

Abstract
The presence of blood pressure (BP) quantitative trait loci (QTL) on rat chromosome (RNO) 10 has been clearly demonstrated by linkage analysis and substitution mapping. Using congenic strains containing the LEW rat chromosomal segments on the Dahl salt-sensitive (S) rat background, further iterations of congenic substrains were constructed and characterized to fine map a chromosome 10 region (QTL1) linked to blood pressure. Comparison of 7 congenic substrains refined QTL1 to a 1.17 Mb segment flanked by D10Mco88 and D10Mco89, which are located at 71,513,116 bp and 72,684,774 bp, respectively. The newly defined QTL1, containing 18 genes, is captured in its entirety within a single congenic substrain. A thorough transcript analysis revealed that three out of these 18 genes, Ccl5, Ddx52 and RGD1559577, had nonsynonymous allelic variations between the S rat and the LEW rat. None of the detected transcripts within the newly defined QTL1 are directly implicated in BP control in humans or model organisms. Therefore, the present work defines a novel blood pressure QTL with three potential quantitative trait nucleotides.
Keywords: Quantitative trait locus, Hypertension, Dahl rats, Ddx52, Ccl5
Introduction
Human essential hypertension is a classic example of a complex trait with documented genetic influence. However, the underlying disease causative genetic components remain unknown. Genetic studies using selectively bred hypertensive rat models provide some advantages over genetic studies of hypertension in humans, one of which is the ability to conduct substitution mapping using consomic/congenic strains [1–3]. Such efforts have not only accumulated evidence for the presence of multiple quantitative trait loci (QTLs) that regulate blood pressure (BP), but also mapped several of these QTLs on the rat genome with high resolution. One such previous study from our laboratory resulted in the identification of the QTL on rat chromosome (RNO) 7 as 11β-hydroxylase [4; 5]. Causative genetic variants within all other BP QTL regions remain unidentified.
The present report is on the genetic analysis of RNO10. Multiple studies, involving ten different rat strains have provided evidence for the existence of multiple loci on this chromosome linked to BP [6–26]. The Dahl salt-sensitive (S) versus LEW and S versus Milan Normotensive Strain (MNS) are the two strain comparisons studied in our laboratory using substitution mapping. A BP QTL was previously located within 12 cM (D10Mco58 to D10Rat24) using S.LEW congenic strains, whereas a BP QTL was located within 2.6 cM (D10Rat27 to D10Rat24) using S.MNS congenic strains [15]. The objective of the present work was to further dissect the 12 cM QTL1 region identified using the S and LEW comparison. For this purpose, new iterations of congenic substrains were developed and studied, which resulted in an 18-fold improved localization of BP QTL1 from the previous localization. The refined region is 1.17 Mb containing 18 annotated genes, none of which are known to play a role in rodent or human BP control. Therefore, BP QTL1 represents a potentially novel underlying genetic factor controlling BP.
The final step from fine-mapping to identifying the underlying genetic determinant by the substitution mapping approach is one of the most difficult steps of QTL mapping for two reasons: (1) the larger the introgressed segment, the greater the number of positional candidates to evaluate, each of which may or may not influence blood pressure; and (2) the smaller the congenic interval, the greater the difficulty in finding animals with informative recombinations to construct congenic substrains with shorter introgressed segments. At this stage, multiple complimentary techniques become necessary to link the observed QTL effect to any locus within the fine-mapped QTL interval. Because nonsynonymous polymorphisms may result in altered protein function, genes with such variants are primary suspects for correlated QTL effects. Therefore, in addition to fine-mapping the BP QTL1, we sought to prioritize gene identification of BP QTL1 based on the presence or absence of nonsynonymous variants within the expressed transcripts of positional candidate genes. Data obtained, which indicate that multiple genes within QTL1 possess potential coding sequence quantitative trait nucleotides (QTNs), will be presented.
Results
RNO10 BP QTL1 region of S.LEWx12
S.LEWx12 was the congenic strain previously used to define QTL1 on the genetic linkage map of RNO10. By defining the ends of the introgressed region of S.LEWx12, a more current and accurate localization of QTL1 on the physical map of RNO10 was determined. The QTL1 region contained within the S.LEWx12 congenic strain was located between D10Got79 (63,481,418 bp) and D10Rat269 (79,137,099 bp) of RNO10, respectively (www.ensembl.org) (Figure 1). This 15,655,681 bp interval of RNO10 encompasses 221 genes (www.ncbi.nlm.nih.gov/genome/guide/rat).
Figure 1. Congenic substrains derived from S.LEWx12 and their BP effects.
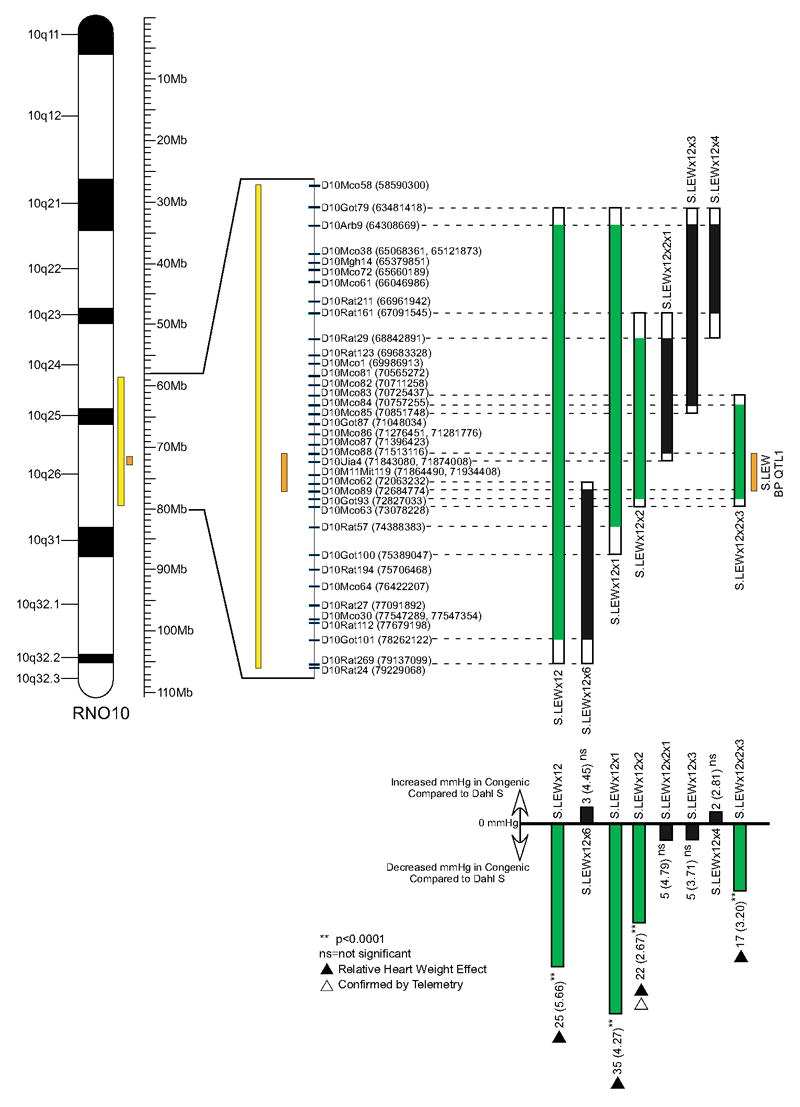
Bars illustrate the LEW segment introgressed onto the background of S. The open bars at the end of each introgressed segment represent the region of recombination. Green colored bars represent the LEW introgressed segment in congenic substrains with a BP lowering effect, whereas the black colored bars represent the LEW introgressed segment in congenic subtrains without a BP lowering effect. Dashed lines point to the markers used to genotype the ends of the congenic substrains. The base pair chromosomal location for the start of each marker was given in parenthesis. The lower portion of the figure illustrates the BP effect observed for each congenic substrain compared to that of the S rat BP (effect = Congenic substrain BP− S rat BP). Also illustrated is the previous (yellow colored bar) and current (orange colored bar) S.LEW RNO10 BP QTL localizations as it relates to the cytogenetic map and the Mb scale of RNO10.
Congenic substrains of S.LEWx12
Figure 1 shows the tiling array of congenic substrains that were developed to span the 15.66 Mb BP QTL1 region trapped within the S.LEWx12 congenic strain. The lack of an effect on BP (Figure 1, Table 1) between the S rat strain and the congenic substrains shown with black color bars (S.LEWx12x6, S.LEWx12x2x1, S.LEWx12x3, and S.LEWx12x4) point to a 1.17 Mb region flanked by markers D10Mco88 and D10Mco89 as the newly refined location of QTL1. In support of this observation, congenic substrains S.LEWx12x1 and S.LEWx12x2 (green color bars), both spanning this region, demonstrated a significant (p<0.0001) decrease in BP compared to the S rat (35 and 22 mmHg, respectively) (Figure 1, Table 1). These data were further corroborated using the congenic substrain S.LEWx12x2x3, which contained the fine-mapped QTL1 within a shorter introgressed segment. The BP of S.LEWx12x2x3 was significantly lower by 17 mm Hg (p<0.0001) compared to the S rat (Figure 1, Table 1). The significant heart weight effects observed with S.LEWx12, S.LEWx12x1, S.LEWx12x2, and S.LEWx12x2x3 correlated with the BP effects that were observed for these congenic substrains.
Table 1.
Effects of rat chromosome 10 congenic strains on blood pressure, body weight, heart weight, and relative heart weight.
| Blood Pressure (mmHg)
|
Body Weight (gm)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Congenic Strain | S | Congenic | **Effect | t-test | S | Congenic | **Effect | t-test |
| S.LEWx12 | 242 [3.70] | 217 [4.28] | −25 (5.66) | <0.0001 | 303 [2.86] | 303 [1.14] | 0 (3.08) | 0.910 |
| S.LEWx12x1 | 218 [3.10] | 183 [2.07] | −35 (4.27) | <0.0001 | 311 [2.11] | 299 [5.81] | −12 (5.81) | 0.110 |
| S.LEWx12x2 | 215 [3.72] | 184 [3.05] | −31 (4.76) | <0.0001 | 297 [3.75] | 299 [3.42] | +2 (5.07) | 0.619 |
| S.LEWx12x3 | 191 [1.58] | 186 [3.21] | −5 (3.71) | 0.444 | 320 [1.52] | 315 [2.77] | −5 (3.59) | 0.314 |
| S.LEWx12x4*† | 190 [1.61] | 192 [2.45] | +2 (2.81) | 0.525 | 314 [2.00] | 306 [2.61] | −8 (3.23) | 0.012 |
| S.LEWx12x6† | 189 [2.92] | 192 [3.89] | +3 (4.45) | 0.842 | 307 [3.23] | 291 [3.33] | −16 (4.49) | 0.002 |
| S.LEWx12x2x1 | 196 [2.53] | 191 [4.07] | −5 (4.79) | 0.315 | 318 [2.25] | 317 [2.48] | −1 (3.35) | 0.697 |
| S.LEWx12x2x3* | 206 [2.23] | 189 [2.28] | −17 (3.20) | <0.0001 | 303 [1.92] | 305 [1.96] | +2 (2.75) | 0.449 |
| Heart Weight (gm)
|
Relative Heart Weight
|
|||||||
| Congenic Strain | S | Congenic | **Effect | t-test | S | Congenic | **Effect | t-test |
| S.LEWx12 | 1.31 [0.015] | 1.22 [0.009] | −0.09 (0.018) | <0.0001 | 4.26 [0.067] | 4.02 [0.035] | −0.24 (0.075) | 0.003 |
| S.LEWx12x1 | 1.23 [0.018] | 1.10 [0.019] | −0.13 (0.028) | <0.0001 | 3.96 [0.053] | 3.68 [0.032] | −0.27 (0.058) | <0.0001 |
| S.LEWx12x2 | 1.26 [0.038] | 1.16 [0.015] | −0.10 (0.039) | 0.023 | 4.26 [0.163] | 3.89 [0.039] | −0.36 (0.156) | 0.025 |
| S.LEWx12x3 | 1.25 [0.010] | 1.21 [0.010] | −0.04 (0.015) | 0.059 | 3.90 [0.024] | 3.86 [0.033] | −0.04 (0.054) | 0.713 |
| S.LEWx12x4*† | 1.21 [0.007] | 1.21 [0.011] | 0 (0.004) | 0..720 | 3.88 [0.029] | 3.98 [0.059] | +0.10 (0.061) | 0.145 |
| S.LEWx12x6† | 1.18 [0.013] | 1.18 [0.011] | 0 (0.016) | 0.751 | 3.85 [0.048] | 4.09 [0.068] | +0.24 (0.083) | 0.222 |
| S.LEWx12x2x1 | 1.26 [0.047] | 1.26 [0.016] | 0 (0.022) | 0.728 | 3.95 [0.048] | 3.99 [0.048] | +0.04 (0.068) | 0.564 |
| S.LEWx12x2x3* | 1.25 [0.013] | 1.21 [0.007] | −0.04 (0.016) | 0.014 | 4.15 [0.050] | 3.99 [0.024] | −0.16 (0.059) | 0.006 |
The values given here were the averages of experiments done on separate days. Two separate experiments for S.LEWx12x4 and 3 separate experiments for S.LEWx12x2x3.
Effect = Congenic value − S value. Negative values indicate a decrease in the congenic effect compared to the S rat, whereas positive values indicate an increase in the congenic effect compared to the S rat.
Heart weight values given were corrected for body weight using the regression between heart weight and body weight. [Standard error of the mean]. (Standard error of the mean difference). Number of rats in each group ranged from 20 to 23 except for the following comparisons: S.LEWx12x4 versus S (32 versus 44 rats), and S.LEWx12x2x3 versus S (30 versus 30 rats). Only male rats were used.
Tail-cuff versus telemetry comparison
Figure 2a shows that tail-cuff BP measurements correlated highly with the telemetry BP measurements. The group averages of the S rat BP was not significantly different between the two BP measurement approaches (tail-cuff versus telemetry, Figure 2b). Similarly, no significant difference was observed between the group averages of S.LEWx12x2. However, the S versus S.LEWx12x2 comparison was significantly different (p<0.0001) for both tail-cuff and telemetry measurements. The 8-day telemetry BP measurements for S and S.LEWx12x2 are shown in Figure 2c. Throughout the 8 day period, the S.LEWx12x2 congenic strain, which contains the QTL1 region, showed a significant (p<0.0001) systolic BP lowering effect when compared to the S rat BP.
Figure 2. Corroboration of tail-cuff measurements by telemetry.
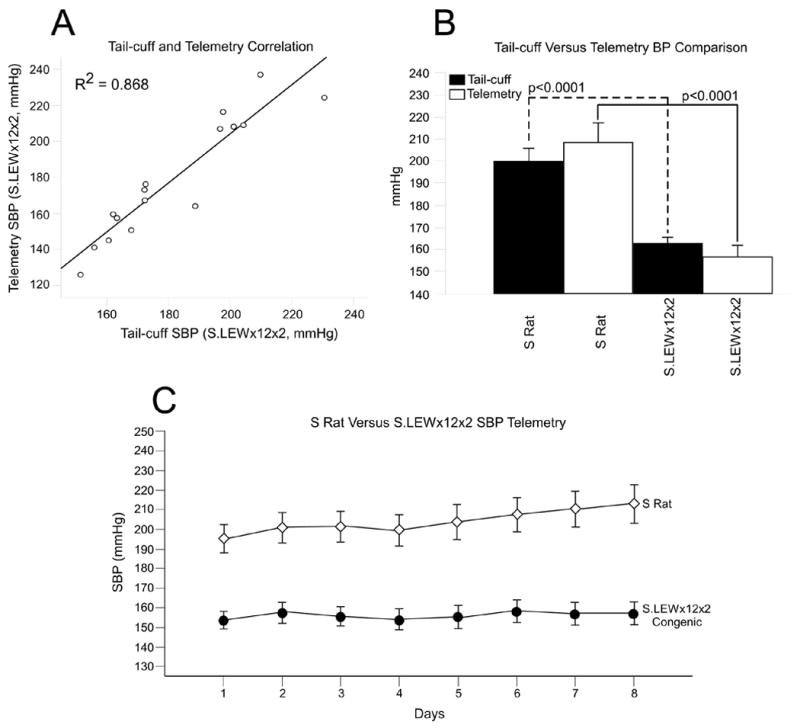
A) Correlation between the blood measurement values obtained by tail-cuff and telemetry on the same group of S and S.LEWx12x2 rats; B) Comparison of the mean values of S rats and S.LEWx12x2 rats measured by tail-cuff and telemetry; C) Daily average systolic blood pressure (SBP) telemetry measurements of S versus S.LEWx12x2.
Genetic composition and comparative mapping of the QTL1 region
The fine-mapped QTL1 region flanked by D10Mco88 to D10Mco89, which are located at 71,513,116 bp and 72,684,774 bp, respectively, corresponds to 1,171,658 bp (Figure 3). This region of RNO10 represents 0.042% of the rat genome with 18 gene annotations. Eleven of these are known genes and seven are predicted genes. Conserved synteny of QTL1 on mouse chromosome 11 and human chromosome 17 (www.ncbi.nlm.nih.gov) is presented in Figure 3. Comparative mapping suggested that 13 out of the 18 gene annotations within QTL1 are orthologous to genes on mouse chromosome 11, whereas only 11 rat gene annotations are orthologous to genes on human chromosome 17. Based on sequence similarity, there is no information currently available for human orthologs of the remaining 7 rat genes that exist anywhere else on the human genome.
Figure 3. Comparative mapping of the newly defined S.LEW RNO10 BP QTL1.
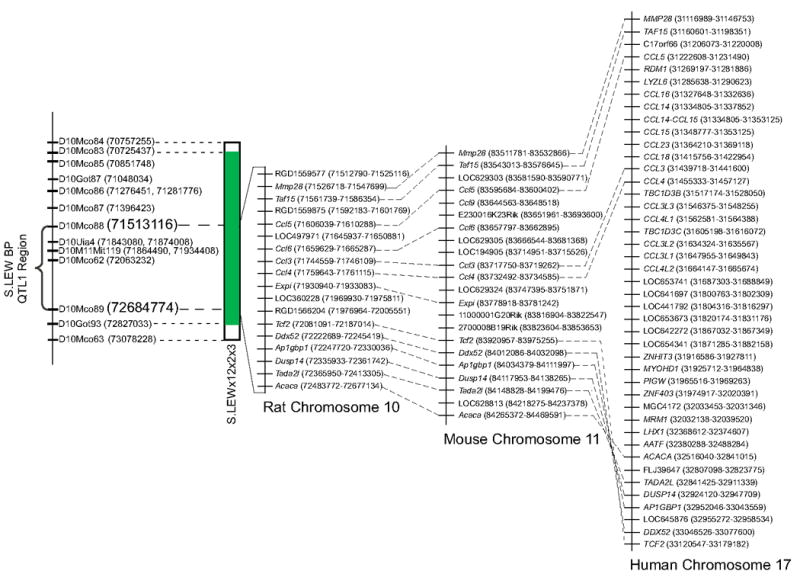
The LEW introgressed segment of the minimal congenic substrain S.LEWx12x2x3 is represented by the green bar. The open segments at the ends of the green bar represent the regions of recombination. The region spanning the 1.17 Mb from 71513116 bp to 72684774 bp is the newly refined S.LEW QTL1 region. Also illustrated are the 18 NCBI (www.ncbi.nlm.nih.gov) annotated genes for the S.LEW QTL1 region. NCBI annotations of the mouse chromosome 11 and human chromosome 17 syntenic regions are also illustrated. The base pair location from the start to end for each gene is given in parentheses.
Features of the S.LEW BP QTL1 gene transcripts
The authenticity of all 18 gene annotations (including both known genes and predicted genes) within QTL1 was assessed by reverse transcription of S and LEW total RNA followed by sequencing of the resultant cDNA. A total of 15 out of the 18 transcripts were confirmed as expressed transcripts. Product sizes of 12 out of the 15 cDNA preparations in both S and LEW rats were as expected based on the annotations by the NCBI database (www.ncbi.nlm.nih.gov). The 12 genes are Mmp28, Ccl5, LOC497971, Ccl6, Ccl3, Ccl4, Expi, LOC360228, RGD1566204, Ddx52, Tada2l, and Acaca (Table 2). cDNA products for the other three genes (Tcf2, Ap1gbp1, and Dusp14) were the same in both S and LEW rats, but the product sizes were not consistent with the predictions by the NCBI database (Table 2).
Table 2.
Nonsynonymous allelic variants of S and LEW genes located within the chromosome 10 QTL1 BP region.
| Transcript Size (ATG to Stop) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene ID | Gene Symbol | Gene Description | Observed S-allele | Observed LEW-allele | Variant RN010 Location | Exon Number | S Rat Variant | LEW Rat Variant |
| 497969 | RGD1559577 | Similar to Riken cDNA 1700020L24 (predicted) | No Product‡ | No Product‡ | 71, 525,070 bp* | 4 | TTC (F326) | TAC (Y326) |
| 303384 | Mmp28 | Matrix metalloproteinase 28 (predicted) | 1577 bp^ | 1577 bp^ | No Variants | - | - | - |
| 287571 | Taf15 | TBP-associated factor 15 (predicted) | No Product‡ | No Product‡ | No Variants | - | - | - |
| 497970 | RGD1559875 | Similar to novel protein (predicted) | No Product‡ | No Product‡ | No Variants | - | - | - |
| 81780 | Ccl5 | Chemokine (C-C motif) ligand 5 | 279 bp | 279 bp | 71 ,61 0,248 bp** | 1 | ATT (114) | GTT (V14) |
| 497971 | LOC497971 | Similar to Ccl9 (predicted) | 354 bp | 354 bp | No Variants | - | - | - |
| 287910 | Ccl6 | Chemokine (C-C motif) ligand 6 | 348 bp | 348 bp | No Variants | - | - | - |
| 25542 | Ccl3 | Chemokine (C-C motif) ligand 3 | 279 bp | 279 bp | No Variants | - | - | - |
| 116637 | Ccl4 | Small inducible cytokine A4 | 279 bp | 279 bp | No Variants | - | - | - |
| 171059 | Expi | Extracellular peptidase inhibitor | 183 bp | 183 bp | No Variants | - | - | - |
| 360228 | LOC360228 | WDNM1 homolog | 192 bp | 192 bp | No Variants | - | - | - |
| 497972 | RGD1566204 | Similar to 2700008B19Rik protein (predicted) | 3759 bp | 3759 bp | No Variants | - | - | - |
| 25640 | Tcf2 | Transcription factor 2 | 1677 bpt†^ | 1677 bp†^ | No Variants | - | - | - |
| 85432 | Ddx52 | ATP-dependent, RNA helicase | 1797 bp | 1797 bp | 72,223,895 bp*** | 2 | AAA (K73) | ACA (T73) |
| 84479 | Ap1gbp1 | AP1 gamma subunit binding protein 1 | 3417 bp†^ | 3417 bp†^ | No Variants | - | - | - |
| 360580 | Dusp14 | Dual specificity phosphate 14 (predicted) | 597 bp^ | 597 bp^ | No Variants | - | - | - |
| 360581 | Tada2l | Transcriptional adapter 2-like | 1332 bp† | 1332 bp† | No Variants | - | - | - |
| 60581 | Acaca | Acetyl-coenzyme A carboxylase alpha | 7038 bp | 7038 bp | No Variants | - | - | - |
Total kidney RNA was used to obtain all the transcripts except for RGD1566204 and Ap1gbp1. A mixed pool of total RNA obtained from S and LEW rat liver, spleen, heart, pancreas, lung, and thyroid was used to obtain the RGD1566204 and Ap1gbp1 transcripts.
Total kidney RNA and the mixed pool of RNA was tried for those few genes that did not give a transcript product. Sequence information for those genes that did not give us a transcript product was obtained by sequencing the genomic DNA. All nonsynonymous variants observed in the amplified transcripts were confirmed by sequencing the genomic DNA.
Chromosomal location is similar to position 12,524,275 of accession number NW_047336 viewed on plus strand;
Chromosomal location is similar to position 12,609,453 of accession numboer NW_047336 viewed on plus strand;
Chromosomal location is similar to position 13,223,100 of accession number NW_047336 viewed on plus strand; The S versus LEW variant nucleotide is underlined; The amino acid and its position within the protein is in parenthesis. Some genes, as designated by †, showed more than one type of transcript variant. All transcript sizes were of expected size, according to NCBI, with some exception as designated by ^. Mmp28 transcript size and sequence was the same as given by NCBI, but Ensembl’s annotation was shorter by seven amino acids at the c-terminal. Tcf2 transcript size and sequence was the same as that given by NCBI, except that the S and LEW transcripts had an additional amino acid (Q403) that the NCBI does not annotate. That amino acid is annotated by Ensembl and is also found in the mouse sequence of this gene’s protein product. Dusp14 transcript obtained was similar to that annotated by Ensembl and overlapped with last segment of the sequence annotated by NCBI. The first 396 bp of the Dusp14 NCBI annotated sequence was confirmed by sequencing the genomic DNA of S and LEW. Ap1gbp1 transcript sequence and size differed from both NCBI’s and Ensembl’s. However, the sequence mostly resembled that of the sequence annotated by Ensembl. Some of the observed transcript variants had sizes and sequences that looked more like a composite between the NCBI and Ensembl deposited sequences for this gene. For all the genes, the gene-specific primers used in amplifying the gene transcripts, genomic DNA amplification, and amplicon sequencing were designed based on the NCBI’s deposited sequences. All S and LEW sequences characterized for these genes were deposited in Genbank with accession numbers ranging from EF121971 to EF122008.
To ensure that the list of candidate transcripts was comprehensive, we also compared our results with the annotations at the Ensembl rat database (www.ensembl.org), which has annotated the rat genome independent of the annotations by NCBI [27]. Our results are in agreement with the Ensembl and NCBI transcript predictions for most of the genes, but with some exceptions. First, the NCBI prediction for the Tcf2 transcript was shorter by three nucleotides compared to the transcript prediction by Ensembl. Tcf2 transcripts from both S and LEW rats were in agreement with the Ensembl prediction indicating that the translated product would result in an additional amino acid, Q403, of Tcf2. Second, NCBI prediction of the Dusp14 transcript is longer than what is predicted by the Ensembl database. Our results are in agreement with that of the Ensembl database. We were not able to reverse transcribe the first 396 bp of the Dusp14 NCBI annotated sequence. Therefore, for completeness that sequence was confirmed by sequencing the genomic DNA of S and LEW. Third, the Ap1gbp1 transcript sequence of both S and LEW mostly resembled that of the sequence annotated by Ensembl, which is different from that of NCBI. Lastly, the Mmp28 transcript size and sequence was the same as given by NCBI, but shorter by 21 nucleotides representing seven amino acids at the c-terminal protein prediction by Ensembl.
Note that out of the 18 genes annotated within the QTL interval, 7 genes are predicted, with no previous evidence for rat transcripts. Amplification of transcripts within the QTL region allowed us to test the authenticity of some of the predicted genes. Regardless of the product sizes, we observed cDNA products from 4 (Mmp28, LOC497971, RGD1566204, and Dusp14) out of these 7 predictions, thus providing evidence that they are indeed true rat transcripts. There are three gene predictions (RGD1559577, Taf15, and RGD1559875) for which we did not detect any transcribed products either in the S or in the LEW rat. This does not necessarily imply that these predicted transcripts do not exist in the rat. Based on the genomic sequence analysis of these predicted transcripts we are able to conclude that there are no allelic variations between these predicted transcripts of S and LEW.
Genetic variants within positional transcripts of the QTL1 region
Table 2 shows the three nonsynonymous variants that were identified between the S and LEW genes located within the QTL1 region. Out of the 18 genes found within the QTL1 region, only three potentially informative nonsynonymous S and LEW allelic variants were detected. The three variants belonged to the chemokine (C-C motif) ligand 5 (Ccl5) , ATP-dependent RNA helicase (Ddx52), and the predicted RGD1559577 genes, respectively. Testing of other inbred strains (i.e., SHRSP, WKY, MHS and MNS) revealed that all four strains carried the LEW variant for all three nonsynonymous polymorphic nucleotides identified in the S.LEW comparison (data not shown). Furthermore, the only other BP QTL that overlaps our QTL region and is corroborated using a congenic strain is the SHRSP.WKY comparison (Figure 4). Since SHRSP and WKY were not polymorphic at any of the three candidate variants that were found between S and LEW, these sites are not QTNs for the BP QTL located using WKY.SHRSP.
Figure 4. A summary of BP QTL mapping on RNO10 .
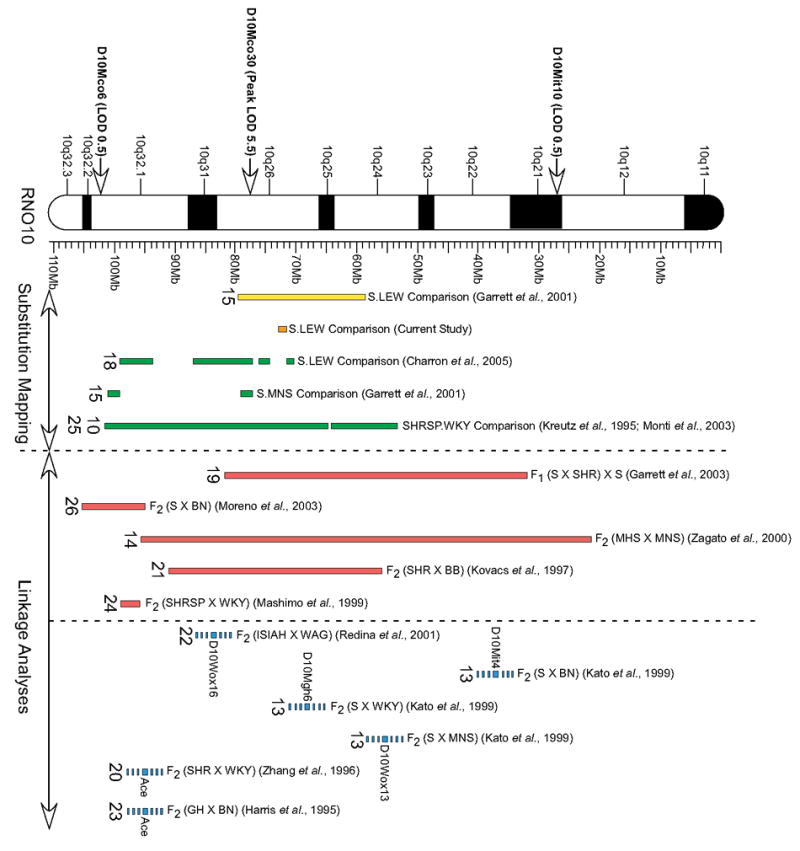
Illustrated to the left of the cytogenetic map are the flank and peak LOD markers previously obtained for the F2 (S X LEW) population [12]. To the right of the cytogenetic map represented in yellow is our previous S.LEW QTL1 localization as it relates to our current (orange) S.LEW QTL1 localization. A collection of other RNO10 substitution mapping (green), linkage analyses with defined flanking markers (red), and linkage analyses with poorly defined flanking markers (blue) are given for comparison. Numbers underneath the mapping locations are citation numbers. Rat strain nomenclature: S = Dahl Salt-sensitive rat, LEW= Lewis, MNS= Milan normotensive strain, SHRSP= Spontaneously hypertensive stroke-prone, WKY= Wistar Kyoto, SHR= Spontaneously hypertensive rat, BN= Brown Norway, MHS= Milan hypertensive strain, BB= Bio-breeding, ISIAH= Inherited stress-induced arterial hypertension, WAG= Wistar Albino Glaxo, GH= Genetically Hypertensive.
Discussion
This study describes the fine-mapping of a BP QTL to a relatively small 1.17 Mb region on RNO10 with only 18 genes. Improved resolution of this magnitude allowed for a comprehensive scanning of all the genes for nucleotide variants or QTNs. This was accomplished systematically, first by collecting all known bioinformatic data for the existence of each of the known/predicted transcripts in S and LEW rats, sequencing all exons and finally confirming each of the variants using genomic DNA from S and LEW rats. Based on the results described here, further analysis of three genes could be prioritized. Thus at this stage of mapping, the possibility for multiple QTNs that contribute to the overall QTL effect of S.LEW QTL1 cannot be ruled out.
Confidence in the current improved localization of QTL1 is supported by two lines of evidence. First, support is provided by the collective BP data for all the congenic substrains that were tested. The congenic substrains that we tested were carefully chosen and constructed in a way as to provide redundancy. The iterations of congenic substrains used in this study allowed for a two- to five-fold redundancy for any given chromosomal segment being tested for its BP effect. Second, the BP lowering effect observed in the S.LEWx12x2x3 congenic substrain, which spans the deduced QTL region of interest with minimal flanking introgressed region. Moreover, relative heart weight has served as a good surrogate indicator of blood pressure. Unlike other reports of heart weight or left ventricular mass QTLs that are under independent control of blood pressure [28–33], the heart weight effects of our study are likely due to the fact that the hypertrophy of the heart develops as a result of increase in blood pressure and peripheral resistance. In any case, the S.LEWx12x2x3 congenic substrain serves as the primary tool for further substitution mapping of the BP QTL.
There are many rat strain comparisons used to locate and identify BP QTLs on RNO10. Among these, a relatively greater degree of fine-mapping has been achieved using the S versus LEW comparison. Independent studies, including the current study, have provided evidence for at least 5 different BP QTLs on RNO10 (Figure 4). The novel BP QTL that we have localized in the S and LEW comparison within 71,513,116 and 72,684,774 bp on RNO10 is designated as S.LEW QTL1. This QTL is flanked by two other mean arterial pressure QTL regions (C10QTL1 and C10QTL4) that do not overlap our current localization [18]. Based on our studies, we cannot replicate the data of Charron et al, who have reported the localization of two BP QTLs, labeled as C10QTL4 within 70.57 Mb and 71.39 Mb of RNO10 and C10QTL1 within 74.86 Mb and 76.03 Mb of RNO10. The exact reasons for this discrepancy are not known. However, the lengths of the introgressed segments within congenic strains used for substitution mapping in the study by Charron et al [18] are different from those used in our study. Complex inter-allelic interactions or closely-linked modifiers of the QTL effect may be present in one congenic strain and not the other, accounting for these differences. Alternately, differences in experimental approaches could be contributing to the observed discrepancies. More importantly however, we would like to point out that coupled with the fact that a minimal congenic strain encompassing our BP QTL region from 71,513,116 bp to 72,684,774 bp, was not tested in the study by Charron et al [18], support for the localization of our BP QTL is also provided by three of the congenic strains (S.LC10, S.L4 and S.L10) used in the study by Charron et al [18].
Our newly defined S.LEW QTL1 region contains 1,171,658 bp. Theoretically, each one of these nucleotides, unless proven otherwise, is a candidate quantitative trait nucleotide (QTN) for S.LEW QTL1. Therefore, an exhaustive search for positional QTNs would include sequencing the S and LEW genome across their 1.17 Mb QTL interval. This approach has the advantage of generating a catalog of sequence variants within genes and also within inter- and intra-genic regions. However, we prioritized the identification of nonsynonymous variants because these variants can potentially directly affect protein structure and function. Therefore, the logical first step for us was to rule out, as comprehensively as possible, any coding sequence variations within candidate transcripts that result in altered primary amino acid sequences.
Out of the 34,005 bp that represent predicted and known transcripts within the QTL region only three exist as nonsynonymous variants between S and LEW. A single nonsynonymous variant was identified within each of three equally prioritized potential BP candidate genes located within QTL1. One of these variants is within Ddx52 gene, which is a member of the DEAD box (Asp-Glu-Ala-Asp/His) protein family of RNA helicases. Mutations within this gene may alter cellular functions related to transcription and translation (mRNA splicing/stability, ribosome assembly, and translation initiation) [34]. A high degree of homology between the rat, mouse, and human Ddx52 proteins (95% rat versus mouse and 88% rat versus human) implies an important evolutionally conserved function for this gene. As a result of the transcript variant detected, the predicted Ddx52 S rat protein had lysine (K) at amino acid position 73. Interestingly, the K at position 73 observed in the S rat is conserved across species, in both mouse and human, the relevance of which to the context of BP QTL effect is not immediately discernable.
There is a cluster of proinflammatory chemokines present as positional candidates within QTL1. Out of these, only one variant was identified within the proinflammatory chemokine Ccl5 gene. Rat Ccl5 protein has 94% and 79% homology to that of the mouse and human, respectively. However, the amino acid position 14, where the valine (V) was substituted with an isoleucine (I) in S rats, was not conserved among species. The V14-I14 variant is within the signal peptide sequence of the Ccl5 protein, which is excised during protein transport. Even single amino acid changes can influence the functionality of the signal peptide of a protein [35]. Whether the detected variant influences any functional aspects of Ccl5 in S compared to the LEW remains to be tested.
The importance of the nonsynonymous variant found within RGD1559577 is difficult to hypothesize because of the anonymity of the gene. Therefore, further studies of this positional candidate remain a challenge. Furthermore, it is not known whether the three genes with nonsynonymous variants operate independent of each other or with other yet undetected genetic factors to exert the observed QTL effect. It is not uncommon for QTL effects to be exerted by a complex concerted action of many underlying genetic factors [36].
Clearly, further substitution mapping is required to assign causation to any/all of these candidate QTNs. Further substitution mapping is possible, but takes considerable amount of time and resources, provided enough hot spots for recombination are available within the 1.17 Mb S.LEW QTL1 region. At this stage of mapping, candidate gene approach based on previously inferred function or differential gene expression analysis are two commonly used approaches to expedite QTL identification [37–44]. Each approach has its advantages and disadvantages. The candidate gene approach is not a comprehensive approach. For example, it is feasible that more than one closely positioned genetic factors may both be quantitative trait loci for the same phenotype [45]. Custom QTL-chips are used to comprehensively enlist differentially expressed positional candidate genes [42], but lack the ability to pin-point candidate QTNs. The comprehensive transcript sequence analysis described in our study certainly has limitations due to the fact that candidate QTNs outside of the coding regions are not detected. Our results do, however, demonstrate that multiple closely positioned genes contain potential QTNs for S.LEW QTL1.
Although large regions of rat chromosome 10 are homologous to human chromosome 17 wherein BP QTLs are reported, homology mapping indicates that the 1.17 Mb S.LEW QTL1 is not within regions of interest for regulating human BP. Therefore, it is conceivable that genetic factors underlying S.LEW QTL1 constitute novel, yet undiscovered mechanisms of BP control in humans.
Materials and Methods
Strains
The inbred Dahl salt-sensitive (SS/Jr) rat strain, designated as S, was from our colony. The LEW/NCrlBR rat strain was originally obtained from Charles River Laboratories (Wilmington, MA) and is referred to as LEW. The congenic substrains used in this study namely S.LEWx12x1, S.LEWx12x2, S.LEWx12x3, S.LEWx12x4, and S.LEWx12x6 were derived from the congenic strain S.LEWx12, previously developed by our group [15]. The congenic substrains S.LEWx12x2x1 and S.LEWx12x2x3 were derived from S.LEWx12x2. All congenic strains/substrains had the LEW chromosomal region of interest introgressed onto the genetic background of the S rat strain. To obtain these congenic substrains, the S.LEWx12 congenic was crossed to the S strain to yield F1 rats that were heterozygous for the original introgressed chromosomal segment. F1 rats were intercrossed to obtain F2 rats that were genotyped for markers in the congenic segment to look for recombinants. Rats with appropriate recombinant chromosomes were crossed to S again to duplicate the recombinant chromosome. The offspring were genotyped to select rats retaining the desired recombinant region. Two rats with the same recombinant chromosomal segment were crossed and the litters were genotyped to obtain rats that were homozygous throughout the recombinant region of interest. The homozygous rats were crossed to fix the recombinant chromosomal segment in a new congenic substrain.
Markers
The markers with D10Mco as the prefix were developed at the University of Toledo Health Science Campus (former Medical College of Ohio) using rat genomic DNA sequence obtained from the ensemble website (www.ensembl.org). These can be accessed on our web site (http://hsc.utoledo.edu/depts/physiology/research/rat/marker.html). The D10Rat marker sequences were obtained from the Whitehead Institute/MIT Center for Genome Research (www.broad.mit.edu/rat/public), Rat Genomic Mapping Project. The D10Arb marker sequences were obtained from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (www.nih.gov/niams/scientific/ratgbase/index.htm). The D10Got marker sequences were obtained from the Wellcome Trust Center for Human Genetics (www.well.ox.ac.uk/rat_mapping_resources). The D10Uia marker sequences were obtained from the University of Iowa [46] and the primer sequences are available at the Rat genome database (http://rgd.mcw.edu/).
Genotyping
DNA was extracted from a tail biopsy using the QIAamp Tissue Kit (Qiagen Inc., Valencia, CA). Polymerase chain reaction (PCR) genotyping with microsatellite markers was done as described earlier [8].
Blood pressure measurement
We employed an experimental design in which the BP of a congenic strain was compared only to its own separate group of control S rats that were bred, housed and studied concomitantly. Rats were weaned at 30 days of age to low-salt (0.3% NaCl) Harlan Teklad diet 7034 (Madison, WI). Twenty male congenic substrain rats were matched by age and weight with twenty male S control rats and were caged in 10 cages each cage containing 2 congenics and 2 S rats. At 40–42 days old, the rats were fed a 2% NaCl Harlan Teklad diet, TD94217 for 24 days. Systolic blood pressure was measured using the tail cuff method on conscious restrained rats warmed to 28°C. The operator did not know the identity of the rat during these measurements. BP was measured on each rat once a day for four consecutive days. The BP value of each day was the average of 3–4 consistent readings. The final BP value used was the averaged BP value of the four days. Rats were killed with CO2 and body and heart weights were measured. Statistical analysis was done using programs the SPSS software (SPSS, Chicago, IL).
Corroboration of tail-cuff BP measurements by telemetry
For the S.LEWx12x2 congenic substrain, BP was also collected using a telemetry system (Data Sciences International, St. Paul, MN). Briefly, after the tail-cuff measurements were completed, eight S and eight congenic rats were randomly selected from the whole group. At 70–73 days of age a transmitter was surgically implanted into the left flank of each rat and the probe was inserted through the femoral artery and advanced to the lower abdominal aorta. The rats continued on the 2% NaCl diet for the duration of the experiment. The animals were allowed to recover for 1 week before BP data was collected for a period of eight days.
Sequencing
mRNA from S and LEW kidney, liver, heart, lung, thyroid, pancreas, and spleen was extracted using TRIzol Reagent (Life Technologies), according to the manufacturer’s recommendation. S and LEW full-length cDNA of the genes within the QTL region was obtained by reverse transcription using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen), according to the provided protocol. Standard polymerase chain reaction, using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) and gene-specific primers, was used to amplify the cDNA. S and LEW gene amplicons were cloned into the Topo-TA-Cloning System (Invitrogen), according to the manufacturer’s recommendation. Plasmid DNA was purified using the QIAprep-miniprep (Qiagen), according to the provided protocol. The full-length S and LEW alleles for each gene within the QTL region were sequenced by MWG (High Point, NC), using vector based and gene-specific primers. cDNA variants were confirmed by sequencing PCR amplicons of their respective exons found on genomic DNA. The three nonsynonymous variants were confirmed in multiple S and LEW rats as well as genomic DNA obtained from MNS, MHS, SHRSP, WKY, and the NGFRx12x2 congenic substrain. We also sequenced PCR amplicons of genomic DNA exons for the few genes that we were not able to obtain cDNA. Intronic gene-specific primers, flanking each exon, were used to PCR amplify and sequence all the exons for those genes. Only the coding regions for all 18 genes were sequenced.
Acknowledgments
This work was supported by an RO1 grant from NIH to Dr. Joe. Authors thank Anna Dominiczak, Professor of Cardiovascular Medicine, Glasgow Cardiovascular Research Center, Glasgow, UK for the generous gift of genomic DNA from the SHRSP rat and John P. Rapp for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Joe B, Garrett MR. Substitution mapping: Using congenic strains to detect genes controlling blood pressure. In: Raizada MKPJ, Kasparov S, Katovich MJ, editors. Cardiovascular Genomics: Gene Mining for Pharmacogenomics and Gene Therapy. Humana Press Inc.; 2004. pp. 39–56. [Google Scholar]
- 2.Garrett MR, Joe B. Genetic analysis of inherited hypertension in the rat. In: Dominiczak A, editor. Handbook of Hypertension. Elsevier Science; 2006. (In press). [Google Scholar]
- 3.Jacob HJ, Kwitek AE. Rat genetics: attaching physiology and pharmacology to the genome. Nat Rev Genet. 2002;3:33–42. doi: 10.1038/nrg702. [DOI] [PubMed] [Google Scholar]
- 4.Cicila GT, Garrett MR, Lee SJ, Liu J, Dene H, Rapp JP. High-resolution mapping of the blood pressure QTL on chromosome 7 using Dahl rat congenic strains. Genomics. 2001;72:51–60. doi: 10.1006/geno.2000.6442. [DOI] [PubMed] [Google Scholar]
- 5.Garrett MR, Rapp JP. Defining the Blood Pressure QTL on Chromosome 7 in Dahl Rats by a 177kb Congenic Segment Containing Cyp11b1. Mamm Genome. 2003;14:268–273. doi: 10.1007/s00335-002-2245-9. [DOI] [PubMed] [Google Scholar]
- 6.Jacob HJ, Lindpaintner K, Lincoln SE, Kusumi K, Bunker RK, Mao YP, et al. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell. 1991;67:213–224. doi: 10.1016/0092-8674(91)90584-l. [DOI] [PubMed] [Google Scholar]
- 7.Hilbert P, Lindpaintner K, Beckmann JS, Serikawa T, Soubrier F, Dubay C, et al. Chromosomal mapping of two genetic loci associated with blood-pressure regulation in hereditary hypertensive rats. Nature. 1991;353:521–529. doi: 10.1038/353521a0. [DOI] [PubMed] [Google Scholar]
- 8.Deng AY, Rapp JP. Cosegregation of blood pressure with angiotensin converting enzyme and atrial natriuretic peptide receptor genes using Dahl salt-sensitive rats. Nature Genetics. 1992;1:267–272. doi: 10.1038/ng0792-267. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y, Rapp JP. Locus for the inducible, but not a constitutive, nitric oxide synthase cosegregates with blood pressure in the Dahl salt-sensitive rat. Journal of Clinical Investigation. 1995;95:2170–2177. doi: 10.1172/JCI117906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreutz R, Hubner N, James MR, Bihoreau MT, Gauguier D, Lathrop GM, et al. Dissection of a quantitative trait locus for genetic hypertension on rat chromosome 10. Proc Natl Acad Sci U S A. 1995;92:8778–8782. doi: 10.1073/pnas.92.19.8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dukhanina OI, Dene H, Deng AY, Choi CR, Hoebee B, Rapp JP. Linkage map and congenic strains to localize blood pressure QTL on rat chromosome 10. Mamm Genome. 1997;8:229–235. doi: 10.1007/s003359900399. [DOI] [PubMed] [Google Scholar]
- 12.Garrett MR, Dene H, Walder R, Zhang QY, Cicila GT, Assadnia S, et al. Genome scan and congenic strains for blood pressure QTL using Dahl salt-sensitive rats. Genome Res. 1998;8:711–723. doi: 10.1101/gr.8.7.711. [DOI] [PubMed] [Google Scholar]
- 13.Kato N, Hyne G, Bihoreau MT, Gauguier D, Lathrop GM, Rapp JP. Complete genome searches for quantitative trait loci controlling blood pressure and related traits in four segregating populations derived from Dahl hypertensive rats. Mamm Genome. 1999;10:259–265. doi: 10.1007/s003359900983. [DOI] [PubMed] [Google Scholar]
- 14.Zagato L, Modica R, Florio M, Torielli L, Bihoreau MT, Bianchi G, et al. Genetic mapping of blood pressure quantitative trait loci in Milan hypertensive rats. Hypertension. 2000;36:734–739. doi: 10.1161/01.hyp.36.5.734. [DOI] [PubMed] [Google Scholar]
- 15.Garrett MR, Zhang X, Dukhanina OI, Deng AY, Rapp JP. Two linked blood pressure quantitative trait loci on chromosome 10 defined by Dahl rat congenic strains. Hypertension. 2001;38:779–785. doi: 10.1161/hy1001.091503. [DOI] [PubMed] [Google Scholar]
- 16.Deng AY, Dutil J, Sivo Z. Utilization of marker-assisted congenics to map two blood pressure quantitative trait loci in Dahl rats. Mamm Genome. 2001;12:612–616. doi: 10.1007/s00335-001-2054-6. [DOI] [PubMed] [Google Scholar]
- 17.Sivo Z, Malo B, Dutil J, Deng AY. Accelerated congenics for mapping two blood pressure quantitative trait loci on chromosome 10 of Dahl rats. J Hypertens. 2002;20:45–53. doi: 10.1097/00004872-200201000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Charron S, Duong C, Menard A, Roy J, Eliopoulos V, Lambert R, et al. Epistasis, not numbers, regulates functions of clustered Dahl rat quantitative trait loci applicable to human hypertension. Hypertension. 2005;46:1300–1308. doi: 10.1161/01.HYP.0000192024.72367.c3. [DOI] [PubMed] [Google Scholar]
- 19.Garrett MR, Dene H, Rapp JP. Time-course genetic analysis of albuminuria in Dahl salt-sensitive rats on low-salt diet. J Am Soc Nephrol. 2003;14:1175–1187. doi: 10.1097/01.asn.0000060572.13794.58. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Summers KM, West MJ. Angiotensin I converting enzyme gene cosegregates with blood pressure and heart weight in F2 progeny derived from spontaneously hypertensive and normotensive Wistar-Kyoto rats. Clinical and Experimental Hypertension. 1996;18:753–771. doi: 10.3109/10641969609081779. [DOI] [PubMed] [Google Scholar]
- 21.Kovacs P, Voigt B, Kloting I. Novel quantitative trait loci for blood pressure and related traits on rat chromosomes 1, 10, and 18. Biochemical and Biophysical Research Communications. 1997;235:343–348. doi: 10.1006/bbrc.1997.6782. [DOI] [PubMed] [Google Scholar]
- 22.Redina OE, Lapteva NE, Khanina SL, Machanova NA, Dymshits GM, Markel AL. The region of rat chromosome 10 (the ngfr gene locus) is associated with blood pressure increase in response to emotional stress. Dokl Biochem Biophys. 2001;380:349–351. doi: 10.1023/a:1012352512006. [DOI] [PubMed] [Google Scholar]
- 23.Harris EL, Phelan EL, Thompson CM, Millar JA, Grigor MR. Heart mass and blood pressure have separate genetic determinants in the New Zealand genetically hypertensive (GH) rat. J Hypertens. 1995;4:397–404. [PubMed] [Google Scholar]
- 24.Mashimo T, Nabika T, Matsumoto C, Tamada T, Ueno K, Sawamura M, et al. Aging and salt-loading modulate blood pressure QTLs in rats. Am J Hypertens. 1999;12:1098–1104. doi: 10.1016/s0895-7061(99)00084-9. [DOI] [PubMed] [Google Scholar]
- 25.Monti J, Plehm R, Schulz H, Ganten D, Kreutz R, Hubner N. Interaction between blood pressure quantitative trait loci in rats in which trait variation at chromosome 1 is conditional upon a specific allele at chromosome 10. Hum Mol Genet. 2003;12:435–439. doi: 10.1093/hmg/ddg041. [DOI] [PubMed] [Google Scholar]
- 26.Moreno C, Kaldunski DPML, Tonellato PJ, Greene AS, Roman RJ, et al. Genomic map of cardiovascular phenotypes of hypertension in female Dahl S rats. Physiol Genomics. 2003;15:243–257. doi: 10.1152/physiolgenomics.00105.2003. [DOI] [PubMed] [Google Scholar]
- 27.Hubbard T, Andrews D, Caccamo M, Cameron G, Chen Y, Clamp M, et al. Ensembl 2005. Nucleic Acids Res. 2005;33:D447–453. doi: 10.1093/nar/gki138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pravenec M, Gauguier D, Schott JJ, Buard J, Kren V, Bila V, et al. Mapping of quantitative trait loci for blood pressure and cardiac mass in the rat by genome scanning of recombinant inbred strains. J Clin Invest. 1995;96:1973–1978. doi: 10.1172/JCI118244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsujita Y, Iwai N, Tamaki S, Nakamura Y, Nishimura M, Kinoshita M. Genetic mapping of quantitative trait loci influencing left ventricular mass in rats. Am J Physiol Heart Circ Physiol. 2000;279:H2062–H2067. doi: 10.1152/ajpheart.2000.279.5.H2062. [DOI] [PubMed] [Google Scholar]
- 30.Di Nicolantonio R, Kostka V, Kwitek A, Jacob H, Thomas WG, Harrap SB. Fine mapping of Lvm1: a quantitative trait locus controlling heart size independently of blood pressure. Pulm Pharmacol Ther. 2006;19:70–73. doi: 10.1016/j.pupt.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 31.Innes BA, McLaughlin MG, Kapuscinski MK, Jacob HJ, Harrap SB. Independent genetic susceptibility to cardiac hypertrophy in inherited hypertension. Hypertension. 1998;31:741–746. doi: 10.1161/01.hyp.31.3.741. [DOI] [PubMed] [Google Scholar]
- 32.Deschepper CF, Masciotra S, Zahabi A, BoutinGanache I, Picard S, Reudelhuber TL. Functional alterations of the Nppa promoter are linked to cardiac ventricular hypertrophy in WKY/WKHA rat crosses. Circulation Research. 2001;88:223–228. doi: 10.1161/01.res.88.2.223. [DOI] [PubMed] [Google Scholar]
- 33.Sebkhi A, Zhao L, Lu L, Haley CS, Nunez DJ, Wilkins MR. Genetic determination of cardiac mass in normotensive rats: results from an F344xWKY cross. Hypertension. 1999;33:949–953. doi: 10.1161/01.hyp.33.4.949. [DOI] [PubMed] [Google Scholar]
- 34.Lamm GM, Nicol SM, Fuller-Pace FV, Lamond AI. p72: a human nuclear DEAD box protein highly related to p68. Nucleic Acids Res. 1996;24:3739–3747. doi: 10.1093/nar/24.19.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutz C, Renner A, Alken M, Schulz K, Beyermann M, Wiesner B, et al. The corticotropin-releasing factor receptor type 2a contains an N-terminal pseudo signal peptide. J Biol Chem. 2006 doi: 10.1074/jbc.M601554200. [DOI] [PubMed] [Google Scholar]
- 36.Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–780. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, Wahid FN, et al. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nature Genetics. 1999;21:76–83. doi: 10.1038/5013. [DOI] [PubMed] [Google Scholar]
- 38.Cicila GT, Lee SJ. Identifying candidate genes for blood pressure quantitative trait loci using differential gene expression and a panel of congenic strains. Hypertension Research. 1998;21:289–296. doi: 10.1291/hypres.21.289. [DOI] [PubMed] [Google Scholar]
- 39.Liang M, Yuan B, Rute E, Greene AS, Zou AP, Soares P, et al. Renal medullary genes in salt-sensitive hypertension: a chromosomal substitution and cDNA microarray study. Physiol Genomics. 2002;8:139–149. doi: 10.1152/physiolgenomics.00083.2001. [DOI] [PubMed] [Google Scholar]
- 40.Wayne ML, McIntyre LM. Combining mapping and arraying: An approach to candidate gene identification. Proc Natl Acad Sci U S A. 2002;99:14903–14906. doi: 10.1073/pnas.222549199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hinojos CA, Boerwinkle E, Fornage M, Doris PA. Combined genealogical, mapping, and expression approaches to identify spontaneously hypertensive rat hypertension candidate genes. Hypertension. 2005;45:698–704. doi: 10.1161/01.HYP.0000156498.78896.37. [DOI] [PubMed] [Google Scholar]
- 42.Joe B, Letwin NE, Garrett MR, Dhindaw S, Frank B, Sultana R, et al. Transcriptional profiling with a blood pressure QTL interval-specific oligonucleotide array. Physiol Genomics. 2005;23:318–326. doi: 10.1152/physiolgenomics.00164.2004. [DOI] [PubMed] [Google Scholar]
- 43.Lee SJ, Cicila GT. Functional genomics in rat models of hypertension: using differential expression and congenic strains to identify and evaluate candidate genes. Crit Rev Eukaryot Gene Expr. 2002;12:297–316. doi: 10.1615/critreveukaryotgeneexpr.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- 44.Lee SJ, Liu J, Qi N, Guarnera RA, Lee SY, Cicila GT. Use of a panel of congenic strains to evaluate differentially expressed genes as candidate genes for blood pressure quantitative trait loci. Hypertens Res. 2003;26:75–87. doi: 10.1291/hypres.26.75. [DOI] [PubMed] [Google Scholar]
- 45.Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc Natl Acad Sci U S A. 2001;98:1787–1792. doi: 10.1073/pnas.031336098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walder RY, Garrett MR, McClain AM, Beck GE, Brennan TMH, Kramer NA, et al. Short tandem repeat polymorphic markers for the rat genome from marker-selected libraries. Mamm Genome. 1998;9:1013–1021. doi: 10.1007/s003359900917. [DOI] [PubMed] [Google Scholar]