

Abstract
Prominent interindividual and sex-dependent differences have been described in responses to sustained pain and other stressful stimuli. Variations in μ-opioid receptor-mediated endogenous opioid neurotransmission may underlie some of these processes. We examined both baseline μ-opioid receptor levels and the activation of this neurotransmitter system during sustained pain using positron emission tomography in a sample of young healthy men and women. Women were studied twice, during low and high estrogen states. The high-estrogen state was associated with regional increases in baseline μ-opioid receptor availability in vivo and a greater activation of endogenous opioid neurotransmission during the pain stressor. The latter did not differ from that obtained in males. During the low estrogen condition, however, significant reductions in endogenous opioid tone were observed at the level of thalamus, nucleus accumbens, and amygdala, which were associated with hyperalgesic responses. Estrogen-associated variations in the activity of μ-opioid neurotransmission correlated with individual ratings of the sensory and affective perceptions of the pain and the subsequent recall of that experience. These data demonstrate a significant role of estrogen in modulating endogenous opioid neurotransmission and associated psychophysical responses to a pain stressor in humans.
Keywords: μ-opioid receptors, pain, stress, sex differences, PET, human
Introduction
A number of persistent painful conditions (e.g., fibromyalgia, temporomandibular pain) are more frequently diagnosed in women, but only after gonadal maturation (Unruh, 1996). They also appear to be influenced by circulating gonadal steroids, with higher ratings of pain during low or rapidly changing levels of estradiol (LeResche et al., 2003). However, our understanding of the underlying neurobiological mechanisms is both limited and confounded by difficulties in isolating the individual effects of each gonadal steroid (i.e., estradiol and progesterone).
We examined the influence of estradiol, the principal female gonadal steroid, on μ-opioid receptor-mediated neurotransmission because of its involvement in the suppression of responses to sustained stressful and painful stimuli (Levine et al., 1978; Watkins and Mayer, 1982; Akil et al., 1984), and in the action of opiate analgesics (Matthes et al., 1996). In human subjects, the regional activation of this system has been associated with the suppression of sensory and affective elements of pain (Zubieta et al., 2001, 2003). Furthermore, sex differences in μ-opioid receptor concentrations have been described in postmortem material (Gross-Isseroff et al., 1990; Gabilondo et al., 1995) and neuroimaging studies, further influenced by age, reproductive status (i.e., premenopause and postmenopause), and circulating levels of estradiol (Smith et al., 1998; Zubieta et al., 1999). More recent work has also shown that during sustained pain, women studied during a low-estradiol, low-progesterone state demonstrated a lower capacity to activate this neurotransmitter system than men, paralleled by higher ratings of pain and a greater negative affective state during the challenge (Zubieta et al., 2002).
The present study was undertaken to determine whether the sex differences observed in μ-opioid receptor concentrations and in the release of endogenous opioids are influenced by circulating levels of estradiol in women. For example, in the ovariectomized animal model, estradiol administration has been shown to increase μ-opioid receptor protein concentrations, both at the level of immunoreactivity and mRNA (Hammer and Bridges, 1987; Dondi et al., 1992; Quinones-Jenab et al., 1997), the concentration of endogenous opioid peptides and their mRNA (Broad et al., 1993; Hammer et al., 1993), and the release of endogenous opioid peptides in cell cultures (Eckersell et al., 1998).
We examined baseline μ-opioid receptor availability in vivo and the activation of μ-opioid receptor-mediated neurotransmission during sustained pain using positron emission tomography (PET) and a μ-opioid receptor selective radiotracer, as described previously (Zubieta et al., 2001, 2002, 2003). The effects of estradiol were studied in regularly cycling women during the early follicular phase, before and after the administration of estradiol. It was hypothesized that estradiol would modulate μ-opioid neurotransmission in regions where sex differences had been previously observed (thalamus, hypothalamus, nucleus accumbens, amygdala) (Zubieta et al., 2002). These are regions receiving dense β-endorphin projections from the arcuate nucleus of the hypothalamus (Gray et al., 1984; Khachaturian et al., 1985; Kineman et al., 1989), where estradiol regulates β-endorphin-containing cell populations and peptide content (Tong et al., 1990; Thornton et al., 1994; Priest and Roberts, 2000).
Materials and Methods
Subjects.
Ten healthy women, 20–30 years of age, were recruited through advertisement. Volunteers were right-handed nonsmokers with no personal history of medical or psychiatric illness, substance abuse or dependence, or family history of inheritable illnesses. Subjects were not taking psychotropic medications or hormone treatments, including hormonal birth control, for at least 6 months and did not exercise in excess of 1 h three times per week. Subjects were instructed not to drink alcohol for at least 24 h, nor to exercise or eat for at least 3 h before the study. Subjects were selected who had regular menstrual cycles of duration 26–32 d, and were scanned during the follicular phase of their menstrual cycles (2–9 d after the onset of menses). The timing of menstrual cycles was followed for at least 2 months before scheduling PET studies. To further standardize the hormonal milieu, the presence of ovulatory cycles before scanning was also determined by a progesterone level of >3 ng/ml during the luteal phase of the preceding menstrual cycle.
An additional series of scans were conducted in the same women after treatment with transdermal micronized estradiol (Estroderm; 0.4 mg daily delivery), applied on the buttocks for 7–9 d and changed every 3 d. As in the studies without treatment, estradiol was administered after ovulatory cycles, as described above. The sequence of hormonal states (no treatment; treatment with estradiol) was randomized and counterbalanced between subjects. Estradiol and progesterone plasma levels were obtained immediately before the PET studies. Estradiol levels reflect both endogenous estradiol production and exogenous administration of estradiol at the time of the study; however, it is likely that there was variability with peaks after patch changes (Khul 2005). Although this does not entirely mimic the natural physiologic state, it allowed us to control the hormonal environment so that the studies could be conducted in relatively low-estradiol and relatively high-estradiol environments.
In one of the subjects, technical problems during the low-estradiol state precluded the analysis of the data. A second subject was then eliminated at random from the analyses to maintain the counterbalancing of the studies. Therefore, a total of 8 women were included in the analyses presented.
A separate sample of five women was studied in the early follicular phase before and after the administration of a placebo with the expectation that it was an analgesic agent, as described previously (Zubieta et al., 2005). These studies were undertaken to ensure that the effects observed after the administration of estradiol were not solely caused by expectations of analgesia and placebo-associated activation of endogenous opioid neurotransmission.
A comparison group of eight men matched by age and educational level to the female group was also studied once to provide a point of comparison. All studies were conducted in the morning, between 8:00 and 11:00 A.M. Subjects were instructed not to drink alcohol or exercise for at least 24 h or eat for 3 h before the studies. Written informed consent was obtained in all cases. All the procedures used were approved by the University of Michigan Investigational Review Board for Human Subject Use and the Radioactive Drug Research Committee.
Neuroimaging methods.
High-resolution magnetic resonance imaging (MRI) images were acquired in all subjects on a 1.5 Tesla scanner (Signa; General Electric, Milwaukee, WI) (TE = 5.5, TR = 14, TI = 300, flip angle = 20o, NEX = 1, 124 contiguous images, 1.5 mm thickness) for structure identification and to aid in the warping of PET images to a standard stereotactic coordinate system [International Conference on Brain Mapping (ICBM)].
PET studies were acquired with a Siemens (Erlangen, Germany) ECAT Exact scanner in three-dimensional mode, with septa retracted and scatter correction. Participants were positioned in the PET scanner gantry and two intravenous (antecubital) lines were placed. A light forehead restraint was used to eliminate intrascan head movement. [11C]carfentanil was synthesized at high specific activity (>2000 Ci/mmol) by the reaction of 11C-methyliodide and a nonmethyl precursor as described previously (Dannals et al., 1985), with minor modifications to improve its synthetic yield (Jewett, 2001). Ten to 15 mCi (370–555 MBq) were administered to each subject for each of the PET studies. Two PET studies were completed during the low-estradiol condition, and two more after treatment with estradiol, as above. Men received only two studies. Each pair of scans included a nonpainful control state and a sustained pain state, separated by at least 2 h to allow for tracer decay. The maximum mass of carfentanil injected was 0.03 μg/kg per scan, ensuring that the compound was administered in tracer quantities (i.e., subpharmacological doses). Receptor occupancy by carfentanil was calculated to be between 0.2 and 0.6% for brain regions with low and high μ-opioid receptor concentrations, based on the mass of carfentanil administered and the known concentration of μ-opioid receptors in the postmortem human brain (Gross-Isseroff et al., 1990; Gabilondo et al., 1995). Fifty-five percent of the [11C]carfentanil dose was administered as a bolus, and the remainder as a continuous infusion using a computer-controlled pump to achieve steady-state tracer levels. For each study, 19 sets of scans were acquired over 70 min with an increasing duration (30 s up to 10 min).
Images were reconstructed using filtered back-projection with a Hanning 0.5 filter, and included both measured attenuation and scatter corrections. Dynamic images were coregistered to each other and the intercommisural line using automated computer routines (Minoshima et al., 1993). Image data were then transformed on a voxel-by-voxel basis into two sets of parametric maps: (1) a tracer transport measure (K1 ratio), and (2) a receptor-related measure [distribution volume ratio (DVR)], the latter using data obtained from 20–70 min post-tracer administration for nonpainful control and pain studies. To avoid the need for arterial blood sampling, the tracer transport and binding measures were calculated using a reference region Logan plot analysis (Logan et al., 1996), using the occipital cortex (an area devoid of μ-opioid receptors) (Titeler et al. 1989) as the reference region. With the tracer administration protocol used, the Logan plot becomes linear by 5–7 min after the start of radiotracer administration, with its slope being the DVR, a measure equal to the (f2Bmax/Kd) + 1 for this receptor site and radiotracer. With f2 being the free radiotracer in extracellular space, the term f2Bmax/Kd (or DVR-1) is the “receptor-related” measure (μ-opioid receptor availability, or binding potential). K1 and DVR images for each experimental period and MR images were coregistered to each other and to the ICBM stereotactic atlas orientation (Meyer et al., 1997).
Sustained pain model.
Pain and saline control conditions were introduced 20 min after radiotracer administration, in a single-blind counterbalanced design (half the volunteers receiving pain first, and half saline control) and maintained from 20 to 40 min post-tracer administration. Pain levels were maintained at a predetermined intensity level by a computer-controlled system through the titrated infusion of medication-grade hypertonic saline (5%) into the masseter (jaw) muscle. In this model of sustained deep somatic pain, the intensity of the painful stimulus is standardized across subjects, as described in detail previously (Stohler and Lund, 1995; Stohler and Kowalski, 1999). Briefly, after a standard bolus administration of 150 μl, injected over 15 s, an electronic version of a 10 cm visual analog scale (VAS) is used by the subject to rate pain intensity once every 15 s. This signal is fed back to the computer via an analog–digital board, fed into an individually adapted pain-response model, which adjusts the infusion rate so that pain is maintained at a target VAS intensity range of 35–45 for the full duration of the challenge. Subjects were informed that the lower end on the VAS scale denoted “no pain,” whereas the upper bound represented the “most intense pain imaginable.” The control condition consisted of isotonic saline infused at the average rate required to achieve VAS intensity in the target range.
The sensory and pain-specific affective qualities of the painful stimulus were assessed after completion of each PET scan with the McGill Pain Questionnaire (MPQ) (Melzack and Katz, 2000). The internal emotional state of the volunteers was rated at the same time with the Positive and Negative Affectivity Scale (PANAS) (Watson et al., 1988). We also examined the recall accuracy of sensory and affective qualities of the pain by asking the volunteers to retrospectively rate the pain experience 24 h after completion of the study using again the MPQ. A measure of pain recall accuracy was obtained by subtracting the MPQ values obtained immediately after the challenge from those acquired 24 h later (negative numbers reflecting overstating pain on recall, and positive values understating the challenge). One subject returned one of the MPQ questionnaires with a date exceeding 48 h from the challenge during the high estradiol condition. That data point was eliminated from the analyses.
Statistics.
Pain and saline control conditions were compared within subjects (two-tailed paired t tests) and sex groups (two-tailed unpaired t tests). These analyses and Pearson correlations with psychophysical variables were calculated voxel-by-voxel with the Statistical Parametric Mapping (SPM) 99 package using the ICBM coordinate system and expressed in standardized z scores, with a statistical threshold of significance of p < 0.0001 for regions in which sex differences had been detected previously (thalamus, hypothalamus, ventral basal ganglia, amygdala). A threshold of z = 4.4 was applied to any other non-a-priori-hypothesized regions (p < 0.05 after correction for multiple comparisons across all brain voxels) identified in the statistical maps. Data for the individual regions was then extracted from the images for voxels achieving thresholds of p < 0.01 within significant clusters and used for the plotting of data, calculation of effect sizes, and regional correlation values (r).
Results
The pain-maintenance protocol used resulted in average VAS ratings of intensity (acquired every 15 s for the duration of the challenge) of 39.2 ± 5.4 during low estradiol and 36.0 ± 7.7 during the high-estradiol condition. The volumes of hypertonic saline required to maintain pain in the target range were 1921 ± 693 and 2231 ± 943 μl during low- and high-estradiol states, respectively, not statistically significant with this sample size (paired two-tailed t test, p > 0.05). Mens’ ratings were 39.5 ± 4.9 with a total infusion volume of 2153 ± 597 μl and were not significantly different from those of women during either low- or high-estradiol phases (unpaired two-tailed t test, p > 0.05).
Estradiol effects on μ-opioid receptor binding potential in vivo
Estradiol plasma levels increased from 38 ± 22 pg/ml (range 16–85) without treatment to 262 ± 105 pg/ml (range 164–503) after patch treatment. Progesterone remained <1 ng/ml under both conditions (ranging from <0.2 to 0.7).
Estradiol administration was associated with significant increases in μ-opioid receptor baseline binding potential (BP) in the medial thalamus [x, y, and z coordinates (in mm): 1, −8, 8, respectively; cluster size, 605 mm3; z = 5.11; p < 0.0001], anterior hypothalamus extending rostral and laterally to the periventricular area of the nucleus accumbens, bilaterally [ipsilateral to pain, x, y, and z coordinates (in mm): 6, 3, −5, respectively; cluster size, 1004 mm3, z = 6.60; contralateral to pain, x, y, and z coordinates: −8, 5, 2, respectively; cluster size, 325 mm3, z = 4.88; p < 0.0001], and the amygdala nuclei bilaterally (ipsilateral, x, y, and z coordinates: 18, −4, −14, respectively; size, 193 mm3, z = 4.16; contralateral, x, y, and z coordinates: −14, −5, −20, respectively; size, 172 mm3, z = 4.15; p < 0.0001), which in the side contralateral to pain extended to a second peak in the subamygdalar temporal cortex (x, y, and z: −26, 9, −34, respectively; size, 623 mm3; z = 4.54; p < 0.0001) (Fig. 1). The mean increases in the receptor binding measure ranged from 15 to 32% across these regions. Significant positive correlations between plasma levels of estradiol before scanning (log10 transformed) and μ-opioid receptor binding were obtained in an area centered around the anterior hypothalamus with rostral extension into the nucleus accumbens bilaterally (x, y, z: 4, 0, −5, respectively; size, 3966 mm3; z = 3.65; p < 0.0001; r = 0.67 for the entire cluster).
Figure 1.

Effect of estradiol on μ-opioid receptor binding and on responses of μ-opioid receptor-mediated neurotransmission to a stress challenge. Top, left, Three-dimensional representation of μ-opioid receptor binding potential values in a representative healthy volunteer, superimposed over an MRI image standardized to ICBM stereotactic coordinates. Binding potential values are represented by the pseudocolor scale in the lower part of the figure. Top, center, Three-dimensional display of significant effects of estradiol treatment on baseline μ-opioid receptor binding potential for the entire sample, superimposed over a magnetic resonance image in ICBM stereotactic coordinates. Standardized z scores are represented by the pseudocolor scale on the left. Significant effects of estradiol treatment were observed in the medial thalamus, ventral basal ganglia, amygdala, and hypothalamus. The graph shows the mean ± SEM of BP values for thalamus, ventral basal ganglia, and amygdala during low and high estradiol states (yellow). BP values for a matched sample of eight men are shown in blue for demonstrative purposes. Bottom, Brain areas in which significant effects of estradiol were observed on stress-induced activation of the μ-opioid system for the entire sample. Standardized z scores are represented by the pseudocolor scale on the left. Significant effects of estradiol condition were observed in the medial thalamus, ventral basal ganglia, and amygdala. The graph shows the mean ± SEM of the change in BP for these regions during the stress challenge (negative values reflecting reductions in μ-opioid neurotransmission, positive values reflecting activation of neurotransmission) during low- and high-estradiol states (yellow). The change in BP values for a matched sample of eight men are shown in blue for demonstrative purposes.
μ-Opioid receptor BP was significantly higher in women during the low-estradiol condition than in men in the ipsilateral nucleus accumbens/anterior hypothalamus region (1.98 ± 0.39 versus 1.68 ± 0.28; one-tail t test, t = 1.76; p = 0.05) and reached trend levels of significance in the contralateral side (2.29 ± 0.37 versus 2.01 ± 0.38; t = 1.52; p = 0.07). These regional sex differences increased in statistical significance during the higher estradiol state (women: ipsilateral 2.00 ± 0.36; contralateral 2.39 ± 0.31; t = 1.97 and 2.18, respectively; p < 0.03) (Fig. 1)
Estradiol effects on μ-opioid system activation during sustained pain
Statistically significant differences between low and high-estradiol states were also observed for the activation of μ-opioid-receptor-mediated neurotransmission from saline control to pain conditions (observed experimentally as reductions in the in vivo availability of the receptors). Greater magnitudes of endogenous neurotransmitter activation were observed during the high-estradiol state in the medial thalamus (x, y, z: −2, −7, 11, respectively; cluster size, 316 mm3; z = 3.71; p < 0.0001), anterior hypothalamus/medial nucleus accumbens ipsilateral to pain (x, y, z: 7, 5, −4, respectively; size 358 mm3; z = 5.15; p < 0.0001), and the amygdala bilaterally (ipsilateral, x, y, z: 19, −5, −15, respectively; size, 261 mm3, z = 4.62; contralateral: −14, −5, −18, respectively; 387 mm3, z = 4.86; p < 0.0001) (Fig. 1).
Sustained pain-induced changes in μ-opioid receptor-mediated neurotransmission correlated positively with plasma levels of estradiol (log10 transformed) for the above regions in the SPM maps (z scores ranging from 4.16 to 4.98; p < 0.0001), corresponding to regional r correlation values, using the data extracted from the significant clusters, of 0.51 in the anterior hypothalamus/medial nucleus accumbens region, r = 0.61, and r = 0.62 in the ipsilateral and contralateral amygdala, respectively, and r = 0.45 in the medial thalamus (Fig. 2). Significant correlations were also observed in the SPM maps for a separate peak in the subamygdalar temporal cortex contralateral to the challenge (z = 4.69; p < 0.0001; regional r = 0.75).
Figure 2.
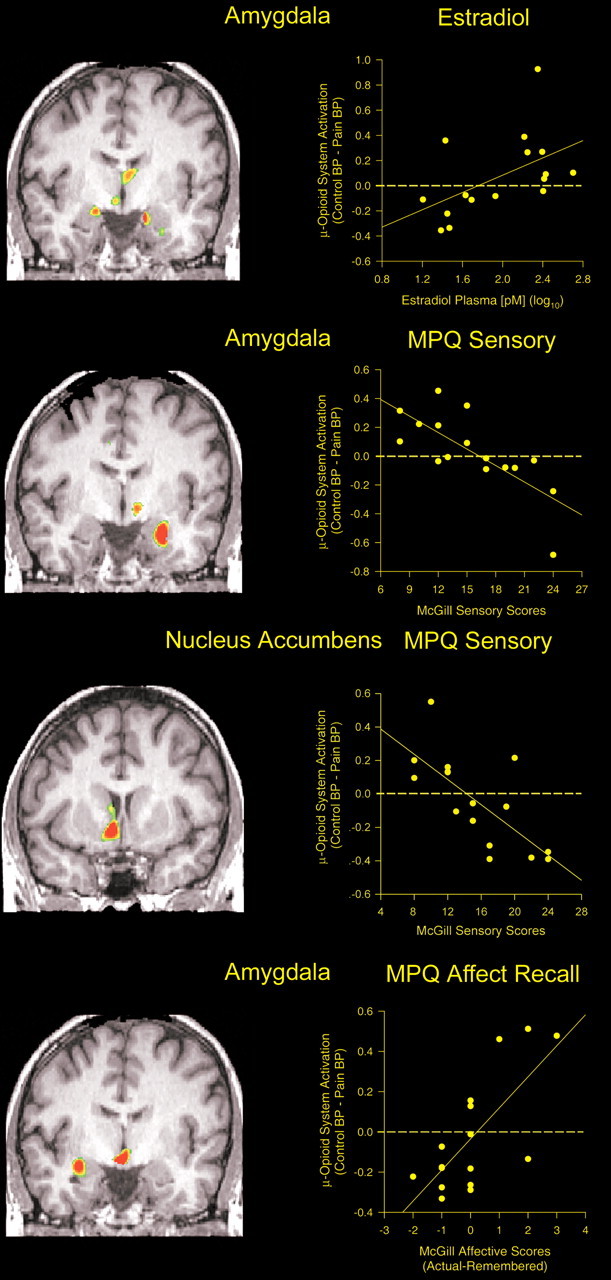
Correlations between the stress-induced changes in μ-opioid neurotransmission, estradiol plasma levels, and other psychophysical measures. Top, Significant correlations between estradiol plasma levels in picomolar (log10) and stress-induced μ-opioid system responses were obtained for the medial thalamus, amygdala (shown), and the ventral basal ganglia (data not shown). A coronal view is shown at the y (anterior–posterior) coordinate, −5. The graph inset shows the individual values for this correlation in the amygdala. Second panel, Significant voxel-by-voxel correlations between stress-induced μ-opioid system responses and MPQ pain sensory scores as rated by the volunteers at the level of the amygdala (coronal view, y = −6). The graph shows the individual values for this correlation. Third panel, Significant voxel-by-voxel correlations between stress-induced μ-opioid system responses and MPQ pain sensory scores as rated by the volunteers at the level of the nucleus accumbens (coronal view, y = 5). The graph shows the individual values for this correlation. Bottom, Correlations between stress-induced μ-opioid system responses and the subsequent recall of the pain experience 24 h after the pain challenge. Significant effects of μ-opioid system activation on the recall of MPQ pain sensory scores were observed in the medial thalamus (data not shown). Effects on the recall of MPQ pain affect scores were detected in the amygdala, medial thalamus, and hypothalamus. The graph shows the individual values for the latter correlation in the amygdala (coronal view, y = −6).
When compared with the male sample, women studied during the low-estradiol condition showed significantly lower levels of neurotransmitter activation in the medial thalamus (x, y, z: −2, −11, 2, respectively; cluster size, 1423 mm3; z = 4.53; p < 0.0001), anterior hypothalamus/medial nucleus accumbens ipsilateral to pain (x, y, z: 10, 14, −4, respectively; size, 2438 mm3; z = 6.60; p < 0.0001), and the amygdala bilaterally (ipsilateral, x, y, z: 19, −1, −19, respectively; size, 1634 mm3; z = 4.86; contralateral: −14, −5, −20, respectively; 2146 mm3; z = 5.71; p < 0.0001). These regions overlapped exactly with those where effects of estradiol were obtained (see above). No significant differences between the male sample and the high-estradiol condition were obtained in any brain region.
Further examination of the changes in μ-opioid receptor BP from the nonpainful control condition to the sustained pain state (Fig. 1) and their correlations with estradiol plasma levels (Fig. 2) additionally showed that the changes in the endogenous opioid neurotransmission occurred in both directions. In the high-estradiol state there was evidence of neurotransmission activation, with reductions in receptor availability of similar magnitude to those of men (Fig. 1). However, during the low-estradiol condition, the response was in the opposite direction, pointing to a deactivation of endogenous opioid neurotransmission.
To confirm these effects, statistical comparisons for data obtained during the nonpainful saline-control state and the sustained pain challenge were then performed separately for each of two hormonal conditions (Table 1). Significant reductions in μ-opioid neurotransmission (observed experimentally as increases in the BP measure from the nonpainful to the sustained pain challenges) were confirmed during the low-estradiol state in the medial thalamus, anterior hypothalamus/medial nucleus accumbens area, bilaterally, and in the amygdala ipsilateral to pain. These increases in BP from control to pain conditions ranged from 11 to 16% for these regions. No evidence of pain-induced μ-opioid system activation (reductions in BP) was observed during the low-estradiol state (Table 1).
Table 1.
Directionality, magnitude, and localization of changes in μ-opioid receptor-mediated neurotransmission as a function of estrogen status
| Regions | Coordinates(x, y, z) (mm) | Cluster size (voxels) | % Δ BP(control − pain) | z score |
|---|---|---|---|---|
| Low estradiol: stress-induced μ-opioid system deactivation | ||||
| Medial thalamus (I) | −11, −13, 10 | 311 | −11.3 | 3.95 |
| Medial thalamus (C) | 5, −5, 14 | 990 | −16.0 | 5.09 |
| Nucleus accumbens, HT (I) | −9, 13, −2 | 752 | −13.7 | 5.43 |
| Nucleus accumbens, HT (C) | 21, 6, −11 | 97 | −11.7 | 3.56 |
| Amygdala (I) | −27, −1, −21 | 356 | −15.8 | 4.11 |
| High estradiol: stress-induced μ-opioid system activation | ||||
| Nucleus accumbens, HT (I) | −7, 4, −4 | 505 | 11.5 | 5.14 |
| Nucleus accumbens, HT (C) | 11, −3, −9 | 74 | 14.9 | 3.33 |
| Amygdala (I) | −20, −7, −15 | 258 | 19.1 | 4.94 |
Data represent the localization (ICBM coordinates) and magnitude of differences in μ-opioid BP from nonpainful control to pain states, a measure of endogenous opioid release and μ-opioid receptor activation, during low- and high-estradiol states. Significant reductions in endogenous opioid neurotransmission were observed in the low-estradiol state, whereas significant enhancements were noted in the high-estradiol state, during the pain stressor. The size of significant clusters are expressed in 13 mm voxels. z scores were deemed significant at p < 0.0001 for regions in which a priori hypothesis had been formulated. Nucleus accumbens, HT refers to peaks that included the nucleus accumbens but also incorporated the anterior portion of the hypothalamus. C, Contralateral to the side of pain; I, ipsilateral to the side of pain.
Contrary to the findings during low-estradiol, the high-estradiol state was associated with significant activation of μ-opioid receptor-mediated neurotransmission in the anterior hypothalamus/nucleus accumbens area bilaterally and in the amygdala ipsilateral to pain (Table 1). These reductions in BP from control to pain conditions ranged from 12 to 19% for the above regions. No significant changes in the opposite direction (i.e., reductions in neurotransmission, increases in BP) were observed during the high-estradiol state.
Psychophysical correlates of activation and deactivation of endogenous μ-opioid receptor-mediated neurotransmission
In the women studied, MPQ sensory ratings of the pain were significantly negatively correlated with the magnitude of μ-opioid system activation in the hypothalamus/nucleus accumbens area, bilaterally (ipsilateral, x, y, z: 8, 10, −8, respectively; cluster size, 2881 mm3, z = 5.96, p < 0.0001; contralateral, x, y, z: −9, −4, −5, respectively; size 327 mm3, z = 4.34, p < 0.0001) corresponding to correlations of r = −0.72 and r = −0.68 for the data in each of these clusters, respectively, as well as the amygdala contralateral to pain (x, y, z: −26, −6, −21, respectively; z = 6.00; p < 0.0001; r = −0.74 for the cluster) (Fig. 2). No significant correlations were obtained for MPQ pain affect or PANAS negative affect scores in this sample.
Sustained pain-induced changes in regional μ-opioid receptor-mediated neurotransmission were additionally associated with the recall of the pain experience 24 h after the challenge. This measure of recall was calculated as the difference between the rating at the time of the study and that recalled 24 h after the challenge (actual minus remembered) (Fig. 2, bottom). Overstating MPQ pain sensory scores was associated with lower magnitudes of μ-opioid system activation, and vice versa, in the medial thalamus (x, y, z: 2, −6, 7, respectively; cluster size, 391 mm3; z = 4.12; p < 0.0001) with a correlation of r = −0.59 for the entire cluster, between the measure of recall and the change in opioid system activity. The recall accuracy of MPQ pain-affect scores was negatively correlated with activation in the amygdala ipsilateral to pain (x, y, z: 31, −6, −14, respectively; cluster size, 621 mm3; z = 5.21; p < 0.0001; cluster r = −0.72) (Fig. 2) and in the anterior hypothalamic region (x, y, z: 3, −4, −11, respectively; size, 924 mm3; z = 7.11; p < 0.0001; cluster r = −0.60).
Placebo effects on μ-opioid system activation during sustained pain
Recent data has shown the capacity to activate endogenous opioid neurotransmission during placebo administration when there is expectation of analgesia (Zubieta et al., 2005). No expectation of analgesia was introduced during the explanation of the studies described in the present study. To confirm that the observed effects were not placebo-mediated, five female subjects were subjected to the same experiment paradigm with the exception that in their case, expectation of analgesia was induced by intravenous infusion of isotonic saline as reported previously (Zubieta et al., 2005). None of the areas that were significantly activated by the expectation of placebo, however, matched those linked to estradiol. Using a lenient statistical threshold of p = 0.01 uncorrected for multiple comparisons, no areas of placebo-induced activation were observed in thalamic or amygdala regions. An area of significant activation was observed in the ventral basal ganglia ipsilateral to the pain challenge (x, y, z coordinates (in mm): −12, 4, −3, respectively; cluster size, 756 mm3; z = 3.77; p < 0.0001, uncorrected), averaging a 10.4% reduction in BP. However, this region was located more lateral (5 mm) to the area where effects of estradiol on endogenous opioid activity were obtained. No significant effects of placebo on baseline BP values were observed.
Discussion
The present study describes for the first time in humans the regulatory effect of estradiol on the functional responses of μ-opioid receptor-mediated endogenous opioid neurotransmission to a sustained pain challenge. During a high-estradiol, low-progesterone state, sustained pain induced the regional activation of the endogenous opioid system as monitored at the level of μ-opioid receptors, in a manner similar to that observed in males. The same women studied during a low-estradiol state, low-progesterone phase of the menstrual cycle demonstrated a reduction in the baseline tonic activity of this neurotransmitter system in the same regions, an effect associated with higher ratings of pain. In addition, we demonstrate that estradiol-induced modulation of endogenous opioid system function was associated with the recall accuracy of the pain experience (overstating and understating past pain) in both its sensory and affective qualities.
The reductions in endogenous opioid tone observed in the low-estradiol state would require that substantial tonic release of this neurotransmitter is present under nonpainful conditions. In this regard, considerable basal β-endorphin tone has been directly described in microdialysis studies in some of these regions (arcuate nucleus, nucleus accumbens) (Zangen et al., 1999). Indirectly, through the assessment of increases in glucose metabolism or c-fos expression after low dose naloxone administration, tonic endogenous opioid activity has been described in the brainstem regions and the amygdala of rodents (Kraus et al., 1996; Gestreau and Besson, 2000). In human subjects, the administration of naloxone has been shown to increase the activity of a number of cortical (e.g., anterior cingulate, prefrontal and insular, entorhinal and parahippocampal cortices) and subcortical (e.g., basal ganglia, hippocampus) regions in a recent functional MRI study (Borras et al., 2004). Microinjection of naloxone in the nucleus accumbens under baseline conditions has also been associated with hyperalgesic responses in animal models (Gear et al., 1999).
The data presented demonstrate a substantial influence of estradiol on a neurochemical system that regulates various aspects of pain and stress responses. Utilizing a model of physical and psychological stress and moderate levels of muscular pain (Stohler and Kowalski, 1999), we observe both pain/stress-permissive and -suppressive responses of the endogenous opioid system through μ-receptors. These opposite responses took place as a function of whether estradiol was present at low or high plasma levels. The psychophysical consequences of this bidirectional regulation included the capacity to both enhance and suppress the individual’s ratings of the pain and the recall accuracy of the sensory and affective qualities of the pain experienced.
The effects of estradiol took place in a series of interconnected brain nuclei known to be potently regulated by endogenous opioid peptides and μ-opioid receptors in animal models and in humans. These regions were a subset of those involved in the supraspinal regulation of pain, previously studied in a predominantly male sample (Zubieta et al., 2001), and conform to the brain distribution of proopiomelanocortin/β-endorphin immunoreactive cell bodies (located in the arcuate nucleus of the hypothalamus) and their terminal fields (Khachaturian et al., 1985; Tong et al., 1990; Thornton et al., 1994; Priest and Roberts, 2000). They included the nucleus accumbens, a dopamine and opioid peptide-rich region involved in responses to salient stimuli, both aversive (Horvitz, 2000) and rewarding (Robinson and Berridge, 2000; Koob and Le Moal, 2001). The hypothalamus, implicated in pain-regulatory mechanisms in animal models (Mayanagi et al., 1982) and humans (Leone et al., 2001), in addition to its more traditional involvement in hypothalamic–pituitary–adrenal axis stress and reproductive neuroendocrine responses (Plotsky, 1986; Smith et al., 1998). Specifically, arcuate nucleus β-endorphin has been identified as a critical factor in endogenous antinociception and stress-induced analgesia, an effect mediated by its activation of the μ-opioid receptor (Rubinstein et al., 1996; Sun et al., 2003), the amygdala and the subjacent periamygdalar temporal cortex, implicated in the regulation of sensory stimulus intensity (Anderson et al., 2003), affective states (Morris et al., 1998; Liberzon et al., 2000) and emotional memory (Cahill et al., 1996), and medial thalamic nuclei. The latter region receives inputs from the basal ganglia and amygdala and gates information flow into higher-order cortical regions (Kudora et al., 1998). Of note, a number of other brain areas involved in the μ-opioid system modulation of the pain experience, most notably the anterior cingulate, prefrontal and insular cortex, implicated in affective, cognitive, and sensory elements of pain (Zubieta et al., 2001, 2005), were not observed to be regulated by estradiol. Nevertheless, this restricted regional involvement should be replicated in larger samples to ensure its accuracy.
Also novel to the present set of results, we demonstrate that larger magnitudes of μ-opioid system activation were associated with an underestimation of sensory and affective qualities of the experience of pain when rated 24 h after the challenge, and vice versa. Substantial correlations with pain-affect ratings were observed in the amygdala, a brain region known to be involved in the processing and consolidation of memories for salient events (i.e., emotional memory) and sex differences in these processes (Cahill et al., 1996, 2004). This is also a region in which μ-opioid receptor-mediated mechanisms modulate emotional memory, possibly through reductions in norepinephrine release (Introini-Collison et al., 1995; McGaugh and Cahill, 1997; Quirarte et al., 1998; Katzen-Perez et al., 2001; McGaugh et al., 2002). Significant correlations between pain recall and μ-opioid system activation during pain were also obtained in the medial thalamus (sensory quality of pain) and hypothalamus (pain affect).
Recent data has also shown that among the regions implicated in estradiol effects, nucleus accumbens endogenous opioid systems are also involved in placebo-induced endogenous opioid activation when there is expectation of analgesia (Zubieta et al., 2005). Although the instructions associated with the study did not suggest that estradiol patches would be associated in any way with a lesser pain experience, we examined whether the introduction of a placebo (with the suggestion of its being an analgesic agent) would induce similar effects to those associated with estradiol. Placebo with expectation of analgesia did induce the activation of endogenous opioid neurotransmission in the ventral basal ganglia, but in a location more lateral to that observed for estradiol. Future studies will need to examine whether these effects could be synergistic under some circumstances (e.g., whether estradiol would increase placebo responses mediated by endogenous opioid neurotransmission in the ventral basal ganglia).
In addition to stress modulation and stress-induced analgesia, dependent on the activation of endogenous opioid neurotransmission, μ-opioid receptors also mediate the effects of exogenous opiate medications used for the treatment of pain. Therefore, changes in their basal concentration, observed as a function of varying estradiol levels, suggest an influence on the individual sensitivity to opiate agonists. In this regard, a higher sensitivity to opiate drugs has been reported in women, as compared with men (Zacny, 2001), including their side-effects (Fillingim et al., 2005a). The observed effect of estradiol on μ-opioid receptors is also consistent with previous PET results showing higher μ-opioid receptor BP in women than in men during reproductive age years, but not after menopause, when gonadal steroid levels decline (Zubieta et al., 1999). Postmortem in vitro data has also shown higher concentrations of μ-opioid receptors in women than in men, without differences in affinity (Gross-Isseroff et al., 1990; Gabilondo et al., 1995).
The enhanced pain perception mediated by the endogenous opioid system during low-estradiol states may represent a mechanism by which female gonadal steroids participate in the development of hyperalgesia and possibly the increased risk for the development of some idiopathic painful states observed in women (Unruh, 1996; LeResche et al., 2003). In this regard, there is substantial evidence showing that physical and mental stressors compromise menstrual cyclicity, leading to anovulatory cycles and consequently lower levels of circulating estradiol and progesterone (Harlow and Matanoski, 1991; Bonen, 1994; Carpenter, 1994; Sanders and Bruce, 1997). These mechanisms could then participate in the creation of a vicious circle by which endogenous antinociceptive mechanisms are compromised as gonadal steroid production is chronically diminished.
The present study was specifically designed to isolate the effects of estradiol from those of progesterone, and therefore the low levels of progesterone present across conditions in this study do not permit any assessments as to the possible effects of this hormone in the sample studied. In this regard, it has been suggested that progesterone may induce opposite effects to those of estradiol on both μ-opioid receptor binding and in the capacity to release endogenous opioids activating those receptors (Ratka et al., 1991; Shen et al., 1995; Ragnauth et al., 2001; Sinchak and Micevych, 2001; Mills et al., 2004). Additional investigation into these processes appears necessary in view of the results obtained here.
The data presented demonstrate that a low-estradiol, low-progesterone condition is associated with a pain vulnerability state by a reduction in endogenous opioid system function, possibly through β-endorphin mediated mechanisms. This hypothesis is consistent with recent observations that the A118G polymorphism of the μ-opioid receptor responded with hyperalgesic responses to heat pain in women studied during the early follicular phase of the menstrual cycle (a low-estradiol, low-progesterone state), but more pronounced analgesia in men (Fillingim et al., 2005b). This polymorphism has been reported to increase three-fold the affinity of the μ-opioid receptor for β-endorphin, with parallel increases in its transduction capacity (Bond et al., 1998). In the context of a more effectively activated system under basal conditions in homozygotes for this polymorphism, reductions in the basal tone of β-endorphin would then produce even more readily apparent hyperalgesic responses. Similarly, other genetic polymorphisms known to reduce the capacity to activate μ-opioid receptor-mediated neurotransmission (e.g., catechol-O-methyl transferase val158met polymorphism) (Zubieta et al., 2003) have been recently associated with a higher probability to develop myogenous temporomandibular pain (TMD) in women (Diatchenko et al., 2005), and with anxiety traits in women (Enoch et al., 2003). TMD is an idiopathic, persistent pain condition that presents substantial overlap with other muscular pain states, such as fibromyalgia syndrome (Plesh et al., 1996; Turp et al., 1997). Anxiety traits have been additionally associated with a higher probability of developing clinical depression (Parker et al., 1999), itself a highly comorbid condition with persistent pain syndromes such as fibromyalgia and TMD (Korszun et al., 1996, 1998; Turp et al., 1997).
We present data demonstrating a bidirectional influence of circulating gonadal steroids, specifically estradiol, on the responses of a stress and pain modulatory neurotransmitter system, the endogenous opioid. Additional investigation of these processes appears to be of importance to examine whether strategies that enhance endogenous opioid activity (presumably mediated through the peptide β-endorphin) could also reduce hyperalgesic responses to injury or stress in women and lower the risk of developing persistent painful states or comorbid conditions.
Footnotes
This work was supported by National Institutes of Health Grants R01 AT 001415 and R01 DA 016423 to J.K.Z. and K23 RR 17043 to Y.R.S. We acknowledge the contributions of the Positron Emission Tomography Center nuclear medicine technologists Jill M. Rothley, Edward J. McKenna, Andrew R. Weeden, Paul Kison, and Shayna Huber.
References
- Akil H, Watson S, Young E, Lewis M, Khachaturian H, Walker J (1984). Endogenous opioids: biology and function. Annu Rev Neurosci 7:223–255. [DOI] [PubMed] [Google Scholar]
- Anderson AK, Christoff K, Stappen I, Panitz D, Ghahremani DG, Glover G, Gabrieli JD, Sobel N (2003). Dissociated neural representations of intensity and valence in human olfaction. Nat Neurosci 6:196–202. [DOI] [PubMed] [Google Scholar]
- Bond C, LaForge K, Tian M, Melia D, Zhang S, Borg L, Gong J, Schuler J, Strong J, Leal S, Tischfield J, Kreek M, Yu L (1998). Single-nucleotide polymorphism in the human mu opioid receptor gene alters β-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA 91:9081–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen A (1994). Exercise-induced menstrual cycle changes. A functional, temporary adaptation to metabolic stress. Sports Med 17:373–392. [DOI] [PubMed] [Google Scholar]
- Borras MC, Becerra L, Ploghaus A, Gostic JM, DaSilva A, Gonzalez RG, Borsook D (2004). fMRI measurement of CNS responses to naloxone infusion and subsequent mild noxious thermal stimuli in healthy volunteers. J Neurophysiol 91:2723–2733. [DOI] [PubMed] [Google Scholar]
- Broad KD, Kendrick KM, Sirinathsinghji DJ, Keverne EB (1993). Changes in pro-opiomelanocortin and pre-proenkephalin mRNA levels in the ovine brain during pregnancy, parturition and lactation and in response to oestrogen and progesterone. J Neuroendocrinol 5:711–719. [DOI] [PubMed] [Google Scholar]
- Cahill L, Haier RJ, Fallon J, Alkire MT, Tang C, Keator D, Wu J, McGaugh JL (1996). Amygdala activity at encoding correlated with long-term, free recall of emotional information. Proc Natl Acad Sci USA 93:8016–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L, Uncapher M, Kilpatrick L, Alkire MT, Turner J (2004). Sex-related hemispheric lateralization of amygdala function in emotionally influenced memory: an fMRI investigation. Learn Mem 11:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter SE (1994). Psychosocial menstrual disorders: stress, exercise and diet’s effect on the menstrual cycle. Curr Opin Obstet Gynecol 6:536–539. [PubMed] [Google Scholar]
- Dannals RF, Ravert H, Frost JJ, Wilson A, Burns H, Wagner HJ (1985). Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int J Appl Radiat Isot 36:303–306. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W (2005). Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet 14:135–143. [DOI] [PubMed] [Google Scholar]
- Dondi D, Limonta P, Maggi R, Piva F (1992). Effects of ovarian hormones on brain opioid binding sites in castrated female rats. Am J Physiol 263:E507–E511. [DOI] [PubMed] [Google Scholar]
- Eckersell C, Popper P, Micevych P (1998). Estrogen-induced alteration of μ-opioid receptor immunoreactivity in the medial preoptic nucleus and medial amygdala. J Neurosci 18:3967–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch MA, Xu K, Ferro E, Harris CR, Goldman D (2003). Genetic origins of anxiety in women: a role for a functional catechol-O-methyltransferase polymorphism. Psychiatr Genet 13:33–41. [DOI] [PubMed] [Google Scholar]
- Fillingim RB, Ness TJ, Glover TL, Campbell CM, Hastie BA, Price DD, Staud R (2005a). Morphine responses and experimental pain: sex differences in side effects and cardiovascular responses but not analgesia. J Pain 6:116–124. [DOI] [PubMed] [Google Scholar]
- Fillingim RB, Kaplan L, Staud R, Ness TJ, Glover TL, Campbell CM, Mogil JS, Wallace MR (2005b). The A118G single nucleotide polymorphism of the mu-opioid receptor gene (OPRM1) is associated with pressure pain sensitivity in humans. J Pain 6:159–167. [DOI] [PubMed] [Google Scholar]
- Gabilondo A, Meana J, Garcia-Sevilla J (1995). Increased density of mu-opioid receptors in the postmortem brain of suicide victims. Brain Res 682:245–250. [DOI] [PubMed] [Google Scholar]
- Gear R, Aley K, Levine J (1999). Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci 19:7175–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestreau C, Besson J (2000). Is there tonic activity in the endogenous opioid systems? A c-Fos study in the rat central nervous system after intravenous injection of naloxone or naloxone-methiodide. J Comp Neurol 427:285–301. [DOI] [PubMed] [Google Scholar]
- Gray TS, Cassell MD, Kiss JZ (1984). Distribution of pro-opiomelanocortin-derived peptides and enkephalins in the rat central nucleus of the amygdala. Brain Res 306:354–358. [DOI] [PubMed] [Google Scholar]
- Gross-Isseroff R, Dillon K, Israeli M, Biegon A (1990). Regionally selective increases in mu opioid receptor density in the brains of suicide victims. Brain Res 530:312–316. [DOI] [PubMed] [Google Scholar]
- Hammer RP Jr, Bridges RS (1987). Preoptic area opioids and opiate receptors increase during pregnancy and decrease during lactation. Brain Res 420:48–56. [DOI] [PubMed] [Google Scholar]
- Hammer RP Jr, Bogic L, Handa RJ (1993). Estrogenic regulation of proenkephalin mRNA expression in the ventromedial hypothalamus of the adult male rat. Brain Res Mol Brain Res 19:129–134. [DOI] [PubMed] [Google Scholar]
- Harlow SD, Matanoski GM (1991). The association between weight, physical activity, and stress and variation in the length of the menstrual cycle. Am J Epidemiol 133:38–49. [DOI] [PubMed] [Google Scholar]
- Horvitz J (2000). Mesolimbic and nigrostriatal dopamine responses to salient non-rewarding stimuli. Neuroscience 96:651–656. [DOI] [PubMed] [Google Scholar]
- Introini-Collison IB, Ford L, McGaugh JL (1995). Memory impairment induced by intraamygdala beta-endorphin is mediated by noradrenergic influences. Neurobiol Learn Mem 63:200–205. [DOI] [PubMed] [Google Scholar]
- Jewett D (2001). A simple synthesis of [11C]carfentanil. Nucl Med Biol 28:733–734. [DOI] [PubMed] [Google Scholar]
- Katzen-Perez KR, Jacobs DW, Lincoln A, Ellis RJ (2001). Opioid blockade improves human recognition memory following physiological arousal. Pharmacol Biochem Behav 70:77–84. [DOI] [PubMed] [Google Scholar]
- Khachaturian H, Lewis ME, Tsou K, Watson SJ (1985). Beta-endorphin, alpha-MSH, ACTH, and related peptides. In: Handbook of chemical neuroanatomy (Bjorklund A, Hokfelt T, eds) pp. 216–272. New York: Elsevier Science.
- Khul H (2005). Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric 8:3–63. [DOI] [PubMed] [Google Scholar]
- Kineman RD, Kraeling RR, Crim JW, Leshin LS, Barb CR, Rampacek GB (1989). Localization of proopiomelanocortin (POMC) immunoreactive neurons in the forebrain of the pig. Biol Reprod 40:1119–1126. [DOI] [PubMed] [Google Scholar]
- Koob G, Le Moal M (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacol 24:97–129. [DOI] [PubMed] [Google Scholar]
- Korszun A, Hinderstein B, Wong M (1996). Comorbidity of depression with chronic facial pain and temporomandibular disorders. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 82:496–500. [DOI] [PubMed] [Google Scholar]
- Korszun A, Papadopoulos E, Demitrack M, Engleberg C, Crofford L (1998). The relationship between temporomandibular disorders and stress-associated syndromes. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 86:416–420. [DOI] [PubMed] [Google Scholar]
- Kraus M, Piper J, Kornetsky C (1996). Naloxone alters the local metabolic rate for glucose in discrete bran regions associated with opiate withdrawal. Brain Res 724:33–40. [DOI] [PubMed] [Google Scholar]
- Kudora M, Yokofujita J, Murakami K (1998). An ultrastructural study of the neural circuit between the prefrontal cortex and the mediodorsal nucleus of the thalamus. Prog Neurobiol 54:417–458. [DOI] [PubMed] [Google Scholar]
- Leone M, Franzini A, Bussone G (2001). Stereotactic stimulation of posterior hypothalamic gray matter in a patient with intractable cluster headache. N Engl J Med 345:1428–1429. [DOI] [PubMed] [Google Scholar]
- LeResche L, Mancl L, Sherman JJ, Gandara B, Dworkin SF (2003). Changes in temporomandibular pain and other symptoms across the menstrual cycle. Pain 106:253–261. [DOI] [PubMed] [Google Scholar]
- Levine JD, Gordon NC, Jones RT, Fields HL (1978). The narcotic antagonist naloxone enhances clinical pain. Nature 272:826–827. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Taylor S, Fig L, Decker L, Koeppe R, Minoshima S (2000). Limbic activation and psychophysiologic responses to aversive visual stimuli. Interaction with cognitive task. Neuropsychopharmacology 23:508–516. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL (1996). Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 16:834–840. [DOI] [PubMed] [Google Scholar]
- Matthes HWD, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, LeMeur M, Dolle P, Tzavara E, Hannoune J, Roques BP, Kieffer BL (1996). Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature 383:819–823. [DOI] [PubMed] [Google Scholar]
- Mayanagi Y, Sano K, Suzuki I, Kanazawa I, Aoyagi I, Miyachi Y (1982). Stimulation and coagulation of the posteromedial hypothalamus for intractable pain, with reference to beta-endorphins. Appl Neurophysiol 45:136–142. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Cahill L (1997). Interaction of neuromodulatory systems in modulating memory storage. Behav Brain Res 83:31–38. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, McIntyre CK, Power AE (2002). Amygdala modulation of memory consolidation: interaction with other brain systems. Neurobiol Learn Mem 78:539–552. [DOI] [PubMed] [Google Scholar]
- Melzack R, Katz J (2000). The McGill pain questionnaire: appraisal and current status. In: Handbook of pain assessment (Turk D, Melzack R, eds) pp. 152–168. New York: Guilford.
- Meyer CR, Boes JL, Kim B, Bland PH, Zasadny KR, Kison PV, Koral K, Frey KA, Wahl RL (1997). Demonstration of accuracy and clinical versatility of mutual information for automatic multimodality image fusion using affine and thin-plate spline warped geometric deformations. Med Image Anal 1:195–206. [DOI] [PubMed] [Google Scholar]
- Mills RH, Sohn RK, Micevych PE (2004). Estrogen-induced μ-opioid receptor internalization in the medial preoptic nucleus is mediated via neuropeptide Y-Y1 receptor activation in the arcuate nucleus of female rats. J Neurosci 24:947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoshima S, Koeppe RA, Mintun MA, Berger KL, Taylor SF, Frey KA, Kuhl DE (1993). Automated detection of the intercommissural line for stereotactic localization of functional brain images. J Nucl Med 34:322–329. [PubMed] [Google Scholar]
- Morris J, Friston K, Büchel C, Frith C, Young A, Calder A, Dolan R (1998). A neuromodulatory role for the human amygdala in processing emotional facial expressions. Brain 121:47–57. [DOI] [PubMed] [Google Scholar]
- Parker G, Wilhelm K, Mitchell P, Austin MP, Roussos J, Gladstone G (1999). The influence of anxiety as a risk to early onset major depression. J Affect Disord 52:11–17. [DOI] [PubMed] [Google Scholar]
- Plesh O, Wolfe F, Lane N (1996). The relationship between fibromyalgia and temporomandibular disorders: prevalence and symptom severity. J Rheumatol 23:1948–1952. [PubMed] [Google Scholar]
- Plotsky PM (1986). Opioid inhibition of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation of rats. Regul Pept 16:235–242. [DOI] [PubMed] [Google Scholar]
- Priest CA, Roberts JL (2000). Estrogen and tamoxifen differentially regulate beta-endorphin and cFos expression and neuronal colocalization in the arcuate nucleus of the rat. Neuroendocrinology 72:293–305. [DOI] [PubMed] [Google Scholar]
- Quinones-Jenab V, Jenab S, Ogawa S, Inturrisi C, Pfaff DW (1997). Estrogen regulation of mu-opioid receptor mRNA in the forebrain of female rats. Brain Res Mol Brain Res 47:134–138. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Galvez R, Roozendaal B, McGaugh JL (1998). Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Res 808:134–140. [DOI] [PubMed] [Google Scholar]
- Ragnauth A, Schuller A, Morgan M, Chan J, Ogawa S, Pintar J, Bodnar RJ, Pfaff DW (2001). Female preproenkephalin-knockout mice display altered emotional responses. Proc Natl Acad Sci USA 98:1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratka A, Hochhaus G, Wissler RN, Simpkins JW (1991). cAMP accumulation in opioid-sensitive SH-SY5Y neuroblastoma cells is modified by estradiol and progesterone. Mol Cell Endocrinol 78:155–162. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (2000). The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 95:Suppl 2, S91–S117. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japon M, Chan EC, Allen RG, Low MJ (1996). Absence of opioid stress-induced analgesia in mice lacking β-endorphin by site directed mutagenesis. Proc Natl Acad Sci USA 93:3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KA, Bruce NW (1997). A prospective study of psychosocial stress and fertility in women. Hum Reprod 12:2324–2329. [DOI] [PubMed] [Google Scholar]
- Shen PJ, Smith AI, Evans RG, Clarke IJ (1995). Effects of ovarian steroids on hypothalamic opioid receptor subtypes in ovariectomized ewes: regional changes in density and affinity. J Endocrinol 145:559–567. [DOI] [PubMed] [Google Scholar]
- Sinchak K, Micevych PE (2001). Progesterone blockade of estrogen activation of μ-opioid receptors regulates reproductive behavior. J Neurosci 21:5723–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith YR, Zubieta JK, Del Carmen M, Dannals RF, Ravert H, Zacur H, Frost JJ (1998). Brain mu opioid receptor measurements by positron emission tomography in normal cycling women: relationship to LH pulsatility and gonadal steroid hormones. J Clin Endocrinol Metab 83:4498–4505. [DOI] [PubMed] [Google Scholar]
- Stohler C, Lund J (1995). Psychophysical and orofacial motor response to muscle pain: validation and utility of an experimental model. In: Brain and oral functions. Oral motor function and dysfunction (Morimoto T, Matsuya T, Takada K, eds) pp. 227–237. Amsterdam: Elsevier Science.
- Stohler C, Kowalski C (1999). Spatial and temporal summation of sensory and affective dimensions of deep somatic pain. Pain 79:165–173. [DOI] [PubMed] [Google Scholar]
- Sun YG, Lundeberg T, Yu LC (2003). Involvement of endogenous beta-endorphin in antinociception in the arcuate nucleus of hypothalamus in rats with inflammation. Pain 104:55–63. [DOI] [PubMed] [Google Scholar]
- Thornton JE, Loose MD, Kelly MJ, Ronnekleiv OK (1994). Effects of estrogen on the number of neurons expressing beta-endorphin in the medial basal hypothalamus of the female guinea pig. J Comp Neurol 341:68–77. [DOI] [PubMed] [Google Scholar]
- Titeler M, Lyon RA, Kuhar MJ, Frost JJ, Dannals RF, Leonhardt S, Bullock A, Rydelek LT, Price DL, Struble RG (1989). Mu opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur J Pharmacol 167:221–228. [DOI] [PubMed] [Google Scholar]
- Tong Y, Zhao HF, Labrie F, Pelletier G (1990). Regulation of proopiomelanocortin messenger ribonucleic acid content by sex steroids in the arcuate nucleus of the female rat brain. Neurosci Lett 112:104–108. [DOI] [PubMed] [Google Scholar]
- Turp JC, Kowalski CJ, Stohler CS (1997). Greater disability with increased pain involvement, pain intensity and depressive preoccupation. Eur J Pain 1:271–277. [DOI] [PubMed] [Google Scholar]
- Unruh A (1996). Gender variations in clinical pain experience. Pain 65:123–167. [DOI] [PubMed] [Google Scholar]
- Watkins L, Mayer D (1982). Organization of endogenous opiate and nonopiate pain control systems. Science 216:1185–1192. [DOI] [PubMed] [Google Scholar]
- Watson D, Clark LA, Tellegen A (1988). Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol 54:1063–1070. [DOI] [PubMed] [Google Scholar]
- Zacny J (2001). Morphine responses in humans: a retrospective analysis of sex differences. Drug Alcohol Depend 63:23–28. [DOI] [PubMed] [Google Scholar]
- Zangen A, Nakash R, Yadid G (1999). Serotonin-mediated increases in the extracellular levels of beta-endorphin in the arcuate nucleus and nucleus accumbens: a microdialysis study. J Neurochem 73:2569–2574. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Dannals RF, Frost JJ (1999). Gender and age influences on human brain mu opioid receptor binding measured by PET. Am J Psychiatry 156:842–848. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn M, Meyer C, Koeppe R, Stohler C (2001). Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 293:311–315. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Woike T, Kilbourn M, Meyer C, Koeppe RA, Stohler CS (2002). μ-Opioid receptor mediated antinociception differs in men and women. J Neurosci 22:5100–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D (2003). COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science 299:1240–1243. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Bueller JA, Jackson LR, Scott DJ, Xu Y, Koeppe RA, Stohler CS (2005). Placebo effects mediated by endogenous opioid neurotransmission and μ-opioid receptors. J Neurosci 25:7754–7762. [DOI] [PMC free article] [PubMed] [Google Scholar]