cyclo-oxygenase 2, increase apoptosis, decrease inflammation, decrease proliferation, and inhibit angiogenesis [4,26,27,105–111]. It has recently been shown that the absolute size of aneuploid clones and clones with TP53 lesions is a risk factor for progression to EA [79]. NSAIDs may act by reducing clone size through increasing apoptosis and decreasing angiogenesis and proliferation. By increasing apoptosis, NSAIDs may decrease the generation of viable clones, thus decreasing diversity and limiting the pool of genetic variants on which natural selection may act. In addition, NSAIDs function to reduce inflammation, which may in turn reduce the mutation rate in evolving clones and decrease the number of cellular variants. The cohort analyzed in the present report is a subset of that described in Vaughan et al. [25]. Of the molecular cohort described in the present study, 65% were current NSAID users—including use in follow-up—and this proportion is comparable with the 63% regular NSAID users (including use in follow-up) in the total cohort. About half of the users took aspirin for reasons concerning cardiovascular health, and the vast majority of the other users primarily took ibuprofen for pain relief. In a previous paper, we reported a strong protective association for EA with current NSAID use (at baseline or during follow-up), a rather rapid diminution of the association among former users, and no evidence of a stronger association with increasing frequency and/or duration of use. Furthermore, we did not find significant differences in risk of EA or aneuploidy according to type of NSAID. Thus for the present report, our patients were classified simply as NSAID user or nonuser (former or never) based on use at least once per week for ≥6 mo any time during follow-up, regardless of daily frequency, duration, or type. By focusing on the salient results from the previous analyses of a larger dataset regarding NSAIDs, we avoided multiple comparisons and could focus on the relationship of NSAID use with the status of molecular abnormalities.
Our results show a protective association between NSAID use and progression to EA in all participants, and particularly among those with multiple somatic genetic abnormalities. The vast majority of patients in this cohort had gastroesophageal reflux disease and were undergoing therapy, predominately using proton pump–inhibitors, to reduce reflux symptoms. It is unclear as to how the frequency or severity of symptoms may affect NSAID use by participants. However, to our knowledge, symptoms from reflux are not associated with intermediate endpoints or cancer in BE, so it is unlikely that symptoms could explain the association of NSAID use with reduced risk of EA. None of the patients had endoscopically visible concomitant conditions of the stomach, such as ulcers, at the baseline endoscopy that could have conceivably altered NSAID use. Given the experimental evidence provided in model systems and in previous studies, we propose that NSAIDs act within the BE tissue to modulate progression to EA. Our study is early translational research, as defined by the National Cancer Institute Translational Research Working Group (http://www.cancer.gov/aboutnci/trwg/presentations). These results advance our understanding of the molecular mechanisms of neoplastic progression as well as the mechanisms by which aspirin and other NSAIDs may prevent cancer. As such, these results are consistent with the National Institutes of Health goals of prevention, prediction, and personalized medicine. Several other types of research will be essential for these results to reach the clinic: multicenter randomized trials with a mechanistic focus to determine the effects of aspirin or other NSAIDs on progression to EA and intermediate endpoints (high-risk biomarkers), health services research to evaluate the effect of the interventions as well as the cost effectiveness of biomarkers that reduce frequencies of endoscopy and numbers of biopsies, and establishment of reimbursement mechanisms to support dissemination and adoption (National Cancer Institute's translational continuum; http://www.cancer.gov/trwg/TRWG-definition-and-TR-continuum).
Our study builds on a body of observational research, a recent meta-analysis, and supportive data in other cancers, most notably colon cancer where several clinical trials have reported a protective association of aspirin and other NSAIDs, although some findings have been inconsistent [9,10,21–25,89–91,112–116]. The present study extends these findings by evaluating the association of NSAID use with somatic genetic events that define progression in BE and demonstrates the benefits of NSAID use in patients at high risk of progressing to EA. Our results are consistent with previous results showing a protective effect on the part of NSAIDs in patients with HGD [25], and with computer models indicating that high-risk patients are the most likely to benefit from NSAID intervention [117]. The incidence of EA is sufficiently low in BE that designing statistically rigorous, adequately powered prevention studies with cancer as an endpoint is not feasible for most research cohorts. Thus, knowledge of the interaction of NSAIDs and somatic genetic abnormalities will help define entry criteria for randomized intervention trials with a cancer endpoint. The combination of a somatic genetic biomarker panel that identifies those patients with BE who are at high risk of progression to EA, combined with an inexpensive, widely available, and relatively safe means of preventing neoplastic progression in such high-risk patients, could have significant public health and economic benefits.
Supporting Information
Accession Numbers
The GenBank (http://www.ncbi.nlm.nih.gov/Genbank) accession numbers for the genes discussed in this paper are TP53 (p53) (7157), CDKN2A (p16, p16-INK4a)/p14ARF (1029), and HIC1 (3090).
Acknowledgments
We thank foremost the study participants who have made this study possible. We thank Janine Kikuchi, Terri Watson, and David Cowan for database support; Christine Karlsen for patient care coordination; Valerie Cerera for research biopsy coordination and flow cytometry; Laura Prevo, Jessica Arnaudo, and Heather Kissel for molecular assays; and Tricia Christopherson for interview management.
Abbreviations
- BE
Barrett's esophagus
- CI
confidence interval
- EA
esophageal adenocarcinoma
- HGD
high-grade dysplasia
- LOH
loss of heterozygosity
- NSAID
nonsteroidal anti-inflammatory drug
- RR
relative risk
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Author contributions. PCG, PLB, CAS, TLV, and BJR contributed to conception and design of the study. PCG, PLB, CAS, KA, TLV, and BJR collected data or did experiments for the study. PCG, XL, CCM, CAS, RDO, PSR, TLV, and BJR contributed to analysis and interpretation of the data. PLB and BJR enrolled patients. PCG, XL, PLB, CCM, CAS, RDO, KA, PSR, TLV, and BJR contributed to drafting the article or revising it critically. All authors approved the final accepted version of the manuscript.
Funding: This study was funded by the US National Institutes of Health (P01 CA91955, R01 CA61202, and R01 CA78828) with interim support from the Fred Hutchinson Cancer Research Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- O'Shaughnessy JA, Kelloff GJ, Gordon GB, Dannenberg AJ, Hong WK, et al. Treatment and prevention of intraepithelial neoplasia: An important target for accelerated new agent development. Clin Cancer Res. 2002;8:314–346. [PubMed] [Google Scholar]
- Kelloff GJ, O'Shaughnessy JA, Gordon GB, Hawk ET, Sigman CC. Counterpoint: Because some surrogate end point biomarkers measure the neoplastic process they will have high utility in the development of cancer chemopreventive agents against sporadic cancers. Cancer Epidemiol Biomarkers Prev. 2003;12:593–596. [PubMed] [Google Scholar]
- Sullivan Pepe M, Etzioni R, Feng Z, Potter JD, Thompson ML, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93:1054–1061. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: Pharmacologic targets for chemoprevention. J Clin Oncol. 2005;23:254–266. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- Hawk E, Lubet R, Limburg P. Chemoprevention in hereditary colorectal cancer syndromes. Cancer. 1999;86:2551–2563. doi: 10.1002/(sici)1097-0142(19991201)86:11+<2551::aid-cncr12>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124:1574–1594. doi: 10.1016/s0016-5085(03)00376-7. [DOI] [PubMed] [Google Scholar]
- Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- Giardiello FM, Yang VW, Hylind LM, Krush AJ, Petersen GM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002;346:1054–1059. doi: 10.1056/NEJMoa012015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, et al. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- Califano J, Westra WH, Meininger G, Corio R, Koch WM, et al. Genetic progression and clonal relationship of recurrent premalignant head and neck lesions. Clin Cancer Res. 2000;6:347–352. [PubMed] [Google Scholar]
- Lee JJ, Hong WK, Hittelman WN, Mao L, Lotan R, et al. Predicting cancer development in oral leukoplakia: Ten years of translational research. Clin Cancer Res. 2000;6:1702–1710. [PubMed] [Google Scholar]
- Rosin MP, Cheng X, Poh C, Lam WL, Huang Y, et al. Use of allelic loss to predict malignant risk for low-grade oral epithelial dysplasia. Clin Cancer Res. 2000;6:357–362. [PubMed] [Google Scholar]
- Mao L, Lee JS, Fan YH, Ro JY, Batsakis JG, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med. 1996;2:682–685. doi: 10.1038/nm0696-682. [DOI] [PubMed] [Google Scholar]
- Hittelman WN, Kim HJ, Lee JS, Shin DM, Lippman SM, et al. Detection of chromosome instability of tissue fields at risk: In situ hybridization. J Cell Biochem. 1996;25:57–62. [PubMed] [Google Scholar]
- Bollschweiler E, Wolfgarten E, Gutschow C, Holscher AH. Demographic variations in the rising incidence of esophageal adenocarcinoma in white males. Cancer. 2001;92:549–555. doi: 10.1002/1097-0142(20010801)92:3<549::aid-cncr1354>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Vizcaino AP, Moreno V, Lambert R, Parkin DM. Time trends incidence of both major histologic types of esophageal carcinomas in selected countries, 1973–1995. Int J Cancer. 2002;99:860–868. doi: 10.1002/ijc.10427. [DOI] [PubMed] [Google Scholar]
- Brown LM, Devesa S. Epidemiologic trends in esophageal and gastric cancer in the United States. Surg Oncol Clin North Am. 2002;11:235–256. doi: 10.1016/s1055-3207(02)00002-9. [DOI] [PubMed] [Google Scholar]
- Sampliner RE. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett's esophagus. Am J Gastroenterol. 2002;97:1888–1895. doi: 10.1111/j.1572-0241.2002.05910.x. [DOI] [PubMed] [Google Scholar]
- Bosetti C, Talamini R, Franceschi S, Negri E, Garavello W, et al. Aspirin use and cancers of the upper aerodigestive tract. Br J Cancer. 2003;88:672–674. doi: 10.1038/sj.bjc.6600820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: A systematic review and meta-analysis. Gastroenterology. 2003;124:47–56. doi: 10.1053/gast.2003.50008. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Garcia Rodriguez LA, Lopez-Ridaura R. Effects of non-steroidal anti-inflammatory drugs on cancer sites other than the colon and rectum: A meta-analysis. BMC Cancer. 2003;3:28. doi: 10.1186/1471-2407-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan TL, Kristal AR, Blount PL, Levine DS, Galipeau PC, et al. NSAID use, BMI, and anthropometry in relation to genetic and cell cycle abnormalities in Barrett's Esophagus. Cancer Epidemiol Biomarkers Prev. 2002;11:745–752. [PubMed] [Google Scholar]
- Vaughan TL, Dong LM, Blount PL, Ayub K, Odze RD, et al. Non-steroidal anti-inflammatory drugs and risk of neoplastic progression in Barrett's oesophagus: A prospective study. Lancet Oncol. 2005;6:945–952. doi: 10.1016/S1470-2045(05)70431-9. [DOI] [PubMed] [Google Scholar]
- Buttar NS, Wang KK, Leontovich O, Westcott JY, Pacifico RJ, et al. Chemoprevention of esophageal adenocarcinoma by COX-2 inhibitors in an animal model of Barrett's esophagus. Gastroenterology. 2002;122:1101–1112. doi: 10.1053/gast.2002.32371. [DOI] [PubMed] [Google Scholar]
- Oyama K, Fujimura T, Ninomiya I, Miyashita T, Kinami S, et al. A COX-2 inhibitor prevents the esophageal inflammation-metaplasia-adenocarcinoma sequence in rats. Carcinogenesis. 2005;26:565–570. doi: 10.1093/carcin/bgh340. [DOI] [PubMed] [Google Scholar]
- Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet. 1999;22:106–109. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GJ, Doak SH, Parry JM, D'Souza FR, Griffiths AP, et al. Genetic pathways involved in the progression of Barrett's metaplasia to adenocarcinoma. Br J Surg. 2002;89:824–837. doi: 10.1046/j.1365-2168.2002.02107.x. [DOI] [PubMed] [Google Scholar]
- Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, et al. Fields of aberrant CpG island hypermethylation in Barrett's esophagus and associated adenocarcinoma. Cancer Res. 2000;60:5021–5026. [PubMed] [Google Scholar]
- Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, et al. p16 INK4a lesions are common, early abnormalities that undergo clonal expansion in Barrett's metaplastic epithelium. Cancer Res. 2001;61:8284–8289. [PubMed] [Google Scholar]
- Bian YS, Osterheld MC, Fontolliet C, Bosman FT, Benhattar J. p16 inactivation by methylation of the CDKN2A promoter occurs early during neoplastic progression in Barrett's esophagus. Gastroenterology. 2002;122:1113–1121. doi: 10.1053/gast.2002.32370. [DOI] [PubMed] [Google Scholar]
- Suspiro A, Pereira AD, Afonso A, Albuquerque C, Chaves P, et al. Losses of heterozygosity on chromosomes 9p and 17p are frequent events in Barrett's metaplasia not associated with dysplasia or adenocarcinoma. Am J Gastroenterol. 2003;98:728–734. doi: 10.1111/j.1572-0241.2003.07411.x. [DOI] [PubMed] [Google Scholar]
- Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci U S A. 1996;93:7081–7084. doi: 10.1073/pnas.93.14.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau PC, Prevo LJ, Sanchez CA, Longton GM, Reid BJ. Clonal expansion and loss of heterozygosity at chromosomes 9p and 17p in premalignant esophageal (Barrett's) tissue. J Natl Cancer Inst. 1999;91:2087–2095. doi: 10.1093/jnci/91.24.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennerty MB, Sampliner RE, Way D, Riddell R, Steinbronn K, et al. Discordance between flow cytometric abnormalities and dysplasia in Barrett's esophagus. Gastroenterology. 1989;97:815–820. doi: 10.1016/0016-5085(89)91483-2. [DOI] [PubMed] [Google Scholar]
- Gimenez A, Minguela A, Parrilla P, Bermejo J, Perez D, et al. Flow cytometric DNA analysis and p53 protein expression show a good correlation with histologic findings in patients with Barrett's esophagus. Cancer. 1998;83:641–651. doi: 10.1002/(sici)1097-0142(19980815)83:4<641::aid-cncr3>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Meltzer SJ, Yin J, Huang Y, McDaniel TK, Newkirk C, et al. Reduction to homozygostiy involving p53 in esophageal cancers demonstrated by the polymerase chain reaction. Biochemistry. 1991;88:4976–4980. doi: 10.1073/pnas.88.11.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarmin L, Yin J, Zhou X, Suzuki H, Jiang HY, et al. Frequent loss of heterozygosity on chromosome 9 in adenocarcinoma and squamous cell carcinoma of the esophagus. Cancer Res. 1994;54:6094–6096. [PubMed] [Google Scholar]
- Menke-Pluymers MB, Mulder AH, Hop WC, van Blankenstein M, Tilanus HW. Dysplasia and aneuploidy as markers of malignant degeneration in Barrett's oesophagus. The Rotterdam Oesophageal Tumour Study Group. Gut. 1994;35:1348–1351. doi: 10.1136/gut.35.10.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan K, Morris AI, Gosney JR, Field JK, Sutton R. Loss of heterozygosity on chromosome 17p predicts neoplastic progression in Barrett's esophagus. J Gastroenterol Hepatol. 2003;18:683–689. doi: 10.1046/j.1440-1746.2003.03048.x. [DOI] [PubMed] [Google Scholar]
- Teodori L, Gohde W, Persiani M, Ferrario F, Tirindelli Danesi D, et al. DNA/protein flow cytometry as a predictive marker of malignancy in dysplasia-free Barrett's esophagus: Thirteen-year follow-up study on a cohort of patients. Cytometry. 1998;34:257–263. doi: 10.1002/(sici)1097-0320(19981215)34:6<257::aid-cyto3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Reid BJ, Levine DS, Longton G, Blount PL, Rabinovitch PS. Predictors of progression to cancer in Barrett's esophagus: Baseline histology and flow cytometry identify low- and high-risk patient subsets. Am J Gastroenterol. 2000;95:1669–1676. doi: 10.1111/j.1572-0241.2000.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid BJ, Prevo LJ, Galipeau PC, Sanchez CA, Longton G, et al. Predictors of progression in Barrett's esophagus II: Baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol. 2001;96:2839–2848. doi: 10.1111/j.1572-0241.2001.04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett's esophagus III: Baseline flow cytometric variables. Am J Gastroenterol. 2001;96:3071–3083. doi: 10.1111/j.1572-0241.2001.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B, Blount P, Rabinovitch P. Biomarkers in Barrett's Esophagus. Gastrointestinal Clin North Am. 2003;13:369–397. doi: 10.1016/s1052-5157(03)00006-0. [DOI] [PubMed] [Google Scholar]
- Reid BJ, Blount PL, Feng Z, Levine DS. Optimizing endoscopic biopsy detection of early cancers in Barrett's high-grade dysplasia. Am J Gastroenterol. 2000;95:3089–3096. doi: 10.1111/j.1572-0241.2000.03182.x. [DOI] [PubMed] [Google Scholar]
- Conio M, Cameron AJ, Romero Y, Branch CD, Schleck CD, et al. Secular trends in the epidemiology and outcome of Barrett's oesophagus in Olmsted County, Minnesota. Gut. 2001;48:304–309. doi: 10.1136/gut.48.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conio M, Blanchi S, Lapertosa G, Ferraris R, Sablich R, et al. Long-term endoscopic surveillance of patients with Barrett's esophagus. Incidence of dysplasia and adenocarcinoma: A prospective study. Am J Gastroenterol. 2003;98:1931–1939. doi: 10.1111/j.1572-0241.2003.07666.x. [DOI] [PubMed] [Google Scholar]
- Cameron AJ, Lomboy CT. Barrett's esophagus: Age, prevalence, and extent of columnar epithelium. Gastroenterology. 1992;103:1241–1245. doi: 10.1016/0016-5085(92)91510-b. [DOI] [PubMed] [Google Scholar]
- O'Connor JB, Falk GW, Richter JE. The incidence of adenocarcinoma and dysplasia in Barrett's esophagus: Report on the Cleveland Clinic. Am J Gastroenterol. 1999;94:2037–2042. doi: 10.1111/j.1572-0241.1999.01275.x. [DOI] [PubMed] [Google Scholar]
- Haggitt RC. Barrett's esophagus, dysplasia, and adenocarcinoma. Hum Pathol. 1994;25:982–993. doi: 10.1016/0046-8177(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Paulson TG, Galipeau PC, Reid BJ. Loss of heterozygosity analysis using whole genome amplification, cell sorting, and fluorescence-based PCR. Genome Res. 1999;9:482–491. [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res. 1997;57:2619–2622. [PubMed] [Google Scholar]
- Wong DJ, Foster SA, Galloway DA, Reid BJ. Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest. Mol Cell Biol. 1999;19:5642–5651. doi: 10.1128/mcb.19.8.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevo LJ, Sanchez CA, Galipeau PC, Reid BJ. p53-mutant clones and field effects in Barrett's esophagus. Cancer Res. 1999;59:4784–4787. [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc (Ser B) 1995;57:289–300. [Google Scholar]
- Farrow DC, Vaughan TL, Hansten PD, Stanford JL, Risch HA, et al. Use of aspirin and other nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev. 1998;7:97–102. [PubMed] [Google Scholar]
- Chow WH, Blot WJ, Vaughan TL, Risch HA, Gammon MD, et al. Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1998;90:150–155. doi: 10.1093/jnci/90.2.150. [DOI] [PubMed] [Google Scholar]
- Hampel H, Abraham NS, El-Serag HB. Meta-analysis: Obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143:199–211. doi: 10.7326/0003-4819-143-3-200508020-00006. [DOI] [PubMed] [Google Scholar]
- Lagergren J, Bergstrom R, Nyren O. Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med. 1999a;130:883–890. doi: 10.7326/0003-4819-130-11-199906010-00003. [DOI] [PubMed] [Google Scholar]
- Gammon MD, Schoenberg JB, Ahsan H, Risch HA, Vaughan TL, et al. Tobacco, alcohol, and socioeconomic status and adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1997;89:1277–1284. doi: 10.1093/jnci/89.17.1277. [DOI] [PubMed] [Google Scholar]
- Lindblad M, Rodriguez LA, Lagergren J. Body mass, tobacco and alcohol and risk of esophageal, gastric cardia, and gastric non-cardia adenocarcinoma among men and women in a nested case-control study. Cancer Causes Control. 2005;16:285–294. doi: 10.1007/s10552-004-3485-7. [DOI] [PubMed] [Google Scholar]
- Lagergren J, Bergstrom R, Lindgren A, Nyren O. The role of tobacco, snuff and alcohol use in the aetiology of cancer of the oesophagus and gastric cardia. Int J Cancer. 2000;85:340–346. [PubMed] [Google Scholar]
- Wu A, Wan P, Bernstein L. A multiethnic population-based study of smoking, alcohol and body size and risk of adenocarcinomas of the stomach and esophagus (United States) Cancer Causes Control. 2001;12:721–732. doi: 10.1023/a:1011290704728. [DOI] [PubMed] [Google Scholar]
- Wu AH, Tseng CC, Bernstein L. Hiatal hernia, reflux symptoms, body size, and risk of esophageal and gastric adenocarcinoma. Cancer. 2003;98:940–948. doi: 10.1002/cncr.11568. [DOI] [PubMed] [Google Scholar]
- Engel LS, Chow WH, Vaughan TL, Gammon MD, Risch HA, et al. Population attributable risks of esophageal and gastric cancers. J Natl Cancer. 2003;95:1404–1413. doi: 10.1093/jnci/djg047. [DOI] [PubMed] [Google Scholar]
- Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, et al. Prospective study of cyclin D1 overexpression in Barrett's esophagus: Association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92:1316–1321. doi: 10.1093/jnci/92.16.1316. [DOI] [PubMed] [Google Scholar]
- Schulmann K, Sterian A, Berki A, Yin J, Sato F, et al. Inactivation of p16, RUNX3, and HPP1 occurs early in Barrett's-associated neoplastic progression and predicts progression risk. Oncogene. 2005;24:4138–4148. doi: 10.1038/sj.onc.1208598. [DOI] [PubMed] [Google Scholar]
- Murray L, Sedo A, Scott M, McManus D, Sloan JM, et al. TP53 and progression from Barrett's metaplasia to oesophageal adenocarcinoma in a UK population cohort. Gut. 2006;55:1390–1397. doi: 10.1136/gut.2005.083295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–246. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- Sharpless NE. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, et al. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Shackney SE, Shackney TV. Common patterns of genetic evolution in human solid tumors. Cytometry. 1997;29:1–27. [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Maley CC, Reid BJ. Barrett's esophagus as an example of the evolution of cell lineages in cancer. Encyclopedia of the Human Genome. London: Nature Publishing Group; 2003. 5000 [Google Scholar]
- Maley CC, Reid BJ. Natural selection in neoplastic progression of Barrett's esophagus. Semin Cancer Biol. 2005;15:474–483. doi: 10.1016/j.semcancer.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, et al. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett's esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, et al. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004;64:7629–7633. doi: 10.1158/0008-5472.CAN-04-1738. [DOI] [PubMed] [Google Scholar]
- McManus DT, Olaru A, Meltzer SJ. Biomarkers of esophageal adenocarcinoma and Barrett's esophagus. Cancer Res. 2004;64:1561–1569. doi: 10.1158/0008-5472.can-03-2438. [DOI] [PubMed] [Google Scholar]
- Paulson TG, Reid BJ. Focus on Barrett's esophagus and esophageal adenocarcinoma. Cancer Cell. 2004;6:11–16. doi: 10.1016/j.ccr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- van Lieshout EM, Jansen JB, Peters WH. Biomarkers in Barrett's esophagus (review) Int J Oncol. 1998;13:855–864. doi: 10.3892/ijo.13.4.855. [DOI] [PubMed] [Google Scholar]
- Reid BJ. p53 and neoplastic progression in Barrett's esophagus. Am J Gastroenterol. 2001;96:1321–1323. doi: 10.1111/j.1572-0241.2001.03844.x. [DOI] [PubMed] [Google Scholar]
- Weston AP, Banerjee SK, Sharma P, Tran TM, Richards R, et al. p53 protein overexpression in low grade dysplasia (LGD) in Barrett's esophagus: Immunohistochemical marker predictive of progression. Am J Gastroenterol. 2001;96:1355–1362. doi: 10.1111/j.1572-0241.2001.03851.x. [DOI] [PubMed] [Google Scholar]
- Fahmy M, Skacel M, Gramlich TL, Brainard JA, Rice TW, et al. Chromosomal gains and genomic loss of p53 and p16 genes in Barrett's esophagus detected by fluorescence in situ hybridization of cytology specimens. Mod Pathol. 2004;17:588–596. doi: 10.1038/modpathol.3800088. [DOI] [PubMed] [Google Scholar]
- Wales MM, Biel MA, el Deiry W, Nelkin BD, Issa JP, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–577. doi: 10.1038/nm0695-570. [DOI] [PubMed] [Google Scholar]
- Zeini M, Traves PG, Lopez-Fontal R, Pantoja C, Matheu A, et al. Specific contribution of p19(ARF) to nitric oxide-dependent apoptosis. J Immunol. 2006;177:3327–3336. doi: 10.4049/jimmunol.177.5.3327. [DOI] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- Nugent KP, Farmer KC, Spigelman AD, Williams CB, Phillips RK. Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br J Surg. 1993;80:1618–1619. doi: 10.1002/bjs.1800801244. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Iwama T, Yoshinaga K, Toyooka M, Taketo MM, et al. A randomized, double-blind, placebo-controlled trial of the effects of rofecoxib, a selective cyclooxygenase-2 inhibitor, on rectal polyps in familial adenomatous polyposis patients. Clin Cancer Res. 2003;9:4756–4760. [PubMed] [Google Scholar]
- Bostwick DG, Shan A, Qian J, Darson M, Maihle NJ, et al. Independent origin of multiple foci of prostatic intraepithelial neoplasia: Comparison with matched foci of prostate carcinoma. Cancer. 1998;83:1995–2002. doi: 10.1002/(sici)1097-0142(19981101)83:9<1995::aid-cncr16>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Saric T, Brkanac Z, Troyer DA, Padalecki SS, Sarosdy M, et al. Genetic pattern of prostate cancer progression. Int J Cancer. 1999;81:219–224. doi: 10.1002/(sici)1097-0215(19990412)81:2<219::aid-ijc9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Wada K. p16 and p53 gene alterations and accumulations in the malignant evolution of intraductal papillary-mucinous tumors of the pancreas. J Hepatobiliary Pancreat Surg. 2002;9:76–85. doi: 10.1007/s005340200007. [DOI] [PubMed] [Google Scholar]
- Feitelson MA, Sun B, Satiroglu Tufan NL, Liu J, Pan J, et al. Genetic mechanisms of hepatocarcinogenesis. Oncogene. 2002;21:2593–2604. doi: 10.1038/sj.onc.1205434. [DOI] [PubMed] [Google Scholar]
- Lindforss U, Papadogiannakis N, Zetterquist H, Lindberg G, Olivecrona H. Distribution of genetic variants in preneoplastic areas of colorectal tumours. Eur J Surg Oncol. 2003;29:491–496. doi: 10.1016/s0748-7983(03)00075-1. [DOI] [PubMed] [Google Scholar]
- Farabegoli F, Champeme MH, Bieche I, Santini D, Ceccarelli C, et al. Genetic pathways in the evolution of breast ductal carcinoma in situ. J Pathol. 2002;196:280–286. doi: 10.1002/path.1048. [DOI] [PubMed] [Google Scholar]
- Euhus DM, Cler L, Shivapurkar N, Milchgrub S, Peters GN, et al. Loss of heterozygosity in benign breast epithelium in relation to breast cancer risk. J Natl Cancer Inst. 2002;94:858–860. doi: 10.1093/jnci/94.11.858. [DOI] [PubMed] [Google Scholar]
- Chow NH, Cairns P, Eisenberger CF, Schoenberg MP, Taylor DC, et al. Papillary urothelial hyperplasia is a clonal precursor to papillary transitional cell bladder cancer. Int J Cancer. 2000;89:514–518. [PubMed] [Google Scholar]
- Bulashevska S, Szakacs O, Brors B, Eils R, Kovacs G. Pathways of urothelial cancer progression suggested by Bayesian network analysis of allelotyping data. Int J Cancer. 2004;110:850–856. doi: 10.1002/ijc.20180. [DOI] [PubMed] [Google Scholar]
- Czerniak B, Li L, Chaturvedi V, Ro JY, Johnston DA, et al. Genetic modeling of human urinary bladder carcinogenesis. Genes Chromosomes Cancer. 2000;27:392–402. [PubMed] [Google Scholar]
- Gazdar AF, Bader S, Hung J, Kishimoto Y, Sekido Y, et al. Molecular genetic changes found in human lung cancer and its precursor lesions. Cold Spring Harb Symp Quant Biol. 1994;59:565–572. doi: 10.1101/sqb.1994.059.01.063. [DOI] [PubMed] [Google Scholar]
- Boland CR, Sato J, Appelman HD, Bresalier RS, Feinberg AP. Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression. Nat Med. 1995;1:902–909. doi: 10.1038/nm0995-902. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Li M, Lotan R, Levin B, Tahara E, Lippman SM, et al. Aspirin induction of apoptosis in esophageal cancer: A potential for chemoprevention. Cancer Epidemiol Biomarkers Prev. 2000;9:545–549. [PubMed] [Google Scholar]
- Souza RF, Shewmake K, Beer DG, Cryer B, Spechler SJ. Selective inhibition of cyclooxygenase-2 suppresses growth and induces apoptosis in human esophageal adenocarcinoma cells. Cancer Res. 2000;60:5767–5772. [PubMed] [Google Scholar]
- Buttar NS, Wang KK, Anderson MA, Dierkhising RA, Pacifico RJ. The effect of selective cyclooxygenase-2 inhibition in Barrett's esophagus epithelium: An in vitro study. J Natl Cancer Inst. 2002;94:422–429. doi: 10.1093/jnci/94.6.422. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- Husain SS, Szabo IL, Tamawski AS. NSAID inhibition of GI cancer growth: Clinical implications and molecular mechanisms of action. Am J Gastroenterol. 2002;97:542–553. doi: 10.1111/j.1572-0241.2002.05528.x. [DOI] [PubMed] [Google Scholar]
- Gupta RA, DuBois RN. Cyclooxygenase-2 inhibitor therapy for the prevention of esophageal adenocarcinoma in Barrett's esophagus. J Natl Cancer Inst. 2002;94:406–407. doi: 10.1093/jnci/94.6.406. [DOI] [PubMed] [Google Scholar]
- Kaur BS, Khamnehei N, Iravani M, Namburu SS, Lin O, et al. Rofecoxib inhibits cyclooxygenase 2 expression and activity and reduces cell proliferation in BE. Gastroenterol. 2002;123:60–67. doi: 10.1053/gast.2002.34244. [DOI] [PubMed] [Google Scholar]
- Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- Cook NR, Lee IM, Gaziano JM, Gordon D, Ridker PM, et al. Low-dose aspirin in the primary prevention of cancer: The Women's Health Study: A randomized controlled trial. JAMA. 2005;294:47–55. doi: 10.1001/jama.294.1.47. [DOI] [PubMed] [Google Scholar]
- Anderson LA, Johnston BT, Watson RG, Murphy SJ, Ferguson HR, et al. Nonsteroidal anti-inflammatory drugs and the esophageal inflammation-metaplasia-adenocarcinoma sequence. Cancer Res. 2006;66:4975–4982. doi: 10.1158/0008-5472.CAN-05-4253. [DOI] [PubMed] [Google Scholar]
- Sturmer T, Glynn RJ, Lee IM, Manson JE, Buring JE, et al. Aspirin use and colorectal cancer: Post-trial follow-up data from the Physicians' Health Study. Ann Intern Med. 1998;128:713–720. doi: 10.7326/0003-4819-128-9-199805010-00003. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, Fennerty MB. Medical decision analysis of chemoprevention against esophageal adenocarcinoma. Gastroenterology. 2003;124:1758–1766. doi: 10.1016/s0016-5085(03)00393-7. [DOI] [PubMed] [Google Scholar]