

Summary
Acireductone dioxygenase (ARD) catalyzes different reactions between O2 and 1,2-dihydroxy-3-oxo-5-(methylthio)pent-1-ene (acireductone) depending upon the metal bound in the active site. Ni+2–ARD cleaves acireductone to formate, CO and methylthiopropionate. If Fe+2 is bound (ARD′), the same substrates yield methylthioketobutyrate and formate. The two forms differ in structure, and are chromatographically separable. Paramagnetism of Fe+2 renders the active site of ARD′ inaccessible to standard NMR methods. The structure of ARD′ has been determined using Fe+2 binding parameters determined by X-ray absorption spectroscopy and NMR restraints from H98S ARD, a metal-free diamagnetic protein that is isostructural with ARD′. ARD′ retains the β-sandwich fold of ARD, but a structural entropy switch increases order at one end of a two-helix system that bisects the β-sandwich and decreases order at the other upon interconversion of ARD and ARD′, causing loss of the C-terminal helix in ARD′ and rearrangements of residues involved in substrate orientation in the active site.
Keywords: metalloenzyme, paramagnetism, X-ray absorption spectroscopy, NMR spectroscopy, cupin, methionine salvage, residual dipolar couplings
Introduction
Stable protein folds typically represent the minimum free energy conformation of the polypeptide chain, and from theory, it is expected that the native conformation of a folded protein is significantly lower in energy than the manifold of other accessible conformations (1, 2). Exceptions are known: The class of serine protease inhibitors known as serpins fold to a kinetically accessible metastable form that rearranges upon cleavage of an exposed peptide bond to a more stable overall fold (3), and some pathological protein folds are the result of kinetic trapping of metastable forms (4). Still, the case of acireductone dioxygenase (ARD), an enzyme in the ubiquitous methionine salvage pathway, is unusual (indeed, perhaps unique) in that two functional enzymes with distinct chemistries are derived from the same polypeptide chain. The two forms differ constitutionally only in the identity of the metal ion bound, but are separable by hydrophobic interaction and strong anion exchange chromatographies, suggestive of significant structural differences between the two isoforms. In this paper, we demonstrate that these structural differences result from differential packing of an integral compact hydrophobic core of the protein, accompanied by extensive changes in secondary structure and ordering of nearby structural features. This repacking has both structural and functional repercussions.
The 179-residue ARD represents a branch point in the methionine salvage pathway of the bacterium Klebsiella ATCC 8724. The methionine salvage pathway returns the thiomethyl group of S-adenosylmethionine 1 (SAM) via methylthioadenosine 2 (MTA) to a new molecule of methionine in which the other carbon atoms are derived from the ribose ring of 1. The penultimate intermediate in the methionine salvage pathway, 1,2-dihydroxy-3-oxo-5-(methylthio)pent-1-ene 4, contains the acireductone functionality C(O)-C(OH)=CH(OH), and is the substrate for ARD (5–7). If Fe+2 is bound in the active site (ARD′), the substrate acireductone reacts with O2 to yield formate and the keto-acid precursor of methionine, 4-methylthio-2-ketobutyrate 5, and formate (Figure 1). The Ni+2-containing ARD catalyzes an off-pathway oxidation of acireductone by O2 with formation of carbon monoxide, formate and 3-(methylthio)propionate 6. ARD and ARD′ are both monomers, and are kinetically stable in that metal replacement is required to switch from one fold to the other. It has been shown (7, 8) that Ni+2 in ARD can be conservatively replaced by Mn+2 or Co+2, giving rise to ARD activity (CO production), while Fe+2 in ARD′ can be replaced (albeit with lower activity) by Mg+2. This promiscuity towards metal cofactors of widely different reduction potentials suggests that the metal is not an activator of triplet O2, but rather acts to control protein structure and ligand orientation so as to direct the reaction between O2 and acireductone towards a particular regiochemistry (9).
Figure 1.
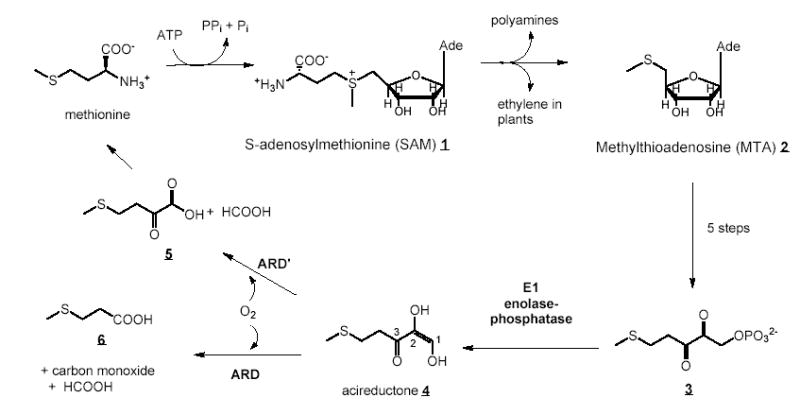
An outline of the methionine salvage pathway in Klebsiella ATCC 8724. Carbon atoms 1, 2 and 3 in acireductone 4 are labeled for reference in the text.
ARD belongs to the cupin structural superfamily. Cupins are a functionally diverse group of proteins distinguished by the presence of an antiparallel β-helix (sometimes called a jellyroll) that forms the central feature of the cupin fold (10, 11). We previously described a structural model of Ni-containing ARD based on NMR-derived restraints and molecular modeling in the vicinity of the metal center (9) (PDB entry 1M4O), and have recently published a refinement of that structure that included the use of residual dipolar couplings as restraints in the calculations (PDB entry 1ZRR) (12). Paramagnetism broadens 1H resonance lines within ~9 Å of the metal center in both Ni- and Fe-containing forms of ARD, making it impossible to determine local structure in the active site by standard NMR methods. Instead, we made use of X-ray absorption spectroscopy (XAS) to establish ligation geometry and bonding of the nickel ion, and combined this information with homology modeling to generate a structural model for the ARD active site. In the original structure (1M4O), this modeling was based on a phylogenetically distant member of the cupin family, jack bean canavalin (13). Recently, a crystallographic structure of a mouse homologue of ARD was deposited in the RCSB PDB by researchers from the Joint Center for Structural Genomics (PDB entry 1VR3) (14). In our refinement of the ARD structure, we replaced the canavalin coordinates used in the original modeling of the active site with those from the corresponding residues in the 1VR3 structure. We note that this did not result in change in the proposed ligands for metal binding (12). Both of the models agree on the identity of His 96, His 98, His 140 and Glu 102 as the metal binding ligands in the active site of ARD (Figure 2). However, the lack of direct structural confirmation of these assignments prompted us to perform site-directed mutagenesis at the positions of these residues. While a complete analysis of these mutations is outside the scope of the present work, mutations at all four proposed ligands result in loss of either protein solubility, loss of tight metal binding, or both. Furthermore, one of these mutations, His 98 to serine (H98S), proved to be interesting for other reasons. The H98S mutation resulted in the formation of a stable soluble protein that while structurally different from ARD shows a high degree of similarity to the ARD′ enzyme as determined by a comparison of 1D 1H NMR and 2D 1H, 15N HSQC spectra (Figure 3). The H98S mutant thus provides a structural model for the Fe-containing form of ARD. ARD′ decomposes during long NMR experiments (possibly by loss of iron) and so has been difficult to characterize structurally. Furthermore, the purified apo-WT ARD also gives broadened resonance lines and precipitation during the course of long NMR experiments, although it is spectroscopically similar to ARD′ and H98S (Tingting Ju, manuscript in preparation). The stably folded H98S mutant, on the other hand, provides us with a tractable model for the ARD′ enzyme, and has allowed us to identify structural differences between the Fe- and Ni-bound isoforms. Furthermore, the absence of a bound paramagnetic metal ion in H98S ARD has allowed us to structurally characterize the ARD active site in detail by NMR for the first time (Figure 2). Combining the information provided by nuclear Overhauser effects (NOEs), residual dipolar couplings, chemical shift and J-coupling-based dihedral angle restraints obtained with the H98S mutant with restraints on Fe+2 ligation provided by X-ray absorption spectroscopy (XAS) performed on ARD′, we have generated a structural model for ARD′ has allowed us to identify the structural differences between the two ARD isoforms. From these differences, we gain insight into the origins of the different chemistries catalyzed by the two enzymes.
Figure 2.

Comparison of the active sites of H98S (left), ARD′ (current work, PDB 2HJI, center), and ARD (PDB 1ZRR, ref. 9) right. The H98S active site structure was determined using experimentally determined NOE and RDC data. The ARD′ active site structure shows Fe+2 as an umber sphere and the ARD active site shows the Ni+2 ion in green. The positions of backbone heavy atoms and β-carbons of ligands His 96, His 98, Glu 102 and His 140 in the ARD′ structure are based on NOE data and residual dipolar couplings from H98S ARD, with metal-ligand bond lengths determined by Fe XAS of ARD′. The corresponding atom positions in the ARD structure are based on the crystal structure of mouse ARD (1VR3, see text) (14) and bond lengths determined from Ni XAS experiments (15). Note that the indole sidechain of Trp 162 partially occludes the active site opening in ARD. In H98S and ARD′, the polypeptide including Trp 162 is disordered and does not occlude the active site. All three sites are shown in approximately the same orientation using the backbone atom positions of the structurally conserved β-sandwich (residues shown in yellow, Pro 137-Phe 142) as reference.
Figure 3.
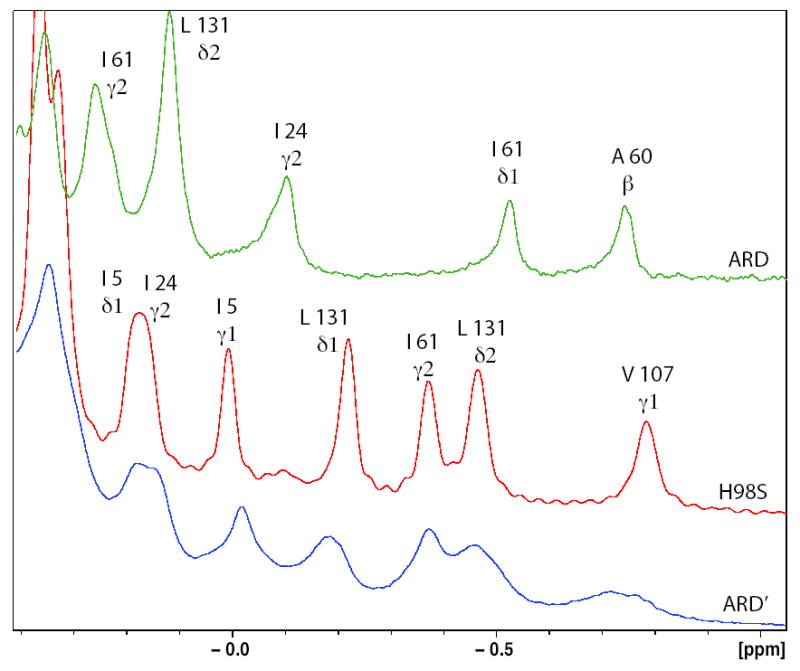

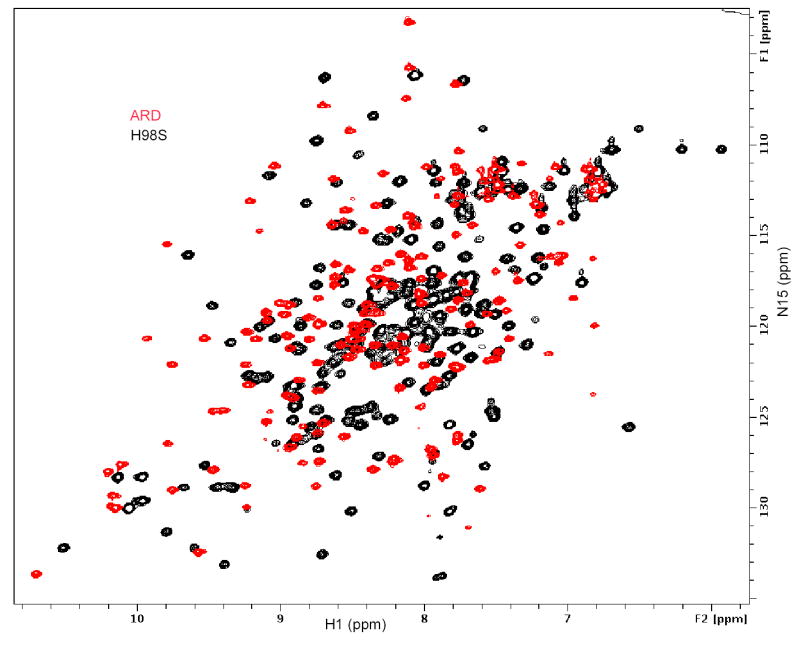
a) Ring current-shifted aliphatic amino acid methyl resonances in the 600 MHz 1H NMR spectra of ARD (green trace), H98S ARD (red trace) and ARD′ (blue trace) with resonance assignments shown. Note the similar patterns for H98S ARD and ARD′, with different shift patterns for ARD. b) Overlay of the 1H, 15N HSQC spectra of ARD′ (peaks shown in red) and H98S ARD (peaks shown in black). Note close correspondence of most peaks in the spectra. Peaks that occur in the H98S spectrum but not in the ARD′ spectrum (labeled) are within ~9 Å of the metal center. The indole NɛH of Trp 162 (lower left) is observed in both ARD′ and H98S ARD (see text for discussion). c) Overlay of the 1H, 15N HSQC spectra of ARD (peaks shown in red) and H98S ARD (peaks shown in black). Different shift patterns indicate significant differences in the tertiary structure. All spectra were taken at 25 °C, pH 7.4 at 600 MHz 1H observe frequency.
Results
H98S ARD as a model for the ARD′ structure
Inspection of both 1D and 2D spectra of the H98S ARD mutant (Figure 3) provides the justification for using the H98S structure as a model for ARD′. In the absence of paramagnetic effects, resonances are shifted upfield in the 1H NMR spectrum of a protein by ring currents resulting from close packing between aromatic (Trp, Phe and Tyr) and aliphatic amino acid side chains. Because ring-current shifts are determined both by the distance between and relative orientations of the interacting side chains, such shifts are sensitive to changes in tertiary structure. The differences between ARD and H98S in the upfield region of the 1H NMR spectrum are obvious (Figure 3A). In ARD, the largest upfield shifts are observed at methyl resonances of Ala 60 and Ile 61 (helix E′) due to close packing with the indole rings of Trp 38 and Trp 73. In H98S, the largest upfield shifts are found to the side chain of Val 107 (helix E), due to close packing with Tyr 57, and to Leu 131, which is proximal to the Trp 38 and Phe 105 side chains. The upfield shift patterns observed for H98S ARD correspond closely with those observed for ARD′, and this correspondence is supported by comparison of 2D TOCSY and NOESY spectra (data not shown) obtained for H98S and for ARD′. Spin system patterns and close contact NOEs observed in both proteins strongly support the assignment of corresponding peaks to the same residues in both H98S ARD and ARD′.
Overlap of the 1H, 15N HSQC spectra of H98S and ARD′ also show very similar fingerprints for the two proteins, while the overlap between H98S and ARD is minimal. The HSQC fingerprint is diagnostic for a particular fold, and it is clear from the comparison in Figure 3B that there is close structural correspondence between H98S and ARD′. Peaks that occur in the H98S spectrum (black) with no corresponding peak in ARD′ have been assigned to residues in the active site or within ~9 Å of the bound Fe+2, and are therefore not observed in the ARD′ spectrum due to paramagnetic broadening. Based on these observations, we expect that structural calculations based on the NMR-derived restraints derived for H98S and the XAS-derived parameters for Fe binding in ARD′ provide a valid structural model for ARD′. The discussion of the ARD′ structure that follows is based upon this assumption.
Metal binding by ARD′
Fe-XAS data, including EXAFS (extended X-ray absorption fine structure) and XANES (X-ray absorption near-edge spectroscopy) obtained for ARD′ are consistent with octahedral ligation of Fe+2 by N/O, with the best fit to EXAFS data obtained for three or four His ligands (see supplementary material). These results are strikingly similar to those obtained from Ni-XAS data (15) obtained for ARD. Of the four protein-based ligands proposed for nickel binding in ARD, His 96, Glu 102 and His 140 are structurally fixed by the conserved β-sandwich in both ARD and ARD′ (vide infra). The other, His 98, is on a loop between two strands (H and I, Fig. 4) of the β-sandwich, and so might be expected to exhibit some conformational flexibility. However, the H98S mutation abolishes the ability of the ARD polypeptide to bind tightly to either Ni+2 or Fe+2, as determined by isothermal calorimetry (T. Ju, unpublished results). As such, we propose that the same four protein-derived ligands His 96, His 98, Glu 102 and His 140 that ligate Ni+2 in ARD also bind Fe+2 in ARD′. As with ARD, the axial ligands are provided by His 96 and Glu 102, and His 98 and His 140 occupying cis equatorial positions. The remaining two cis equatorial positions are occupied by water or a solvent-derived ligand in the absence of substrate.
Figure 4.
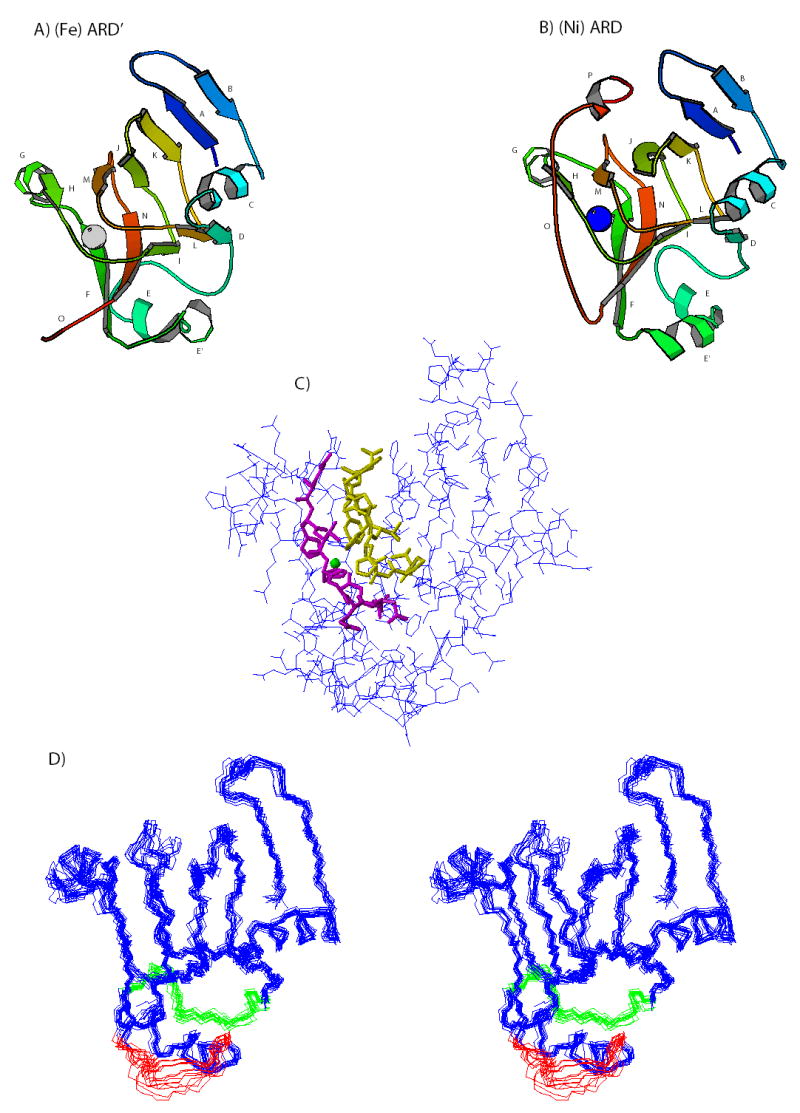
Comparison of the structures of ARD′ (A) and ARD (B). Letters reference to the ARD sequence as follows: A (Ala 2-Phe 6), B (Leu 15-Ser 18), C (Glu 23-Lys 31), E (Thr 50-Tyr 57), E′ (Ile 61-Lys 68), F (Ser 72-Leu 78), G (Lys 85-Glu 90), H (Phe 92-Glu 95), I (Arg 104-Val 107), J (Gly 111-Ile 117), K (Glu 120-Leu 125), L (Asn 129-Ile 132), M (His 140-Met 144), N (Phe 150-Phe 156), O (Gly 161-Gly 168), P (Ile 171-Ala 174). The positions of metal ions are indicated by gray (Fe+2) and blue (Ni+2) spheres. Residues 157-175 (loop O and helix P in ARD) are disordered in ARD′, and so for clarity are not shown in Figure 4A. C) Positions of heavy atoms in structure 10 of the ensemble shown in Fig. 4D. This structure is closest to the average of the ensemble. Residues close to the active site are shown in neon, Phe 92-Arg 104 in magenta and Val 134-Asp 143 in yellow. The position of the iron is indicated by a green sphere. D) Superposition of 14 accepted structures of ARD′. Statistics are shown in Table 1. Residues 39-49 are shown in green, and residues 65-73 are shown in red.
Comparison of the ARD and ARD′ structures: a conserved structural core
Like ARD (Figure 4B), the structure of ARD′ (Figure 4A) centers on the conserved β-helical cupin fold. We refer to this fold as a β-sandwich, because of the flattened appearance of the structure as viewed in Figs. 4A and 4B. The register of inter-strand hydrogen bonds that form the β-sandwich in H98S (and in ARD′) is identical to that in ARD where direct comparisons can be made. Most of the residues that make up the active site are positionally fixed within the β-sandwich, and are expected to occupy similar positions in ARD and ARD′. The absence of paramagnetic broadening in H98S allows the active site to be characterized in detail by NMR methods for the first time. Multiple NOEs are detected between the side chains of His 96, Ser 98 and His 140, confirming the close association of those side chains. Only Glu 102 could not be confirmed as being in close association with the proposed metal binding tetrad, but this is not surprising, since only the carboxylate of Glu 102 is likely to be close to the metal binding site and this functionality does not lend itself to characterization by NOEs. However, inter-strand NOEs confirm the placement of Glu 102 antiparallel with Val 134 in the β-sandwich structure, placing the side chain carboxylate of Glu 102 in the appropriate position for metal ligation. Arg 104, which is conserved across the ARD family (9, 16), extends toward the metal binding tetrad, and NOEs between the side chains of Arg 104 and Arg 154 (also conserved across the ARD family) (16) indicate that the side chains of these residues are roughly parallel. We have proposed that Arg 104 interacts with substrate acireductone via hydrogen bonding interactions, and/or acts as the base that deprotonates the substrate upon binding to the enzyme (7). This deprotonation has been shown to be functionally required in a model of the ARD reaction (17). Although arginine residues are usually too basic to be useful in catalysis at physiological pH (18), the close interaction between Arg 104 and Arg 154 could modulate the pKa of the diad so that either guanidine group could act as proton acceptor.
Structural differences between ARD and ARD′: disorder at the C-terminal polypeptide in H98S and ARD′
Along with the conserved cupin core fold, helices C and G are not significantly displaced between ARD and ARD′. These helices occupy positions related by a pseudosymmetric C2 axis normal to the long axis of the β-sandwich, and cap the two open ends of the sandwich (Fig. 4). However, inspection of Figure 4 reveals an obvious difference between the ARD′ (Fig. 4A) and ARD (Fig. 4B) structures at the C-terminus of the polypeptide. In ARD, residues 158 to 164 form a tight turn (Asn 158-Pro-159-Glu 160-Gly 161) and a short antiparallel extended structure stabilized by hydrogen bonds to the backbone C=O and NH of Thr 97 (12). This effectively places the side chain indole of Trp 162 near the entrance to the active site. Residues 171 to 175 form a short 3,10-helix (P) in ARD that packs onto the upper surface of the β-sandwich as viewed in Fig. 4B.
In H98S and ARD′, however, the last 22 residues (Asp 157-Ala 179) are disordered, with resonances in this stretch exhibiting narrow line widths and no non-sequential NOEs. Furthermore, two sets of resonances can be assigned to residues extending from Gly 161 to Ala 172 in H98S ARD, suggesting that both cis- and trans-isomers of the Asn 158-Pro 159 peptide bond are occupied. The NɛH resonance of the Trp 162 indole ring, which is not detected in ARD due to proximity to the active site metal, can be identified in 1H, 15N HSQC spectra of ARD′, indicating that it is no longer adjacent to the active site (Figure 3B). The disordering of the C-terminal peptide in ARD′ effectively opens the active site, rendering it more accessible to substrate. The mechanistic implications of this will be discussed below.
A structural entropy switch between ARD and ARD′
All of the structural differences between ARD and ARD′ can be related either to the decrease in order at the C-terminus of ARD′ relative to ARD described above, or to an opposed increase in order near the N-terminal of the E helix in ARD′ relative to ARD. In turn, the collective changes in order are related by displacements of the E and E′ helices that bisect the β-sandwich and the accompanying repacking of hydrophobic residues projecting from the bottom of the β-sandwich that interact with the E and E′ helices.
In both structures, the long axes of the E and E′ helices meet at a ~130° angle. The side chain amide of Gln 58 at the C-terminal end of the E helix provides a capping hydrogen bond to the carbonyl oxygen of Ile 54, introducing the break between the E and E′ helices. Gln 58 remains in approximately the same position relative to the β-sandwich in both ARD and ARD′, so the bend between the two helices acts as a pivot point for the relative displacements of the E and E′ helices observed in the two structures (see Figure 5). The long axes of both helices shift by ~20° relative to the β-sandwich long axis between ARD and ARD′. Accompanying this shift is a significant repacking of hydrophobic residues in this region. It is this repacking that gives rise to the different chemical shift patterns observed in the upfield regions of the 1H spectra of ARD and ARD′ (Fig. 3a). In ARD, the methyl groups of Ala 60 and Ile 61 on the E′ helix pack tightly against the indole rings of Trp 38 and Trp 73, respectively. In ARD′, this packing is largely absent. Instead, the Trp 38 indole in ARD′ packs more tightly with Leu 131 on strand L of the β-sandwich, resulting in the increased upfield shift of the Leu 131 methyl resonance (Fig. 3A). Trp 73 on strand F interacts less strongly with all of its neighbors in ARD′ than in ARD. This loosening of structural integrity results in fraying at the C-terminal of the E′ helix in ARD′, with a loss of NOEs consistent with regular helical structure past Val 65. In ARD, the E′ helix is well defined up to Lys 68. 1H NMR resonances of residues between Val 65 and Trp 73 in H98S (and by inference, in ARD′) are either broadened or absent: The backbone resonances of Glu 67, Lys 68 and Tyr 70 in H98S are sufficiently broadened that they could not be assigned in standard 3D triple-resonance experiments, and are assigned based solely on NOE data, while those of Glu 71 could not be assigned at all. The broadening of resonance lines indicates that this region of the polypeptide occupies multiple conformations that interchange on the millisecond time scale.
Figure 5.
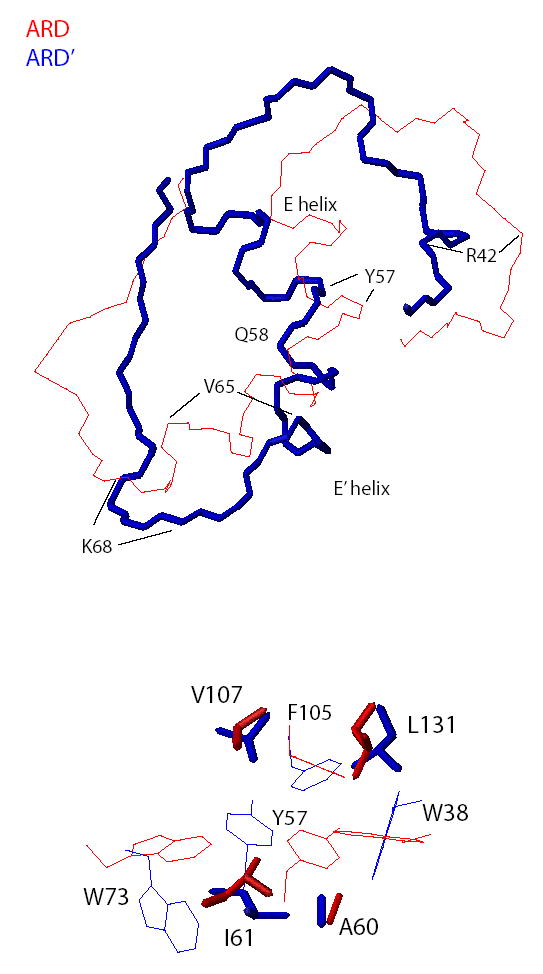
Top) Relative displacement of the E and E′ helices in ARD and ARD′. For this comparison, the backbone coordinates of the conserved β-sandwiches of both proteins were superimposed, but only the helices and leading/following peptides are shown. ARD is shown in red, ARD′ in blue neon. Bottom) Side chain packing differences in ARD and ARD′ using the same superposition as in the top figure. Aromatic residues that give rise to ring current shifts observed in Fig. 3a are shown in light lines, shifted aliphatic side chains are shown in neon. Red corresponds to ARD, blue to ARD′.
The opposite phenomenon is observed near the N-terminal end of E helix. In ARD, residues 39-50 form a largely disordered loop between strand D of the β-sandwich and the beginning of the E helix, with few NOEs between residues in this loop and other parts of the protein (9, 12). In ARD′, however, this loop becomes more ordered, particularly those residues near the side chain of Tyr 57 on the E helix. Ala 40, Arg 42 and Leu 44 form a pocket around the Tyr 57 side chain in ARD′, and Phe 105 and Val 107 from strand I of the β-sandwich close pack with the Tyr 57 side chain. The large upfield shift of the Val 107 methyl 1H resonance in H98S and ARD′ (Fig. 3A) is due to this repacking. It is likely that either the carbonyl of Arg 42 or the carboxylate of Glu 108 forms a hydrogen bond with the Tyr 57 OηH, and the orientation of the carbonyls of Ala 40 and Arg 42, along with the carboxylate of Glu 108 and the Tyr 57 phenoxyl oxygen suggest a potential cationic binding site involving these groups.
The differential packing of the E and E′ helices, and the attendant reordering of the polypeptide at the N-terminal of the E helix and C-terminal of the E′ helix represents a structural entropy switch between ARD and ARD′. In other words, a decrease in order at one end of the E, E′ helix structure is accompanied by an increase in order at the other, resulting in relocation of structural entropy accompanying the interconversion of the two isozymes.
Discussion
Cause and effect: How are structural differences between ARD and ARD′ propagated?
The structural differences between ARD and ARD′ are doubtless due to metal binding in the active site (6). Based on comparison of NMR data, the Fe-containing ARD′ is structurally similar to apo-WT ARD and H98S, suggesting that the ARD′ fold is the “default”, with little perturbation resulting from binding of iron, while the binding of Ni+2 triggers the structural switch described here. This is consistent with the experimentally observed lability of iron in samples of ARD′ and the fact that Fe+2 can be removed from ARD′ by dialysis against EDTA-containing buffers, but that Ni+2 removal from ARD requires that the protein be denatured first (8). Perturbations accompanying the binding of Ni+2 are likely propagated to the rest of the protein via the formation of the antiparallel hydrogen bonding arrangement between Thr 97 and Ile 163 (present in ARD, absent in ARD′) and the salt bridge formed by Glu 100 and Lys 68 on the E′ helix (frayed in ARD′). In turn, these interactions depend on the backbone conformation in the Thr 97-His 98-Gly 99-Glu 100 loop. However, how information regarding the presence or absence of Ni+2 is transmitted to the Thr 97-Glu 100 backbone is not clear. The Fe-N bonds in ARD′ are slightly longer (2.18 Å) than the Ni-N bonds in ARD′ (2.07 Å) based on fitting of EXAFS data, as are the metal-oxygen bonds (2.00 Å for Fe-O vs. 1.91 Å for Ni-O). With the caveat that the active site structure of ARD is based on modeling from the mouse ARD crystal structure, while the ARD′ active site structure is derived from NOE and RDC constraints obtained from the H98S mutant, comparison of the two active sites suggests that the expansion of the ligand sphere might tilt the equatorial ligand plane (His 140, His 98 and the two solvent-derived ligands) downward in ARD′ as viewed in Figure 2. This in turn could force a change in conformation for the Thr 97-Glu 100 loop, transmitting the perturbations described above.
Another possibility for driving the conformational change is a switch at His 98 between ligation via the Nɛ or the Nδ imidazole nitrogens upon changing metal. While introduction of Nδ ligation of the iron at His 98 into our structural calculations for ARD′ resulted in multiple NOE violations in the active site, such a switch made little difference in calculations of Ni binding in ARD using modeled coordinates (9). It is possible that nickel prefers Nδ ligation at His 98. Although the crystal structure of mouse ARD shows all-Nɛ ligation of the metal, patterns of H/D exchange in hyperfine shifted imidazole 1H resonances in ARD are consistent with ligation of Ni+2 via two His Nɛ and one His Nδ (15). Current progress in our laboratory towards a crystallographic structure of ARD may clarify this issue.
Mechanistic implications
The reaction between the des-thiomethyl homologue of acireductone 4 and O2 proceeds at a significant rate under physiological conditions even in the absence of enzyme (kobs ~ 0.025 s−1, G. Pagani, personal communication), and yields the same products, keto-acid and formate, that are observed from the ARD′-catalyzed reaction. As such, the activation of dioxygen, an important role for most oxygenases, is likely less important for ARD, and no significant binding of O2 is observed to the substrate-free enzyme (7). At physiological pH, acireductone is predominantly a monoanion, and acireductone binding to either ARD and ARD′ involves the loss of a second proton, yielding the dianionic species (7).
We have proposed that the mechanism of both enzymes involves initial formation of a radical pair by single electron transfer from the metal-bound acireductone dianion to O2, forming superoxide (O2−), followed by collapse of the radical pair to form the peroxide species at C1 (compounds 4a and 4c in Figure 6) (9). In ARD, this peroxide cyclizes via intramolecular nucleophilic attack at C3, forming the five-membered peroxy compound 4d that undergoes electrocyclic reaction leading to formate, CO and methylthiopropionate. In ARD′, the acyclic peroxide attacks at C2, leading to the dioxetane 4b, electrocyclic decomposition of which yields the α-keto-methylthiobutyrate and formate. These mechanisms are supported by the results of isotope incorporation experiments using 18O2. (5) The role of the metal in either case would be to activate a particular carbon (O2 in ARD′ and O3 in ARD) to the nucleophilic ring closure via Lewis acid interactions with the attached oxygen. As such, one would expect that substrate 4 would chelate the Fe+2 in ARD′ via the oxygens at positions 1 and 2 of the acireductone, while in ARD, the Ni+2 would be chelated via the oxygens at positions 1 and 3 (see Fig. 6). The proposed mechanism for ARD in particular is strongly supported by recent work from Berreau and coworkers, who have shown that a hexacoordinate N,O complex of Ni+2 indeed ligates a sterically bulky acireductone in a bidentate fashion via the C1 and C3-bound oxygens. Furthermore, treatment of this complex with one equivalent of base and O2 results in the formation of CO and two carboxylate products, in precise analogy with the enzymatic reaction (17).
Figure 6.

Proposed mechanisms for regioselectivity in ARD′ (left) and ARD (right). Bidentate ligation of the metal by the substrate via the O1 and O2 positions (ARD′) versus O1 and O3 positions (ARD) results in Lewis acid activation of the O2 position in ARD′ and O3 position in ARD for intramolecular nucleophilic attack by peroxide anion, giving ring closure to the 4-membered cyclic peroxide in the case of ARD′ and the 5-membered cyclic peroxide in the case of ARD. For more detail concerning these mechanisms, see references 7 and 9.
The next question that arises is whether differential ligation of 4 in ARD′ versus ARD is the result of geometric requirements of the ligating metal or whether ligation geometry is enforced by the structures of the active sites. The most obvious difference between the active sites of ARD and ARD′ is the more restricted access of the Ni+2-bound ARD active site relative to that of ARD′. The presence of the Trp 162 indole in the vestibule of the active site of ARD is enforced by the hydrogen bonding between Ile 163 and Thr 97 backbone, the tight turn Asn 158-Gly 161 and packing of helix P onto the top face of the β-sandwich. We have modeled how substrate might fit into the active sites of the two isoforms (Figure 7). In the case of ARD′, the open active site permits an extended conformation of the acireductone ethylthiomethyl group, with the guanidinium groups of Arg 104 and Arg 154 arranged near the O1 and O2 metal ligand bonds, respectively. In ARD, the acireductone ethylthiomethyl group packs against the Trp 162 indole side chain, and the sterically less demanding O1, O3 ligation of the acireductone is favored. Although the positions of the Arg 104 and 154 side chains in the ARD active site are less well defined due to paramagnetic effects than in the ARD′ structure, it is likely that at least Arg 154 is in a position to hydrogen bond with O2 of the acireductone in ARD.
Figure 7.

Comparison of the active sites of ARD′ (left) and ARD (right) modeled with substrate acireductone 4 bound. Modeled structures were generated using AMBER 8.0 (30). Initial structure for ARD′ was from the current work, starting structure for ARD was from PDB entry 1ZRR (12). In both cases, the two equatorial water ligands were replaced by oxygens from acireductone (shown in blue in both structures). In ARD′, 4 is ligated to the Fe+2 via O1 and O2 (ACRC), while in ARD, 4 is ligated to Ni+2 via O1 and O3 (ACRT). Complexes were subjected to 4000 steps of minimization to remove close contacts. The two structures are not in the same orientations, but are rotated to obtain clear views. Due to paramagnetic broadening, the positions of side chains within the active site of ARD (1ZRR) are not as precisely defined as in the ARD′ structure. Figures were generated using MOLMOL (31).
Conclusions
The validity of the model of ARD′ presented here is based on the assumption that the structural similarity observed by 1D and 2D NMR methods between ARD′ and H98S extends to the active site, which cannot be observed in ARD′ by standard NMR methods due to paramagnetism, and that any structural perturbations arising from Fe binding are minimal beyond the immediate vicinity of the metal binding domain. As noted above, this assumption is supported by the similarity between spectra of H98S, apo-WT ARD and ARD′ in regions where comparisons can be made. However, until such time as crystallographic structures of the active sites of ARD and ARD′ are determined with defined metals, the active site models for both of these enzymes should be used cautiously as guides to further experiments.
Materials and Methods
Mutagenesis and expression of H98S ARD
The wild-type ARD gene in pET-3a was used as a template for site-directed mutagenesis experiments. All mutant primers were designed in-house for use with the Quik-Change (Stratagene) kit and protocol. Primers are listed in the supplementary material. The H98S ARD gene was placed in the appropriate orientation behind the T7 promoter of pET-3a (Novagen) between the NdeI and BamHI restriction sites. Mutant plasmids were transformed into electrocompetent E. coli XL1-Blue cells (Stratagene) for amplification, miniprepped and the correct mutant sequence was confirmed by DNA sequencing. For expression, purified mutant plasmid was transformed into E. coli strain BL21(DE3)pLysS, with induction and purification following the published procedure for WT ARD (6). For isotopically enriched H98S used in NMR experiments, the growth and expression procedures of Mo et al. were used (19).
XAS Methods
Samples of ARD′ were reconstituted with Fe+2 in order to insure complete occupancy of the metal site by Fe. The reconstitution procedure and XAS sample preparation are described in the supplementary material. XAS data were acquired at beamline X9B at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory. The samples were placed in polycarbonate holders that were inserted into a slotted aluminum holder and held near 50 K using a He displex cryostat. The XAS data were collected under dedicated conditions at ca. 2.8 GeV and 160–260 mA as previously described,(20) except that a sagitally focusing Si(111) double-crystal monochromator was used. The X-ray energy of the focused monochromatic beam was internally calibrated to the first inflection point of Fe foil (7112.0 eV).
X-ray fluorescence data were collected using a 13-element Ge detector (Canberra). X-ray absorption data were collected over the range from ca. 6.9–8.1 KeV. Harmonic rejection was achieved by use of a Ni mirror. An average of 10 scans were used for EXAFS analysis by WinXAS.(21) The summed, energy-calibrated data files were background-corrected and normalized using a two third-order polynomial fits. The data were converted to k space using the relationship [2me(E − E0)/ħ2]½ (where me is the electron mass, E is the photon energy, ħ is Planck’s constant divided by 2π, and E0 is the threshold energy of the absorption edge and defined here as 7125.0 eV). A least-squares fitting procedure was employed over a k range of 2–12.5 Å−1. The fitting procedure minimized (where σ is an estimate of the experimental error and yexp and ytheo are experimental and theoretical data points, respectively, and N is the number of data points).(21)
| (Eq. 1) |
Theoretical phases and amplitudes for EXAFS analyses were obtained from FEFF 8.2 calculations of crystallographically characterized model compounds catena-(hexakis(μ-2-imidazoyl-N-N′)bis(imidazole)tri-iron) (22) and (Et4N)2[Fe(SC6H4CH3-p)4] (23). The EXAFS analysis of Fe-ARD data was carried out as previously described.(24) Integer values for the number of scattering atoms in a shell were used in the fits without refinement. This led to the following running parameters for each shell in the first coordination sphere: the distance (r), the disorder parameter (σ2) and phase shift parameter (ΔE0). Comparison of these parameters and residual values(21) (Equation 1) were used to select the best fits of the data (see supplementary material).
NMR data acquisition and analysis
Samples for NMR experiments were prepared by buffer exchange using a desalting column pre-equilibrated with 90/10 H2O/D2O containing 50 mM potassium phosphate buffer (pH 7.5). Typical sample concentrations were ~ 1 mM. The NMR experiments and methodology used for sequential assignment of H98S ARD are similar to those previously described for ARD (19). All NMR experiments were performed on a 14 T (600 MHz 1H) Varian Unity Inova NMR spectrometer equipped with an inverse-detection triple resonance probe, z-axis pulsed field gradients and three RF channels. Three-dimensional triple-resonance experiments performed include HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH and HNCO. Three-dimensional double-resonance experiments performed include two versions of HCCH-TOCSY, one optimized for aliphatic carbons and one for aromatic carbons, a 15N-edited NOESY and two versions of 13C-edited NOESY, one for aliphatic protons and one for aromatic protons. All NOESY experiments were acquired with a 70 ms mixing time to minimize the contribution of spin diffusion. With modifications (e.g., carrier frequencies optimized for aliphatic versus aromatic carbons) and parameter optimization, the implementations of all experiments are those found in the standard Varian BioPack release. Quadrature detection in the 15N indirect dimension for all NH-detected experiments was obtained using the Rance-Kay method (25), with pulsed field gradient coherence selection for magnetization transfer through 15N. Quadrature detection in indirectly detected 13C dimensions was obtained using States-TPPI and coherence selection was obtained by phase cycling. All NMR datasets were initially processed using XWinNMR (Bruker Biospin Inc.) and data analysis performed using XEASY (26). Backbone sequential assignments were made for most of the polypeptide backbone using correlations identified in through-bond coherence transfer experiments (HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH). Side chain assignments were made using a combination of HCCH-TOCSY and HNCACB data. NOE restraints were obtained by analysis of 3D isotope-edited NOESY experiments.
One-bond 1H-15N residual dipolar couplings (RDCs) were measured in two aligning media, filamentous phage (fd) and asymmetric alkylpolyol HO-(CH2O)5-(CH2)11CH3 (C12E5) (Aldrich) in a 0.96 molar ratio with 1-hexanol (final conentration in NMR samples 5% w/w). Final fd concentration in NMR samples was 32 mg/mL. RDCs were obtained and analyzed using the methods recently described for ARD (12). Reference 1H, 15N HSQC spectra were acquired without 1H decoupling in the indirect dimension at 25 °C without alignment, followed immediately by acquisition of identical spectra under aligning conditions. RDCs were obtained by comparison of the reference and aligned spectra. Maximum estimated error in measurement is +/− 2 Hz, although most measurements are precise to within +/−1 Hz.
Structural calculations
Structural calculations were performed on a Dell computer with an Intel 386 processor running a RedHat Linux operating system. Restrained molecular dynamics/simulated annealing using torsional dynamics were performed with the NIH implementation of XPLOR version 2.10 (27, 28) as described recently for ARD (12). Restraints on the metal center were based on bond lengths and geometry for metal ligation obtained from best fits of XAS data, with planarity of atoms and ligation geometry enforced using improper angle restraints. Due to the lack of defined structure for the C-terminal residues Asp 157 to Ala 179 (vide supra), these residues were not constrained in the calculations in any fashion, and thus are not included in the deposited coordinates. Energies were calculated using bond, angle, van der Waals, improper, NOE, dihedral angle, J-coupling, RDC (SANI), 13C chemical shift, hydrogen bonding and database torsional terms. Electrostatic terms were not included in the refinement. Restraints used in the calculations are summarized in Table 1. Starting values of anisotropy Da and rhombicity R = Dr/Da for fitting of RDCs from each alignment medium were determined by fitting RDCs to structures obtained without RDC restraints using SSIA (29). As refinement progressed, values of Da and R were recalculated to improve fits.
Table 1.
Experimental and modeling restraints used to generate the fourteen accepted structures shown in Figure 4c and statistical analysis.
| Experimental restraints | |
|---|---|
| long range NOEs (i − j > 10)a | 540 |
| short range interresidue NOEs (i − j </= 10)a | 492 |
| restrictive intraresidue NOEsa | 53 |
| residual dipolar couplings (C12E5) | 58 |
| residual dipolar couplings (phage) | 102 |
| J-coupling restraints (periodic) | 119 |
| dihedral angle restraintsb | 256 |
| 13C chemical shift restraints | 155 |
| Metal-ligand bond lengths & angles from XAS | 14 |
| total number of experimental restraints | 1789 |
| Modeling restraints | |
| hydrogen bonds in regular secondary structurec | 137 |
| Salt bridges and non-regular hydrogen bondsc | 10 |
| total number of modeling restraints | 147 |
| Structural statistics | |
| RMS deviations from experimental restraints | |
| ave. distance restraint violation (Å)d | 0.071 |
| ave. no. of distance violations > 0.5 Å per structured | 0.46 |
| ave. no. dihedral violations > 5° per structure | 0.53 |
| Goodness of fit of RDCs to anisotropy tensors | |
| phage | R=0.99, Q=0.14 |
| C12E5 | R=0.96, Q=0.26 |
| RMS deviations from ideal geometry | |
| Bonds (Å) | 0.003 |
| Angles (degrees) | 0.536 |
| Ramachandran analysise | |
| residues in most favored regions | 87.7% (85.8%) |
| residues in additional allowed regions | 12.0% (12.6%) |
| residues in generously allowed regions | 0.3% (1.3%) |
| residues in dissallowed regions | 0.0% (0.2%) |
| Coordinate precision (Å) | |
| backbone (residues 1-64, 73-157) | 0.68 |
| heavy atoms (residues 1-64, 73-157) | 1.07 |
NOEs were classified by relative intensity, with strong NOEs restrained to ≤ 3 Å, medium NOEs to ≤ 4 Å and weak NOEs to ≤ 5 Å. Intraresidue NOEs were used only if they provided a meaningful restriction. Ambiguities due to methyl protons and non-stereospecific assignments were accounted for using an r−6 averaging of distances between sets of protons defined by an NOE restraint. i and j refer to residue numbers.
Dihedral angle restraints were obtained by chemical shift analysis using TALOS (32).
Hydrogen bonds were determined by repeated occurrence in unrestrained structures. Salt bridges were inferred from close approach of oppositely charged residues.
Structures with no more than one NOE violation > 0.5 Å in the multiconformer loop between Leu 64 and Trp 73 were accepted as long as the Ramachandran diagram was reasonable.
Ramachandran statistics were calculated using PROCHECK_NMR (33) and excludes disordered C-terminal residues 157-179, which are not included in the PDB deposition. Values calculated without residues 65-72, for which minimal experimental restraints are available, are shown first, and values calculated including residues 65-72 are shown in parentheses.
After an initial (2000 step) energy minimization using bond, angle and harmonic restraints with reduced van der Waals repulsion terms, structures were heated to 3000 K for 5,000 steps of torsional dynamics using a 2 fs step size. The simulation temperature was then reduced gradually during annealing (12.5 K temperature changes of 50 steps each, 2 fs step size, final temperature = 25 K), while van der Waals repulsive terms, experimental database and harmonic restraints were gradually increased to their full values. Accepted structures gave no more than one NOE violation (see Table 1), good fits of RDCs to calculated order tensors and reasonable Ramachandran plots. Structural statistics are summarized in Table 1.
Structural data deposition
Coordinates and structural restraints for ARD′ (residues 1-156) have been deposited with the RCSB Protein Data Base (entry 2HJI). NMR resonance assignments for H98S ARD are available through the BioMagResBank database, accession number 7103.
Supplementary Material
Acknowledgments
This work was supported in part by the U. S. Public Health Service (R01-GM067786, TCP). The 600 MHz NMR spectrometer used was purchased via a grant from the National Science Foundation (DBI-9871130). Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this research (MJM). Trainee funding (SCC) was provided by the NIH Chemistry-Biology Interface Program, T32 GM08515.
References
- 1.deAraujo AFP, Pochapsky TC. Monte Carlo simulations of protein folding using inexact potentials: How accurate must parameters be in order to preserve the essential features of the energy landscape? Folding & Design. 1996;1:299–314. doi: 10.1016/S1359-0278(96)00043-0. [DOI] [PubMed] [Google Scholar]
- 2.Shakhnovich EI. Proteins with selected sequences fold into unique native conformation. Phys Rev Lett. 1994;72:3907–3910. doi: 10.1103/PhysRevLett.72.3907. [DOI] [PubMed] [Google Scholar]
- 3.Devlin GL, Bottomley SP. A protein family under ‘stress’ - Serpin stability, folding and misfolding. Frontiers In Bioscience. 2005;10:288–299. doi: 10.2741/1528. [DOI] [PubMed] [Google Scholar]
- 4.Malolepsza E, Boniecki M, Kolinski A, Piela L. Theoretical model of prion propagation: A misfolded protein induces misfolding. Proc Natl Acad Sci USA. 2005;102:7835–7840. doi: 10.1073/pnas.0409389102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wray JW, Abeles RH. A bacterial enzyme that catalyzes formation of carbon monoxide. J Biol Chem. 1993;268:21466–21469. [PubMed] [Google Scholar]
- 6.Dai Y, Wensink PC, Abeles RH. One protein, two enzymes. J Biol Chem. 1999;274:1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 7.Dai Y, Pochapsky TC, Abeles RH. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae. Biochemistry. 2001;40:6379–6387. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- 8.Dai Y. Brandeis University; Waltham, MA.: 1999. Ph. D. thesis. [Google Scholar]
- 9.Pochapsky TC, Pochapsky SS, Ju TT, Mo HP, Al-Mjeni F, Maroney MJ. Modeling and experiment yields the structure of acireductone dioxygenase from Klebsiella pneumoniae. Nature Struc Biol. 2002;9:966–972. doi: 10.1038/nsb863. [DOI] [PubMed] [Google Scholar]
- 10.Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Evolution of functional diversity in the cupin superfamily. TIBS. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- 11.Dunwell JM, Purvis A, Khuri S. Cupins: the most functionally diverse protein superfamily? Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Pochapsky TC, Pochapsky SS, Ju TT, Hoefler C, Liang J. A refined model for the structure of acireductone dioxygenase from Klebsiella ATCC 8724 incorporating residual dipolar couplings. J Biomol NMR. 2006;34:117–127. doi: 10.1007/s10858-005-5735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ko TP, Day J, Mcpherson A. Apr) The refined structure of canavalin from jack bean in two crystal forms at 2.1 and 2.0 Angstrom resolution. Acta Crystallogr D-Biol Crystallogr. 2000;56:411–420. doi: 10.1107/s0907444900002237. [DOI] [PubMed] [Google Scholar]
- 14.Xu Q, et al. Crystal structure of acireductone dioxygenase (ARD) from Mus musculus at 2.06 Å resolution. Proteins-Struct Funct Gen. 2006;64:808–813. doi: 10.1002/prot.20947. [DOI] [PubMed] [Google Scholar]
- 15.Al-Mjeni F, Ju T, Pochapsky TC, Maroney MJ. XAS investigation of the structure and function of Ni in acireductone dioxygenase. Biochemistry. 2002;41:6761–6769. doi: 10.1021/bi012209a. [DOI] [PubMed] [Google Scholar]
- 16.Sauter M, Lorbiecke R, OuYang B, Pochapsky TC, Rzewuski G. The immediate-early ethylene response gene OsARD1 encodes an acireductone dioxygenase involved in recycling of the ethylene precursor S-adenosylmethionine. Plant J. 2005;44:718–729. doi: 10.1111/j.1365-313X.2005.02564.x. [DOI] [PubMed] [Google Scholar]
- 17.Szajna E, Arif AM, Berreau LM. Aliphatic carbon-carbon bond cleavage reactivity of a mononuclear Ni(II) cis-beta-keto-enolate complex in the presence of base and O-2: A model reaction for acireductone dioxygenase (ARD) J Am Chem Soc. 2005;127:17186–17187. doi: 10.1021/ja056346x. [DOI] [PubMed] [Google Scholar]
- 18.Schlippe YVG, Hedstrom L. Is Arg418 the catalytic base required for the hydrolysis step of the IMP dehydrogenase reaction? Biochemistry. 2005;44:11700–11707. doi: 10.1021/bi048342v. [DOI] [PubMed] [Google Scholar]
- 19.Mo HP, Dai Y, Pochapsky SS, Pochapsky TC. H1, C13 and N15 NMR assignments for a carbon monoxide generating metalloenzyme from Klebsiella pneumoniae. J Biomol NMR. 1999;14:287–288. doi: 10.1023/a:1008396624784. [DOI] [PubMed] [Google Scholar]
- 20.Bagyinka C, Whitehead JP, Maroney MJ. An X-ray-absorption spectroscopic study of nickel redox chemistry in hydrogenase. J Am Chem Soc. 1993;115:3576–3585. [Google Scholar]
- 21.Ressler T. WinXAS: A new software package not only for the analysis of energy-dispersive XAS data. J De Physique IV. 1997;7:269–270. [Google Scholar]
- 22.Lehnert R, Seel F. Crystal structure of iron(II) derivative of imidazole. Z Anorg Allgem Chem. 1978;444:91–96. [Google Scholar]
- 23.Kang B, Cai J. Formation and crystal structure of (Et4N)2[Fe(SC6H4CH3-p)4] Jiegou Huaxue. 1985;4:119–122. [Google Scholar]
- 24.Davidson G, Clugston SL, Honek JF, Maroney MJ. An XAS investigation of product and inhibitor complexes of Ni-containing GlxI from Escherichia coli: Mechanistic implications. Biochemistry. 2001;40:4569–4582. doi: 10.1021/bi0018537. [DOI] [PubMed] [Google Scholar]
- 25.Muhandiram DR, Farrow NA, Xu GY, Smallcombe SH, Kay LE. Dec) A Gradient C13 NOESY-HSQC Experiment for recording NOESY spectra of C13-labeled proteins dissolved in H2O. J Magn Reson B. 1993;102:317–321. [Google Scholar]
- 26.Bartels C, Xia TH, Billeter M, Guntert P, Wuthrich K. The program Xeasy for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 27.Kuszewski J, Gronenborn AM, Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Prot Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuszewski J, Clore GM. Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J Magn Reson. 2000;146:249–254. doi: 10.1006/jmre.2000.2142. [DOI] [PubMed] [Google Scholar]
- 29.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 30.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comp Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 32.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, Rullmann JAC, Macarthur MW, Kaptein R, Thornton JM. Aqua and Procheck-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 1.Dai Y, Wensink PC, Abeles RH. One protein, two enzymes. Journal Of Biological Chemistry. 1999;274:1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 2.Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L. X-Ray Absorption-Spectroscopy of Iron-Tyrosinate Proteins. Journal of the American Chemical Society. 1984;106:1676–1681. [Google Scholar]
- 3.Randall CR, Zang Y, True AE, Que L, Charnock JM, Garner CD, Fujishima Y, Schofield CJ, Baldwin JE. X-Ray-Absorption Studies of the Ferrous Active-Site of Isopenicillin N-Synthase and Related Model Complexes. Biochemistry. 1993;32:6664–6673. doi: 10.1021/bi00077a020. [DOI] [PubMed] [Google Scholar]
- 4.Bertini I, Briganti F, Mangani S, Nolting HF, Scozzafava A. X-Ray-Absorption Studies on Catechol 2,3-Dioxygenase from Pseudomonas-Putida Mt2. Biochemistry. 1994;33:10777–10784. doi: 10.1021/bi00201a027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.