

Abstract
Protein arginine deiminase 4 (PAD4) is a transcriptional coregulator that catalyzes the calcium-dependent conversion of specific arginine residues in proteins to citrulline. Recently, we reported the synthesis and characterization of F-amidine, a potent and bioavailable irreversible inactivator of PAD4. Herein, we report our efforts to identify the steric and leaving group requirements for F-amidine-induced PAD4 inactivation, the structure of the PAD4–F-amidine·calcium complex, and in vivo studies with N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide (Cl-amidine), a PAD4 inactivator with enhanced potency. The PAD4 inactivators described herein will be useful pharmacological probes in characterizing the incompletely defined physiological role(s) of this enzyme. In addition, they represent potential lead compounds for the treatment of rheumatoid arthritis because a growing body of evidence supports a role for PAD4 in the onset and progression of this chronic autoimmune disorder.
Protein arginine deiminase 4 (PAD4)1 is a 663-amino acid, 74 kDa, human protein whose deiminating activity [Arg → Cit (Figure 1)] appears to be dysregulated in rheumatoid arthritis (RA). Specific evidence linking the dysregulation of this enzyme to RA includes the following: (i) The PAD4 gene has been identified as a RA susceptibility locus in the Japanese and Korean population (1, 2). (ii) PAD4 and Cit-containing proteins colocalize in RA synovial tissues, and the level of their expression is correlated to the severity of the disease (3). (iii) A RA-associated major histocompatibility complex II molecule (HLA-DRB1*0401) binds preferentially to Cit-containing peptides (4). (iv) RA patients produce autoantibodies that recognize Cit-containing peptides (5–11), and these autoantibodies can often be detected prior to the onset of clinical symptoms (12). Although speculative, it has been suggested that an elevated PAD4 activity causes an overproduction of deiminated proteins that initially leads to a break in self-tolerance and eventually causes the immune system to attack its own tissues (13). On the basis of this model, we and others have suggested (14–17) that PAD4 represents a novel therapeutic target for RA and that inhibition of PAD4 would reduce the levels of deiminated proteins and consequently suppress the immune response directed toward these antigens.
Figure 1.
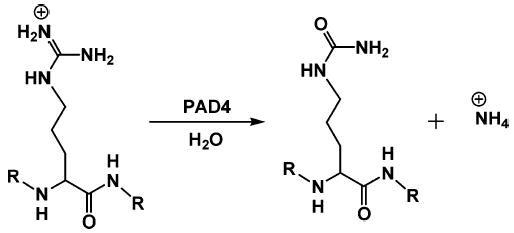
PAD4 hydrolyzes the guanidinium group of an Arg residue to form citrulline (Cit) and ammonia, which is subsequently protonated to form the ammonium ion. R = peptide backbone.
In addition to its presumed role in RA, PAD4 is thought to play a regulatory role in a number of human cell signaling pathways, including differentiation, apoptosis, and gene transcription (18–22). Of these various pathways, the best characterized is its incompletely defined role in human gene regulation. For example, PAD4 deiminates multiple transcriptional regulators, including p300, a histone acetyltransferase (HAT) (23) that acts as a transcriptional coactivator, and histones H2A, H3, and H4 (24, 25). Interestingly, the deimination of p300 and the histone proteins appears to have opposite effects on transcriptional regulation; i.e., the modification of p300 enhances its ability to activate the expression of an artificial reporter construct (23), whereas the deimination of histones H3 and H4 represses the expression of genes under the control of the estrogen receptor (20, 21).
As a part of our ongoing efforts to develop PAD4-targeted therapeutics, we recently reported the synthesis and characterization of N-α-benzoyl-N5-(2-fluoro-1-iminoethyl)-l-or-nithine amide [1, F-amidine (Figure 2A)], a PAD4 inactivator that is significantly more potent than either paclitaxel (IC50 ~5 mM) (26) or 2-chloroacetamidine [kinact/KI = 35 M−1 min−1 (16)], each a known PAD inhibitor. In vitro studies with F-amidine revealed that it irreversibly inactivates PAD4 in a calcium-dependent manner via the specific modification of Cys645 (vide infra), an active site residue that is critical for catalysis; Cys645 acts as a nucleophile to form a thiouronium intermediate that is ultimately hydrolyzed to form Cit. The inhibitory properties of F-amidine have also been evaluated in vivo, and the results indicate that this compound is bioavailable (17). In an effort to identify the effects of warhead positioning and the identity of the leaving group, we synthesized a series of analogues (compounds 2–9) in which the length of the side chain and the identity of the halide were systematically varied. Herein, we report the results of these studies, as well as the identification of a significantly more potent PAD4 inactivator, N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide [2, Cl-amidine (Figure 2A)], that like the parent compound is bioavailable. The structural basis for the inactivation of PAD4 by F-amidine is also reported.
Figure 2.
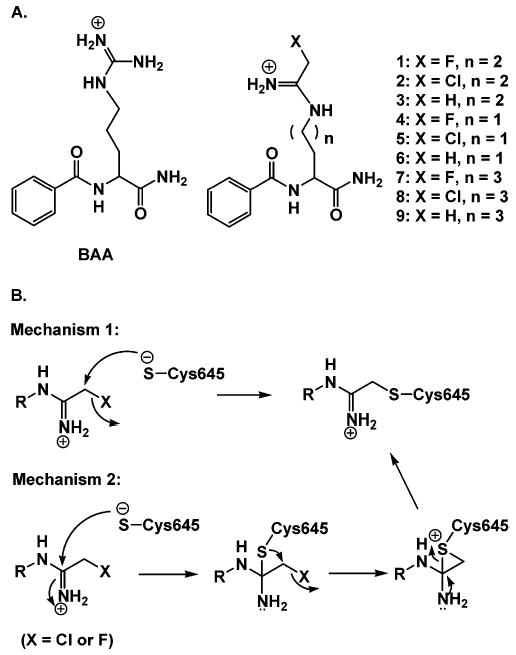
Structures of (halo)acetamidine-based PAD4 inhibitors and/or inactivators and potential mechanisms of inactivation. (A) Structure of BAA and the (halo)acetamidine-based inhibitors and inactivators of PAD4. (B) Two potential mechanisms of PAD4 inactivation. Mechanism 1 involves direct substitution of the halide, whereas mechanism 2 involves the formation of a tetrahedral intermediate, which first evolves into a three-membered sulfonium ring and subsequently rearranges to the thioether with the collapse of the tetrahedral intermediate. The latter mechanism is invoked to account for the poor leaving group potential of fluoride.
EXPERIMENTAL PROCEDURES
Chemicals
HOBt, HBTU, and Rink Amide AM resin (200–400 mesh) were purchased from Novabiochem. Fmoc-Orn(Dde)-OH, Fmoc-Lys(Dde)-OH, and Fmoc-Dab(Dde)-OH were obtained from AnaSpec Inc. (San Jose, CA). Fluoroacetonitrile, chloroacetonitrile, hydrogen chloride (1.0 M in ether), dithiothreitol (DTT), benzoyl l-arginine ethyl ester (BAEE), N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 1,3-diisopropylcarbodiimide (DIC), and benzoyl chloride were acquired from Sigma-Aldrich (St. Louis, MO). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was obtained from Fluka.
Synthesis of PAD4 Inhibitors and/or Inactivators
Complete synthetic methods for compounds 2–11, as well as their structural characterization, are available as Supporting Information. Briefly, the (halo)acetamidine-based PAD4 inhibitors and/or inactivators described herein were synthesized in a manner analogous to that of the synthesis of F-amidine (17). Briefly, a 9-fluorenylmethyloxycarbonyl (Fmoc)-protected (main chain) and 1-(4,4-dimethyl-2,6-dioxocyclohex-1-yldine)ethyl (Dde)-protected (side chain) diamino acid, e.g., ornithine, is coupled to Rink Amide AM resin via a standard uronium-based coupling method. Subsequently, the Fmoc group is removed with 20% piperidine and the resulting free α-amino group is benzoylated with benzoyl chloride. The side chain amine was then deprotected with 2% hydrazine and reacted with either ethyl fluoroacetimidate, ethyl chloroacetimidate, or ethyl acetimidate hydrochloride to form the (halo)acetamidine-based warhead. The acetimidates are readily derived from their corresponding acetonitrile derivatives in a one-step synthesis that involves a reaction between the acetonitrile derivative and ethanol in acidified ether (1 M HCl in ether) (27). Final compounds were subsequently cleaved from the resin and purified by reverse phase HPLC, and their structures were confirmed by NMR (1H and 13C) and HR-ESI-MS.
Protein Purification
Recombinant human PAD4 was expressed and purified using a previously described expression system and established methodologies (14).
IC50 and Time Course Assays
IC50 values for compounds 2–11 were determined in a manner identical to the methods used to determine the IC50 for F-amidine, 1 (17). IC50 values were determined by fitting the concentration–response data to eq 1
| (1) |
using Grafit version 5.0.11 (28). The concentration of an inhibitor that corresponds to the midpoint (fractional activity = 0.5) is termed the IC50; [I] is the concentration of inhibitor or inactivator. The calcium dependence of the IC50 was determined identically, except that CaCl2 was omitted during the initial preincubation step and then added (final concentration of 10 mM) with BAEE to initiate the reaction.
To initially evaluate the inhibitory properties of compounds H2-, H-, and H4-amidine, progress curves were generated. For these experiments, inhibitors were preincubated for 10 min at 37 °C in assay buffer containing 2 mM BAEE. Reactions were then initiated by addition of PAD4 to a final concentration of 0.2 μM. At various time points, a 60 μL aliquot was withdrawn from an individual reaction, enzyme activity quenched by flash-freezing, and the amount of Cit produced quantified. The data obtained for H2-, H-, and H4-amidine were fit to a simple linear equation. For Cl-amidine, values for kinact, KI, and kinact/KI were obtained by multiplying the apparent kobs.app values by the transformation 1 + [S]/Km to obtain the pseudo-first-order rate constant, kobs (29), and these values were plotted versus inhibitor concentrations and fit to eq 2
| (2) |
using GraFit version 5.0.11. kinact is the maximal rate of inactivation; KI is the concentration of inactivator that yields half-maximal inactivation, and [I] is the concentration of inactivator.
Rapid Dilution Time Course Assays
To determine whether F2-, F4-, Cl2-, Cl-, and Cl4-amidine are irreversible PAD4 inactivators, rapid dilution time course experiments were performed to test for the recovery of enzymatic activity after rapid 100-fold dilution of preformed enzyme–inactivator complexes into assay buffer. For these experiments, the preformed enzyme–inactivator complex was generated by incubating PAD4 with F2-, F4-, Cl2-, Cl-, and Cl4-amidine (6 mM for F2-, F4-, and Cl2-amidine, 250 μM for Cl-amidine, and 3 mM for Cl4-amidine) at 37 °C for 30 min. Reactions were then initiated by adding 6 μL of the preformed complex to assay buffer containing 10 mM BAEE (final volume of 600 μL). At various time points (0, 2, 4, 6, 10, and 15 min), 60 μL of the reaction mixture was withdrawn and quenched by flash-freezing in liquid nitrogen. Cit production was then quantified according to previously established methodologies (14).
Dialysis Experiments
To verify that Cl-amidine is an irreversible PAD4 inactivator, preformed enzyme–Cl-amidine complexes were generated and then dialyzed against 20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 500 mM NaCl, 1 mM DTT, and 10% glycerol. Aliquots were taken at 0, 3.5, and 20 h, and the activity present in these samples was quantified and compared to that of control reaction mixtures that were treated identically.
H-Amidine Inhibition Assays
Initial rates were determined in the absence and presence of various amounts of H-amidine (0, 5, and 10 mM), using BAEE as the substrate. BAEE and H-amidine were preincubated in assay buffer for 10 min at 37 °C. Reactions were initiated by the addition of PAD4 to a final concentration of 0.2 μM. After incubation at 37 °C for 15 min, the reactions were quenched and the amount of Cit produced was quantified. The initial rates derived from these experiments were fit by a nonlinear least-squares fit to equations representing linear competitive inhibition (eq 3) and linear noncompetitive inhibition (eq 4), using GraFit version 5.0.11.
| (3) |
| (4) |
where Kii represents the Ki intercept and Kis represents the Ki slope. Comparisons of the standard errors derived from fits of the data to eqs 3 and 4 are most consistent with H-amidine being a linear competitive inhibitor.
In Vivo Studies
Transient transfection assays were performed in a manner analogous to previously described methods (17, 23). Briefly, transfections were allowed to proceed for 3 h, at which point, the medium was removed and replaced with fresh DMEM and 10% FBS. Cl-amidine, dissolved in 10 mM Na-HEPES (pH 7.0), was then added over a range of concentrations (from 0 to 200 μM). Luciferase activity present in cell extracts was then quantified after 40 h. Note that these assays were performed in parallel to those reported in ref 17.
Crystal Preparation and Structure Determination
Crystals of F-amidine in complex with wild-type PAD4 were prepared by soaking this compound into previously prepared crystals of wild-type PAD4. Crystals of the wild-type enzyme were prepared according to previously established methods (30,31). Subsequently, these crystals were transferred to fresh crystallization buffer [0.1 M imidazole (pH 8.0), 0.2 M Li2-SO4, and 10% PEGMME] containing 5 mM CaCl2 and 5 mM F-amidine for 8 h. Diffraction data were then collected on BL41XU at SPring-8, indexed, and then scaled using HKL2000 (32). Crystallographic data are listed in Table 1. The initial structure of the PAD4–F-amidine·calcium complex was derived from the atomic coordinates of the PAD4C645A·BAA·calcium complex (Protein Data Bank entry 1WDA). The structure was refined at 2.3 Å resolution using CNS (33), and manual construction was performed using O (34). At this stage, the F-amidine moiety in the complex was identified on the |Fo| − |Fc| maps. The structure was further refined by simulated annealing, energy minimization, and B-factor individual refinement using CNS and finally converged after several further cycles of refinement with REFMAC (35). The final refinement statistics are listed in Table 1.
Table 1.
Crystallographic Data and Refinement Statistics
| crystallographic data | |
| space group | C2 |
| cell dimensions | a = 146.3 Å, b = 60.5 Å, c = 115.0 Å, β = 124.2° |
| resolution range (Å) | 50.00–2.30 |
| no. of total observations | 109379 |
| no. of unique observations | 31719 |
| completeness (%) | 85.0 (47.6)a |
| Rmerge (%)b | 3.4 (19.5)a |
| 〈I/σ(I)〉 | 15.9 |
| refinement statistics | |
| resolution (Å) | 50.00–2.30 |
| Rwork/Rfree (%)c | 19.8/25.4 |
| root-mean-square deviation | |
| bond lengths (Å) | 0.017 |
| bond angles (deg) | 1.706 |
| mean B value (Å2) | 56.5 |
Values in parentheses are for the highest-resolution shell.
Rmerge = ∑h∑i|I(h)i − 〈I(h)〉|/∑h∑iI(h)i.
Rwork/Rfree = ∑||Fo| − |Fc||/∑ |Fo|, where Rwork and Rfree are calculated by using the working and free reflection sets, respectively. Rfree reflections (5% of the total) were held aside throughout the refinement.
RESULTS
Design of PAD4 Inhibitors and/or Inactivators
The initial design of F-amidine was based in part on its structural homology to N-α-benzoyl Arg amide [BAA (Figure 2A)], one of the best small molecule PAD4 substrates [kcat/Km = 1.1 × 104 M−1 s−1 (14)], and can be considered to consist of two major moieties, a fluoroacetamidine-based warhead and a specificity determinant that was expected to target the warhead to the active site of PAD4, where it will react with C645 to form a stable thioether adduct via one of two potential mechanisms (Figure 2B) (14). To identify PAD4 inhibitors with enhanced potency and to gain insights into the steric and leaving group requirements for PAD4 inactivation, we synthesized a series of compounds in which both the length of the side chain and the leaving group were varied. The lengths of the side chains ranged from two to four methylene units, thereby allowing us to evaluate the importance of positioning to inactivation, and the fluoro group was replaced with a chloro group. The fluoro group was also replaced with hydrogen to evaluate the requirement for a leaving group.2 Three potential scenarios were envisioned for H2-, H-, and H4-amidine [compounds 6, 3, and 9, respectively (Figure 2A)], which are detailed here. (i) The iminium carbon of the acetamidine moiety would not possess sufficient reactivity with the active site thiolate, and the compounds would be competitive inhibitors. (ii) The iminium carbon would react with the active site thiolate to form the first tetrahedral intermediate and thereby generate a transition state analogue. (iii) The iminium carbon would react with the active site thiolate to form the first tetrahedral intermediate. Subsequently, the intermediate would collapse, resulting in the loss of benzoylated ornithine, and the formation of an irreversible imidothioic acid adduct.
Structure–Activity Relationships
The inhibitory properties of compounds 2–9 were initially evaluated by determining the concentration of compound that yielded the half-maximal activity, i.e., the IC50, and comparing the results of these studies to the IC50 value obtained for F-amidine; IC50 values were determined under conditions that were identical to those used to determine the IC50 for F-amidine. The results of these initial studies (Table 2) indicated that Cl-amidine is a significantly more potent inhibitor than F-amidine; the detailed inhibitory properties of Cl-amidine are discussed below. Interestingly, H-amidine, the acetamidine-containing isostere of F-amidine (and BAA), is a very poor inhibitor of PAD4 (IC50 > 1000 μM). Time course experiments with H-amidine, and related compounds H2- and H4-amidine (compounds 6 and 9, respectively), were linear with respect to time (see Figure S1 of the Supporting Information), thereby indicating that on the time scale of these experiments the acetamidine-bearing compounds are reversible PAD4 inhibitors. Note that even extended incubations (2 h) of H-amidine with PAD4 did not result in PAD4 inactivation, and full activity could be restored after overnight dialysis (not shown). The reversible nature of the inhibition rules out the possibility that these compounds react with the active site Cys to form the postulated imidothioic acid adduct. The fact that these compounds are such poor inhibitors suggests that an additional electron-withdrawing group is required to promote reactions between Cys645 and the iminium carbon, i.e., form the first transition state.
Table 2.
IC50 Values of PAD4 Inhibitors and Inactivatorsa
| compound no. | IC50 (μM) | |
|---|---|---|
| F-amidine | 1 | 21.6 ± 2.1 |
| Cl-amidine | 2 | 5.9 ± 0.3 |
| H-amidine | 3 | >1000 |
| F2-amidine | 4 | >1000 |
| Cl2-amidine | 5 | 585 ± 65 |
| H2-amidine | 6 | >1000 |
| F4-amidine | 7 | 655 ± 100 |
| Cl4-amidine | 8 | 640 ± 10 |
| H4-amidine | 9 | >1000 |
| F-acetamideb | 10 | >500 |
| Cl-acetamideb | 11 | >500 |
IC50 is the concentration of the inhibitor or inactivator that yields half-maximal activity. IC50 values were determined by preincubating PAD4 and the inhibitor or inactivator in the presence of 10 mM calcium for 15 min prior to the addition of 10 mM BAEE to initiate the enzyme assay. See Experimental Procedures for assay details.
No inhibition was noted with these compounds even at the highest concentration that was tested. Higher concentrations could not be tested because of solubility issues.
Cl2-, F4-, and Cl4-amidine were also relatively poor PAD4 inhibitors with IC50 values in the 500 μM range. To determine if these compounds were irreversible inactivators, Cl2-, F4-, and Cl4-amidine were preincubated with PAD4 and then rapidly diluted into assay buffer containing an excess of substrate. The results of these experiments (see Figure S2 of the Supporting Information) show no recovery of activity, suggesting that these compounds are, like F-amidine, irreversible PAD4 inactivators. The fact that compounds with side chains of two or four methylene units are significantly less potent inhibitors and/or inactivators than either F-amidine or Cl-amidine indicates that the proper positioning of the reactive warhead is critical for reaction with the active site thiolate.
In contrast to the results obtained for Cl2-amidine, its isostere, F2-amidine, which contains the fluoroacetamidine rather than the chloroacetamidine warhead, was a significantly less potent PAD4 inhibitor; at 1000 μM F2-amidine, the observed activity was 93% of the control. Consistent with its lack of potency is the fact that time courses were linear with respect to time (not shown) and the rapid dilution time course experiments demonstrated complete recovery of activity (Figure S2), thereby indicating that this compound is neither a slow binding nor irreversible inactivator of PAD4. The finding that F2-amidine does not irreversibly inactivate PAD4 likely reflects the fact that the side chain of this compound is too short to appropriately position the fluoroacetamidine warhead for reaction of the iminium carbon with Cys645 via mechanism 2 in Figure 2B; this compound is unlikely to react via mechanism 1 due to the intrinsically poor leaving group potential of fluoride. The fact that Cl2-amidine is an irreversible inactivator could reflect the intrinsic reactivity of the chloro group or alternatively that chloroacetamidines can react with the active site thiolate through either mechanism 1 or 2 in Figure 2B whereas fluoroacetamidines can react through only mechanism 2.
The importance of a positively charged warhead for inactivator potency was also evaluated by synthesizing the neutral isosteres of F- and Cl-amidine, i.e., N-α-benzoyl-N5-(2-fluoroacetyl)ornithine amide, compound 10, and N-α-benzoyl-N5-(2-chloroacetyl)ornithine amide, compound 11. IC50 assays were performed with these compounds; however, no inhibition was noted at even the highest concentration of compound that was tested, i.e., 500 μM, and higher concentrations were not tested because of solubility issues. The simplest explanation for the lack of potency is that these compounds do not form high-affinity interactions with the PAD4 active site. This lack of affinity is most likely due to the lack of a positively charged warhead. The presence of hydrogen bond acceptors in the warhead, rather than hydrogen bond donors, as is the case in the acetamidine warhead, could also account for the lack of affinity.
In Vitro Characterization of H- and Cl-Amidine
The detailed inhibitory properties of H-amidine and Cl-amidine were also determined (see below) in an effort to gain additional insights into both their potency and mechanism of inhibition. We focused on characterizing these compounds because they are both isosteric with F-amidine and Cl-amidine is significantly more potent, whereas H-amidine is significantly less potent.
To identify the mechanism of H-amidine inhibition, the steady state kinetic parameters for the deimination of BAEE were determined in the absence and presence of increasing amounts of H-amidine. The results of these studies indicate that H-amidine is a linear competitive inhibitor of PAD4 (see Figure S3 of the Supporting Information) with a Kis in the low millimolar range (Kis = 3.2 ± 0.9 mM). The reversible nature of the inhibition and the lack of potency clearly demonstrate the significant gains in binding energy that can be achieved through covalent bond formation.
A series of experiments were also performed on Cl-amidine. These experiments included assays to (i) evaluate the calcium dependence of inactivation, (ii) determine if substrate can protect against inactivation, (iii) confirm the irreversible nature of the enzyme–Cl-amidine complex, and (iv) more fully characterize the kinetics of inactivation. Initially, the calcium dependence of the IC50 was determined. For these studies, Cl-amidine was preincubated with PAD4 in the absence and presence of calcium, prior to the addition of BAEE to initiate the enzyme assay; calcium was also added to the sample preincubated in the absence of this metal ion to activate PAD4. These experiments were performed because binding of calcium to PAD4 triggers a conformational change that moves Cys645 into a position that is competent for catalysis; thus, if Cl-amidine reacts with an active site residue, one would expect it to preferentially inactivate the calcium-bound form of the enzyme. The results of these experiments, which are depicted in Figure 3A, indicate that Cl-amidine preferentially inactivates the calcium-bound form of the enzyme by >10-fold. This result is consistent with previous results reported for F-amidine (17).
Figure 3.
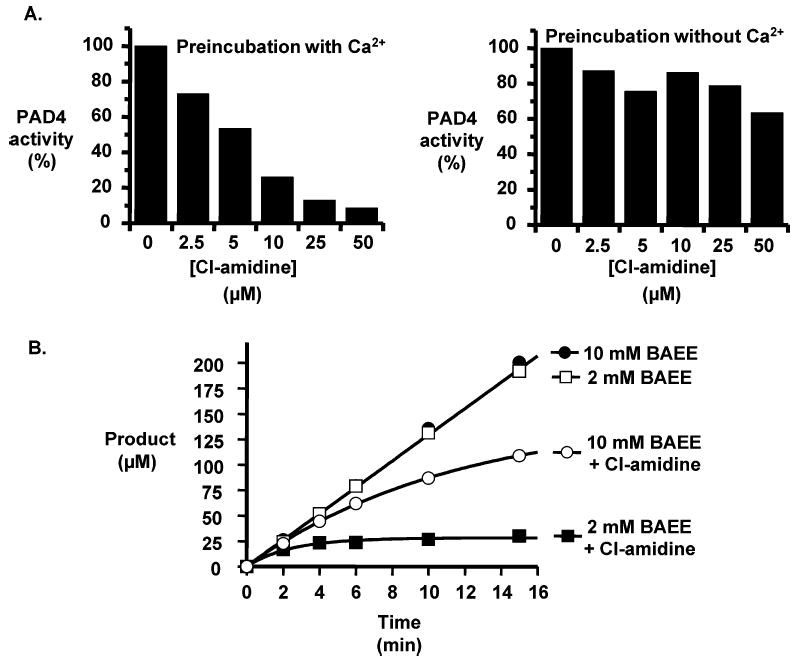
Calcium and substrate dependence of Cl-amidine-induced inactivation. (A) PAD4 was preincubated with increasing concentrations of Cl-amidine in the absence and presence of calcium and then BAEE added to initiate the IC50 assay. Calcium was also added at this time to the sample preincubated in the absence of this metal ion to upregulate the activity of PAD4. (B) Substrate protection was assayed by observing the time-dependent inactivation properties of Cl-amidine (100 μM). For these studies, product formation in the presence and absence of Cl-amidine was quantified as a function of time using two different concentrations of BAEE (2 and 10 mM) as the substrate.
Substrate protection experiments were also performed by monitoring product formation as a function of time for two different concentrations of BAEE (2 and 10 mM) in the absence and presence of Cl-amidine. The results of these experiments (Figure 3B) clearly establish that the rate of inactivation is higher at the lower substrate concentration. Thus, substrate can protect against inactivation, consistent with the preferential modification of an active site residue, which on the basis of the precedents obtained for F-amidine, and the related compound 2-chloroacetamidine (16), is most likely Cys645.
To confirm that Cl-amidine is an irreversible inactivator of PAD4, rapid dilution time course experiments were performed with this compound. Briefly, preformed PAD4–Cl-amidine complexes were diluted 100-fold into assay buffer containing 10 mM BAEE (7.5Km), and product formation was monitored as a function of time (Figure 4A). The time courses showed no recovery of PAD4 activity, consistent with the irreversible inactivation of PAD4. Dialysis experiments were also performed on preformed PAD4–Cl-amidine complexes to assay for the recovery of enzymatic activity over a longer time frame and, thereby, exclude the possibility that Cl-amidine is a reversible slow binding inhibitor with a very long half-life. Briefly, PAD4 was incubated with Cl-amidine to effect enzyme inactivation, at which point the enzyme–inhibitor complex was dialyzed for 3.5 h and the buffer exchanged and then dialyzed for an additional 16.5 h (20 h total). Activity measurements performed before and after dialysis demonstrated that there was no recovery of activity at either the 3.5 or 20 h time point, again consistent with the irreversible inactivation of PAD4 (Figure 4B) and ruling out the possibility that Cl-amidine is a slow binding inhibitor.
Figure 4.
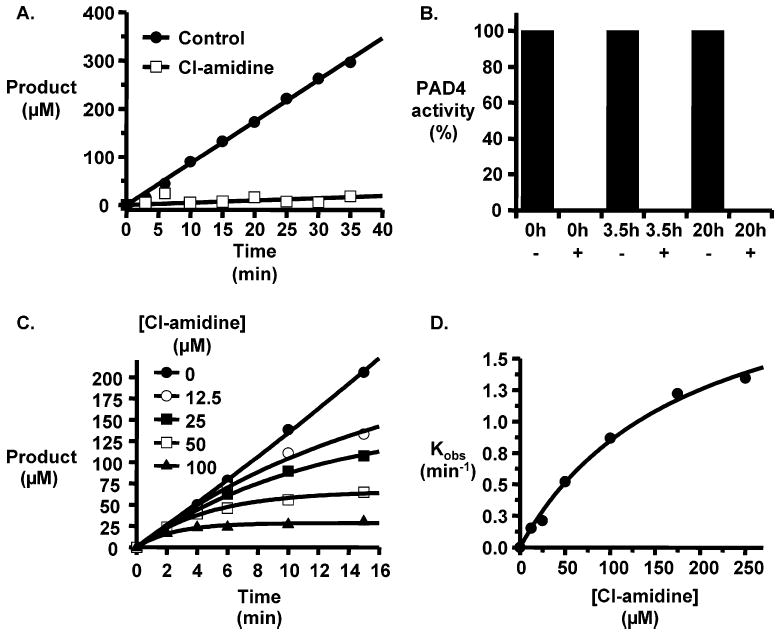
Cl-amidine is an irreversible time- and concentration-dependent inactivator of PAD4. (A) Rapid dilution of the preformed PAD4·Cl-amidine complex, and controls containing no inhibitor, into assay buffer containing excess substrate did not show any recovery of activity. (B) Dialysis of preformed PAD4–Cl-amidine complexes for 0, 3.5, and 20 h did not show any recovery of activity. (C) Plots of product formation vs time in the absence and presence of increasing concentrations of Cl-amidine. (D) Plot of kobs vs the concentration of Cl-amidine.
To further define the inhibitory properties of Cl-amidine, the rate constants for the inactivation process, i.e., KI, kinact, and kinact/KI, were determined. For these studies, product formation was monitored as a function of time in the absence and presence of different concentrations of Cl-amidine (Figure 4C). The nonlinear progress curves were fit to eq 5
| (5) |
where vi is the initial velocity, kobs is the apparent pseudo-first-order rate constant for inactivation, and [Cit] refers to the concentration of Cit produced during the time course. The pseudo-first-order rate constants were then plotted versus the concentration of Cl-amidine and the resulting curves fit to eq 2. Using this analysis (Figure 4D), values for kinact and KI of 2.4 ± 0.2 min−1 and 180 ± 33 μM, respectively, were determined. The second-order rate constant (kinact/KI = 13 000 M−1 min−1) is 4.3-fold higher than that observed for F-amidine (17), roughly consistent with the 3.6-fold decrease in the IC50 of Cl-amidine relative to that of the fluoroacetamidine-containing compound. The kinact/KI obtained for Cl-amidine is also 370-fold higher than that obtained for 2-chloroacetamidine, i.e., the warhead alone; the kinact/KI for this compound (35 M−1 min−1) was recently reported by the Fast group (16). Because the increase in kinact/KI is mostly driven by a decrease in KI (20 mM for 2-chloroacetamidine vs 180 μM for Cl-amidine), the improved inactivation kinetics most likely result from the inclusion of the benzoylated ornithine portion in the molecule, which would be expected to provide additional binding energy for the formation of the initial enzyme·Cl-amidine complex, thereby suggesting that further improvements to potency could be gained by tailoring this portion of the molecule to maximize its interactions with the active site of PAD4.
In Vivo Studies with Cl-Amidine
The PAD4-catalyzed deimination of the GRIP1 binding domain of p300 (p300GBD) is known to enhance interactions between p300 and GRIP1, a nuclear receptor coactivator (23). Because we previously reported that F-amidine could antagonize this effect in vivo (17) and because Cl-amidine displays enhanced in vitro potency, we evaluated its ability to interfere with the PAD4-mediated enhancement of the p300GBD–GRIP1 interaction. For these studies, we utilized a previously described mammalian two-hybrid assay that monitors the efficiency of the p300GBD–GRIP1 interaction as well as the effects of PAD4 on this system (Figure 5) (17, 23). Briefly, CV-1 cells were transiently transfected with plasmids encoding a luciferase reporter construct, p300GBD fused to the Gal4 DNA binding domain, the p300 binding domain of GRIP1 (i.e., the AD1 domain) fused to the VP16 activation domain (AD), and either wild-type PAD4 or the catalytically defective C645S mutant. Cl-amidine (0–200 μM) was then added to the cell culture medium and the amount of luciferase activity in cell extracts determined. The results clearly indicate that Cl-amidine antagonizes the PAD4-mediated enhancement of the the p300GBD–GRIP1 interaction in a dose-dependent manner, and it is noteworthy that Cl-amidine treatment caused only a minimal reduction in the efficiency of the interaction in Cys645S-transfected cells, thereby indicating that the inhibitory effect of this compound is not a nonspecific one but is targeted at the active PAD4 enzyme. The small, though significant, reduction in the efficiency of the p300GBD–GRIP1 interaction observed in cells transfected with the C645S mutant, as well as the reduction below baseline observed with the wild-type construct, likely represents inhibition of endogenous PAD4 because the efficiency of the interaction is nearly identical for both wild-type and mutant PAD4 transfections when identical amounts (100 μM) of Cl-amidine are added (Figure 5). The results depicted here demonstrate that Cl-amidine is significantly more potent than F-amidine, consistent with its improved in vitro potency.
Figure 5.
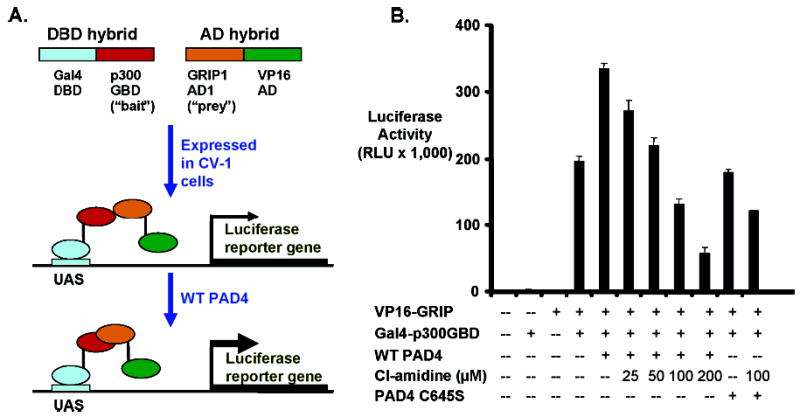
Cl-amidine inhibits the PAD4-mediated enhancement of the p300GBD–GRIP1 interaction in CV-1 cells. (A) Schematic depiction of the mammalian two-hybrid assay used to evaluate the efficiency of the p300GBD–GRIP1 interaction. (B) CV-1 cells were transfected with a luciferase reporter construct as well as plasmids encoding the proteins depicted in this panel. The indicated concentrations of Cl-amidine were added to the cell culture medium and the mixtures incubated for 40 h. Cell extracts were then prepared, and the luciferase activity present in these extracts was quantified. The catalytically defective PAD4 C645S mutant was also transfected into this system to demonstrate that the decrease in the strength of the p300GBD–GRIP1 interaction is not a nonspecific effect.
Structural Basis for Inactivation
To gain further insights into the inactivation properties of F-amidine, we determined the structure of the wild-type PAD4–F-amidine·calcium complex (Figure 6). Crystals of the calcium-bound enzyme–inactivator complex were obtained by soaking calcium-free crystals in crystallization buffer containing 5 mM CaCl2 and 5 mM F-amidine. Diffraction data were then collected, and the initial structure was derived by molecular replacement using the coordinates of the previously determined PAD4· BAA·calcium complex.
Figure 6.
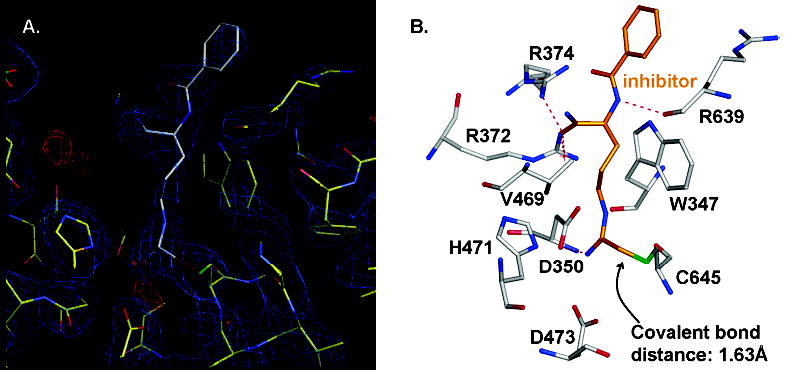
Structure of the PAD4–F-amidine·calcium complex. (A) The 2Fo − Fc (blue) and Fo − Fc (red) electron density maps of the active site, with contour levels of 1.0σ and 3.0σ, respectively. PAD4 and F-amidine are shown in stick format, colored yellow and white, respectively. The green dotted line indicates the electron density for the covalent bond formed between Cys645 and F-amidine. (B) Active site structure of the PAD4–F-amidine·calcium complex. F-Amidine and PAD4 are shown in stick format (colored orange and white, respectively). Red dashed lines indicate potential hydrogen bonds.
The overall structure of the PAD4–F-amidine·calcium complex is comparable to the previously determined structure of PAD4 bound to calcium and BAA; PAD4 consists of three contiguous domains that include two immunoglobulin-like folds that are present in the N-terminal half of the enzyme and a C-terminal catalytic domain. The major difference between these two structures is electron density corresponding to a 1.63 Å covalent bond between Sγ in Cys645 and the Cη atom of F-amidine (Figure 6); the presence of the electron density for this covalent bond was confirmed by analyzing the 2Fo − Fc and Fo − Fc difference Fourier maps with contour levels of more than 2σ. In contrast, no electron density for the fluoride atom was apparent in the structure. Note that electron density for F-amidine was only detected at the active site of the enzyme; thus, this compound specifically inactivates PAD4 by modifying Cys645.
In addition to the presence of electron density for an ideal 1.63 Å Cη–Sγ covalent bond, comparisons of the structures bound to BAA and F-amidine revealed subtle conformational changes in the side chain of F-amidine relative to BAA. For example, in the PAD4·BAA·calcium complex, Asp350 forms hydrogen bonds with both Nδ and Nω1 of the guanidinium moiety, whereas in the PAD4–F-amidine·calcium complex, Asp350 is hydrogen bonded to only Nω1. Moreover, the distance between Asp473 and Nω1 and Nω2 of the guanidinium group is increased in the F-amidine-containing structure, resulting in the loss of the bifurcated hydrogen bond network observed in the PAD4·BAA·calcium complex (Figure 7). The loss of these interactions between the enzyme and the acetamidine moiety is likely caused by differences in the torsion and dihedral angles of the warhead that result upon reaction with the active site Cys; the plane of the covalently bonded acetamidine moiety is roughly perpendicular to the plane of the guanidinium in the enzyme–substrate complex. These conformational changes are transmitted through the rest of the molecule and result in the loss of an additional hydrogen bond between the guanidinium group of Arg374 and the main chain carbonyl of F-amidine; Arg374 hydrogen bonds to the two main chain carbonyls of the substrate in the PAD4·BAA·calcium complex.
Figure 7.
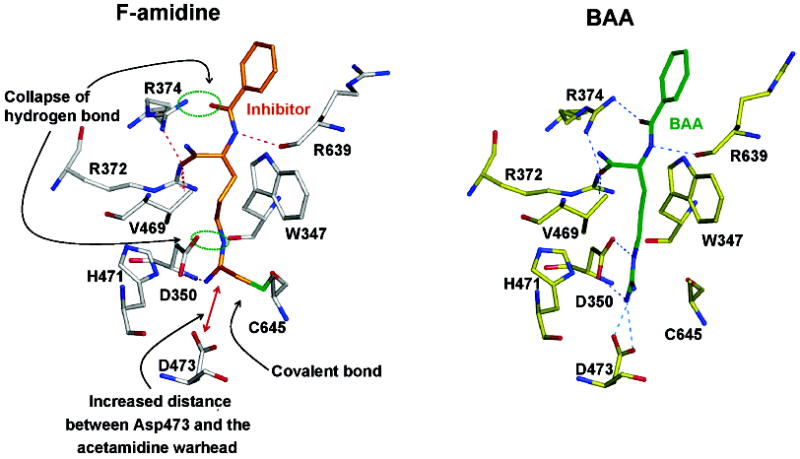
Structural comparison of the PAD4·Ca2+·F-amidine and PAD4·Ca2+·BAA complexes.
DISCUSSION
The deiminating activity of PAD4 has generated widespread interest from both the academic and pharmaceutical industry because current models predict that this enzyme is dysregulated in RA, i.e., overactive, and further suggests that PAD4 inhibitors will treat an underlying cause of the disease rather than merely treating its symptoms. Building on our initial success in developing F-amidine, a potent and bio-available PAD4 inactivator, we synthesized a series of analogues to identify the effects of positioning and leaving group identity on inactivation.
The results of our studies clearly indicate that both factors are critical determinants of inactivator potency. For example, the fact that F2-, F4-, Cl2-, and Cl4-amidine are significantly poorer inhibitors than either F- or Cl-amidine is consistent with the idea that the correct positioning of the warhead is important for inactivation. The decreased potency observed for F4- and Cl4-amidine is most likely the result of a reduction in affinity for these inactivators because the side chains are too long, which results in disruptions to the hydrogen bond network that is formed between the backbone of the inactivator and the main chain carbonyl of Arg639 and the guanidinium group of Arg374. In contrast, the relative lack of potency observed for Cl2-amidine is likely due to the inability to correctly position the warhead for optimal reaction with the active site Cys while maintaining the aforementioned hydrogen bonding network.
The role of warhead positioning in inactivator potency is also highlighted by the fact that Cl2-amidine is an irreversible inactivator whereas F2-amidine is not. These differences likely reflect the very different leaving group potentials of fluoride and chloride and suggest that the correct orientation of the fluoroacetamidine warhead, relative to the active site Cys, is absolutely required for the reaction of the latter warhead with this residue. While not definitive, these results are most consistent with our initial hypothesis (17) that fluoroacetamidines inactivate PAD4 via an initial attack on the iminium carbon to form a tetrahedral intermediate that evolves into a three-membered sulfonium ring prior to its rearrangement to form the thioether observed in the structure of the PAD4–F-amidine·calcium complex. In contrast, the fact that Cl2-amidine is an irreversible inactivator, combined with the expected increase in distance between Cys645 and the chloroacetamidine warhead, suggests that this compound can inactivate PAD4, albeit with reduced efficiency, via the direct displacement of the halide, i.e., mechanism 1 in Figure 2. In total, these results indicate that the requirement for a correctly positioned warhead will have to be taken into account during the design of inhibitors with improved potency. This will be especially important for compounds containing a fluoroacetamidine warhead.
The results described herein also highlight the key requirement for an electron-withdrawing leaving group. For example, the fact that H2-, H-, and H4-amidine are reversible inhibitors demonstrates that reaction with the active site thiolate requires an electron-withdrawing group to enhance the electrophilicity of the iminium carbon. The lack of potency observed for these compounds also highlights the inherent challenges in developing reversible inhibitors targeting this enzyme, thereby providing a strong rationale for the use of the haloacetamidine-based warhead in the development of a PAD4-targeted therapeutic.
The identity of the leaving group also plays an important role in inactivator potency. For example, Cl-amidine is a significantly, ~4-fold, more potent inactivator than F-amidine. This likely reflects the greater leaving group potential of chloride. However, it is interesting to note that the increase in potency does not fully reflect the greater than 105-fold difference in leaving group potential. The lack of a more significant effect may reflect the larger size of the chloro group, which could sterically hinder optimum reaction with the active site thiolate. Consistent with such a possibility is the fact that methylated Arg residues are very poor in vitro substrates for PAD4 (14), presumably because the added bulk of the methyl group prevents the guanidinium group from adopting an orientation that maximizes its reaction with the active site Cys. Thus, the lack of a more significant improvement in inhibitor potency may be due to an improperly oriented warhead.
Although structural studies with F-amidine have demonstrated that this compound specifically modifies Cys645, the identification of the residue modified by Cl-amidine has remained elusive; mass spectrometry experiments have failed to definitively identify the residue modified by the latter compound. However, our results with F-amidine, combined with the Fast group’s finding that 2-chloroacetamidine modifies the active site Cys in DDAH, strongly suggest that Cl-amidine inactivates PAD4 via the modification of Cys645. The modification of this active site residue is further supported by the fact that inactivation is substrate- and calcium-dependent.
As noted above, the calcium dependence of inactivation likely arises because PAD4 is a calcium-dependent enzyme whose in vivo activity can be regulated by the addition of a calcium ionophore (19). In vitro, calcium binding is known to cause a conformational change that moves Cys645 and His471 into positions that are competent for catalysis and presumably reaction with these inactivators. From a therapeutic standpoint, this discovery is highly significant because it suggests that compounds bearing either the fluoro- or chloroacetamidine warhead would preferentially modify the enzyme only in those regions of the body, e.g., the synovial joints of RA patients, in which PAD4 is activated, thereby limiting the toxicity that might be expected to result if both the active and inactive forms of the enzyme were to be inactivated. The ability to preferentially modify the active form of the enzyme should also facilitate the development of biotin-tagged activity-based protein profiling reagents that can be used to selectively enrich the active form of the enzyme and thereby identify the numbers and types of post-translational modifications that this enzyme undergoes in vivo. These latter experiments are significant because they will undoubtedly help identify the signaling pathways in which PAD4 participates and should give clues about how dysregulation of PAD4 could give rise to RA.
It is also noteworthy that the ease and versatility of the solid phase synthetic methodology described in this report will enable the identification of PAD4 inactivators with enhanced potency and selectivity from focused chemical libraries, thereby meeting our ultimate goal of developing a PAD4-targeted pharmaceutical for RA treatment. In conclusion, the haloacetamidine-based inactivators of PAD4, described herein, are not only lead compounds for the treatment of RA but also powerful chemical probes that will ultimately help to identify and decipher the physiological roles of this enzyme in human cell signaling and how this relates to the onset and progression of RA.
Supplementary Material
Footnotes
This work was supported in part by the startup funds from the University of South Carolina Research Foundation (P.R.T.) and National Institutes of Health Grant DK55274 to M.R.S.
Coordinates and structure factors have been deposited with the Protein Data Bank as entry 2DW5.
SUPPORTING INFORMATION AVAILABLE
Figures S1–S3 and synthetic procedures and spectral characterizations. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: PAD, protein arginine deiminase; Cit, citrulline; RA, rheumatoid arthritis; BAEE, benzoyl l-arginine ethyl ester; BAA, benzoyl l-arginine amide; DTT, dithiothreitol; GST, glutathione S-transferase; TCEP, tris(2-carboxyethyl)phosphine hydrochloride; HEPES, N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid; Orn, ornithine; Fmoc, 9-fluorenylmethyloxycarbonyl; Dde, 1-(4,4-dimethyl-2,6-dioxocyclohex-1-yldine)ethyl.
Note that the compounds’ names are based on the identity of the leaving group, i.e., Cl, F, or H, and the number of methylene units in the side chain (two to four). Compounds containing three methylene units are simply termed Cl-, F-, and H-amidine to remain consistent with the nomenclature described in ref 17.
References
- 1.Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 2.Kang CP, Lee HS, Ju H, Cho H, Kang C, Bae SC. A functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in Koreans. Arthritis Rheum. 2006;54:90–6. doi: 10.1002/art.21536. [DOI] [PubMed] [Google Scholar]
- 3.Lundberg K, Nijenhuis S, Vossenaar ER, Palmblad K, van Venrooij WJ, Klareskog L, Zendman AJ, Harris HE. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res Ther. 2005;7:R458–67. doi: 10.1186/ar1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: The conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. 2003;171:538–41. doi: 10.4049/jimmunol.171.2.538. [DOI] [PubMed] [Google Scholar]
- 5.Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, Simon M, Senshu T, Masson-Bessiere C, Jolivet-Reynaud C, Jolivet M, Serre G. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol. 1999;162:585–94. [PubMed] [Google Scholar]
- 6.Masson-Bessiere C, Sebbag M, Durieux JJ, Nogueira L, Vincent C, Girbal-Neuhauser E, Durroux R, Cantagrel A, Serre G. In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin Exp Immunol. 2000;119:544–52. doi: 10.1046/j.1365-2249.2000.01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–81. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nogueira L, Sebbag M, Vincent C, Arnaud M, Fournie B, Cantagrel A, Jolivet M, Serre G. Performance of two ELISAs for antifilaggrin autoantibodies, using either affinity purified or deiminated recombinant human filaggrin, in the diagnosis of rheumatoid arthritis. Ann RheumDis. 2001;60:882–7. [PMC free article] [PubMed] [Google Scholar]
- 9.Union A, Meheus L, Humbel RL, Conrad K, Steiner G, Moereels H, Pottel H, Serre G, De Keyser F. Identification of citrullinated rheumatoid arthritis-specific epitopes in natural filaggrin relevant for antifilaggrin autoantibody detection by line immunoassay. Arthritis Rheum. 2002;46:1185–95. doi: 10.1002/art.10229. [DOI] [PubMed] [Google Scholar]
- 10.Vincent C, Nogueira L, Sebbag M, Chapuy-Regaud S, Arnaud M, Letourneur O, Rolland D, Fournie B, Cantagrel A, Jolivet M, Serre G. Detection of antibodies to deiminated recombinant rat filaggrin by enzyme-linked immunosorbent assay: A highly effective test for the diagnosis of rheumatoid arthritis. Arthritis Rheum. 2002;46:2051–8. doi: 10.1002/art.10436. [DOI] [PubMed] [Google Scholar]
- 11.Masson-Bessiere C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, Serre G. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the α- and β-chains of fibrin. J Immunol. 2001;166:4177–84. doi: 10.4049/jimmunol.166.6.4177. [DOI] [PubMed] [Google Scholar]
- 12.Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–63. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Utz PJ, Genovese MC, Robinson WH. Unlocking the “PAD” lock on rheumatoid arthritis. Ann Rheum Dis. 2004;63:330–2. doi: 10.1136/ard.2003.015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kearney PL, Bhatia M, Jones NG, Luo Y, Glascock MC, Catchings KL, Yamada M, Thompson PR. Kinetic characterization of protein arginine deiminase 4: A transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry. 2005;44:10570–82. doi: 10.1021/bi050292m. [DOI] [PubMed] [Google Scholar]
- 15.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays. 2003;25:1106–18. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 16.Stone EM, Schaller TH, Bianchi H, Person MD, Fast W. Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: Dimethylargininase and peptidylarginine deiminase. Biochemistry. 2005;44:13744–52. doi: 10.1021/bi051341y. [DOI] [PubMed] [Google Scholar]
- 17.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. A Fluoro-Acetamidine Based Inactivator of Protein Arginine Deiminase 4 (PAD4): Design, Synthesis, and in vitro and in vivo Evaluation. J Am Chem Soc. 2006;128:1092–3. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asaga H, Yamada M, Senshu T. Selective deimination of vimentin in calcium ionophore-induced apoptosis of mouse peritoneal macrophages. Biochem Biophys Res Commun. 1998;243:641–6. doi: 10.1006/bbrc.1998.8148. [DOI] [PubMed] [Google Scholar]
- 19.Nakashima K, Hagiwara T, Ishigami A, Nagata S, Asaga H, Kuramoto M, Senshu T, Yamada M. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1α,25-dihydroxyvitamin D3. J Biol Chem. 1999;274:27786–92. doi: 10.1074/jbc.274.39.27786. [DOI] [PubMed] [Google Scholar]
- 20.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–53. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA. Human PAD4 Regulates Histone Arginine Methylation Levels via Demethylimination. Science. 2004;306:279–83. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 22.Liu GY, Liao YF, Chang WH, Liu CC, Hsieh MC, Hsu PC, Tsay GJ, Hung HC. Overexpression of peptidylarginine deiminase IV features in apoptosis of haematopoietic cells. Apoptosis. 2006;11:183–96. doi: 10.1007/s10495-006-3715-4. [DOI] [PubMed] [Google Scholar]
- 23.Lee YH, Coonrod SA, Kraus WL, Jelinek MA, Stallcup MR. Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc Natl Acad Sci USA. 2005;102:3611–6. doi: 10.1073/pnas.0407159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagiwara T, Hidaka Y, Yamada M. Deimination of Histone H2A and H4 at Arginine 3 in HL-60 Granulocytes. Biochemistry. 2005;44:5827–34. doi: 10.1021/bi047505c. [DOI] [PubMed] [Google Scholar]
- 25.Hagiwara T, Nakashima K, Hirano H, Senshu T, Yamada M. Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem Biophys Res Commun. 2002;290:979–83. doi: 10.1006/bbrc.2001.6303. [DOI] [PubMed] [Google Scholar]
- 26.Pritzker LB, Moscarello MA. A novel microtubule independent effect of paclitaxel: The inhibition of peptidylarginine deiminase from bovine brain. Biochim Biophys Acta. 1998;1388:154–60. doi: 10.1016/s0167-4838(98)00175-7. [DOI] [PubMed] [Google Scholar]
- 27.Hughes LR, Jackman AL, Oldfield J, Smith RC, Burrows KD, Marsham PR, Bishop JAM, Jones TR, O’Connor BM, Calvert AH. Quinazoline antifolate thymidylate synthase inhibitors: Alkyl, substituted alkyl, and aryl substituents in the C2 position. J Med Chem. 1990;33:3060–7. doi: 10.1021/jm00173a024. [DOI] [PubMed] [Google Scholar]
- 28.Leatherbarrow RJ. Grafit, version 5.0.11. Erathicus Software; Staines, UK: 2004. [Google Scholar]
- 29.Copeland R. Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmacologists. Vol. 46. John Wiley & Sons; Hoboken, NJ: 2005. [PubMed] [Google Scholar]
- 30.Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M. Structural basis for Ca2+-induced activation of human PAD4. Nat Struct Mol Biol. 2004;11:777–83. doi: 10.1038/nsmb799. [DOI] [PubMed] [Google Scholar]
- 31.Arita K, Hashimoto H, Shimizu T, Yamada M, Sato M. Crystallization and preliminary X-ray crystallographic analysis of human peptidylarginine deiminase V. Acta Crystallogr. 2003;D59:2332–3. doi: 10.1107/s0907444903022741. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54(Part 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47(Part 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.