

Abstract
Mitochondria are principal mediators of apoptosis and thus can be considered molecular targets for new chemotherapeutic agents in the treatment of cancer. Inhibitors of mitochondrial complex I of the electron transport chain have been shown to induce apoptosis and exhibit antitumor activity. In an effort to find novel complex I inhibitors which exhibited anti-cancer activity in the NCI’s tumor cell line screen, we examined organized tumor cytotoxicity screening data available as SOM (self-organized maps) (http://spheroid.ncifcrf.gov) at the Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI). Our analysis focused on an SOM cluster comprised of compounds which included a number of known mitochondrial complex I (NADH:CoQ oxidoreductase) inhibitors. From these clusters ten compounds whose mechanism of action was unknown were tested for inhibition of complex I activity in bovine heart submitochondrial particles (SMP) resulting in the discovery that five of the ten compounds demonstrated significant inhibition with IC50's in the nM range for three of the five. Examination of screening profiles of the five inhibitors toward the NCI’s tumor cell lines revealed that they were cytotoxic to the leukemia subpanel (particularly K562 cells). Oxygen consumption experiments with permeabilized K562 cells revealed that the five most active compounds inhibited complex I activity in these cells in the same rank order and similar potency as determined with bovine heart SMP. Our findings thus fortify the appeal of mitochondrial Complex I as a possible anti-cancer molecular target and provide a data mining strategy for selecting candidate inhibitors for further testing.
Keywords: Apoptosis, Mitochondria, NADH: Coenzyme Q Oxidoreductase, Enzyme Inhibition, Drug Discovery, Self-Organized Maps, K562 leukemia cells
1. INTRODUCTION
Mitochondria are central to apoptosis induction and the modulation of mitochondria with the intent to promote apoptosis has been projected as a target for mechanism-based cancer therapeutics [1,2]. Pro-apoptotic signals transduced to mitochondria from chemotherapeutic agents and/or cell surface receptors allow permeabilization of the mitochondrial outer membrane (MOM) and the release of intermembrane space proteins (IMS) (such as cytochrome c) into the cytosol, events which initiate, enable or amplify the cascade of caspase proteases to propagate apoptosis [3,4]. Cytochrome c has historically been recognized for its participation in the “shuttle” of electrons from complex III to complex IV within the mitochondrial electron transport system. However, the egress of cytochrome c to the cytosol, its participation in formation of the apoptosome and initiation of caspases may effect a decline in the efficiency of mitochondrial electron transport. For example, a major subunit of complex I (NADH: coenzyme Q oxidoreductase [EC 1.6.5.3]) [5] is specifically targeted for degradation by caspases [6]. In normal functioning cells, the production of ROS may result from the escape of single electrons from the ETS, primarily at the level of coenzyme Q, and their combination with molecular oxygen, a process successfully attenuated by anti-oxidant mechanisms within the cell. During the apoptotic process, however, damage to ETS complexes may generate elevated levels of reactive oxygen species (ROS) [7], augmenting concurrent caspase action [8], while subjecting the ETS to further insults and increases of ROS [9–11]. During this process mitochondrial DNA may be damaged by ROS and this may generate additional bioenergetic alterations [12]. These changes in mitochondrial homeostasis are especially critical in cancer cells, since these metabolically active cells are already under oxidative stress due to intrinsic production of ROS [13], and thus are more vulnerable to added stress by exogenous ROS-generating agents. Increases in ROS and the induction of apoptosis are also invoked by inhibitors of complex I enzyme activity, such as rotenone [14,15] , MPP+ [16], or the Annonaceous acetogenins, annonacin [17], squamocin [18], and bullatacin [19]. Tamoxifen also induces increases in ROS [20] and mitochondrial failure upon interaction with Complex I [21].
Complex I is the largest (~1000kD) and most complicated (41–46 subunits) of the electron transport components within the mitochondrial inner membrane [5,22]. Although no high-resolution 3D structure has been determined, low-resolution images have revealed that the complex I is boot (or L) shaped, and divided into the hydrophilic “promontory” or “peripheral” part which projects into the matrix, a connecting region, a hydrophobic membrane part and accessory subunits [5,23]. Complex I initiates mitochondrial electron transport by catalyzing the oxidation of water-soluble NADH and transfer of electrons via a flavin mononucleotide cofactor and several iron-sulfur clusters to reduce the lipid- soluble ubiquinone (Coenzyme Q or “Q”) . This process is concomitantly coupled to translocation of protons to the intermembrane space, contributing to the formation of the electrochemical proton gradient required for and resulting in synthesis of ATP [24].
The size and complexity of complex I offer multiple sites for interaction with inhibitors of enzyme action which could effect apoptosis. Sixty different families of compounds are known to inhibit complex I, some naturally occurring but many of commercial or synthetic origin, supporting the claim that it is difficult to precisely classify inhibition of this enzyme solely in terms of chemical structure [25,26]. Compounds of natural origin include the rotenoids (rotenone the most potent), piericidins, Annonaceous acetogenins, myxobacterial antibiotics (myxothiazol and stigmatellin), vanilloids (Capsaicin), ubiquinones (idebenone), and Tamoxifen [21]. Synthetic compounds include fenpyroximate, pyridaben, pethidine, and MPP+(1-methyl-4-phenylpyridinium ion) [26]. Evidence suggests that complex I possesses a unique wide (cavity-like) [27] inhibitor binding domain, with binding sites overlapping (at least) two ubiquinone reaction centers. A functional classification groups the inhibitors according to their kinetic behavior as either Type A /I (quinone antagonists-Annonaceous acetogenins, peiricidin A, Idebenone), Type B/II (semiquinone antagonists-rotenone), or Type C (quinol antagonists-stigmatellin,capsaicin, myxothiazol) [28–30].
Recently, the Developmental Therapeutics Program of the National Cancer Institue (NCI) used a unique set of computational tools to analyze and sort the complete set of NCI’s publicly available antitumor drug-screening data, incorporating structural chemotypes as well as biological data into self-organized maps (SOM’s). Data mining of the SOM reorganized data revealed clusters (as loci) of compounds which exhibit similar toxicity profiles toward the NCI’s panel of ~80 cell lines. For some compounds within clusters sharing similar cytotoxic profiles, experimental investigation has validated that they manifest a common mechanism of action toward a particular molecular target [31]. Cases where compounds exhibit similar cytotoxic profiles, but lack experimental verification, represent potential opportunities for novel drug discoveries. Towards this end, we have identified SOM clusters which contained acknowledged complex I inhibitors, such as acetogenins, and proposed for investigation here studies to determine whether previously untested compounds within these clusters also inhibit mitochondrial complex I. Using this procedure, we have selected ten compounds sharing cytotoxic profiles with acetogenins and examined their capacity to inhibit mitochondrial complex I activity. We have herein used bovine heart submitochondrial particles (SMP) as the enzyme source for testing inhibition of NADH: CoQ oxidoreductase. Our data reveal that five of the ten chosen compounds exhibited significant inhibition of complex I activity, a statistic which strikingly attests to the utility of the SOM clustering for predicting a compound’s biological response and thereby dramatically improving the efficiency of discovery of compounds sharing a particular mode of action.
2. MATERIALS AND METHODS
2.1 .Reagents
Beef hearts were obtained from J.W. Trueth &Co., Baltimore, MD. Potassium cyanide was obtained from Mallinkrodt Baker, Inc., Magnesium chloride solution from Quality Biological Inc., Gaithersburg, MD., and Coenzyme Q1, Antimycin A, rotenone and all other chemicals were purchased from Sigma-Aldrich, St. Louis, MO.
2.2. Methods
a. SOM Analysis of NCI’s Tumor Cell Screen
The NCI’s tumor cell screen has functioned for over a decade to select potentially active compounds based on underlying mechanisms of growth inhibition (GI50)and cell killing [32–36]. This database consists of GI50 profiles for ~42 thousand compounds measured against ~80 immortalized tumor cell lines. Data of this and other types have been the focus of considerable statistical mining efforts [37–40] aimed largely at the identification of novel compounds, as potential candidates for development as anticancer agents. GI50 patterns across the NCI60 have been previously found to be an information rich source for establishing a compound’s mechanism of action (MOA) [37].
Recently our group extended the pioneering work of Paull et al. [37] by the development of a suite of data mining tools((http://spheroid.ncifcrf.gov) for addressing a range of bioinformatic and chemoinformatic issues [31,41,42]. Self-Organizing Maps (SOMs) [43] were used to cluster the NCI’s tumor screening data into major response categories [31] involving agents active in the cellular processes of mitosis, nucleic acid metabolism, and membrane function. The results generated by our SOM analysis provide a basis for assessing compounds in terms of apparent dependencies associated with a specific structural class and associated GI50 profiles and an experimentally verifiable MOA. In principle, the current (http://spheroid.ncifcrf.gov) web tools allow user-driven queries into each SOM cluster for visualization of cytotoxicity profiles and chemical (2D) structures. Utilizing these tools, the analysis proposed herein focused on the GI50 responses observed for the class of acetogenins and additional compounds that also shared this cytotoxic profile. The projected design constitutes a practical test of whether compounds sharing a similar response in a cell-based screen might also be revealed to share a similar biological MOA. Ten compounds were identified in this scheme, and proposed for testing as inhibitors of mitochondrial complex I.
b. Preparation of beef heart mitochondria (BHM)
Preparation of beef heart mitochondria is essentially that of Smith [44,45]. Beef heart is obtained from a slaughterhouse within 1–2hr of slaughter and all procedures are carried out at 2–4 °C. Briefly, the heart muscle (left ventricle) is trimmed and 300g is diced and passed through a precooled meat grinder (holes 0.5mm). The resulting mince is suspended in 400 ml 0.25M sucrose, 0.01 M Tris-Cl, pH 7.8, and the pH adjusted to ~pH 7.8 by addition of 1M Tris (pH 10.8). The initial buffer is removed by squeezing the suspension through cheesecloth, and 200g of the ground, neutralized heart tissue is resuspended in 400 ml of 0.25M sucrose, 1 mM Tris-succinate, 0.2 mM EDTA, 0.01 M Tris-Cl, pH 7.8 (Smith Sucrose solution). The suspension is placed in a pre-cooled Waring blender and homogenized in three bursts of 5 sec, 20 sec, and 5 sec. The homogenate is readjusted to ~ph 7.8, and is centrifuged for 20 min at 1200 X g (Sorvall RC5B, SS34 rotor) to pellet the unruptured tissue and nuclei. The supernatant is decanted, filtered through cheesecloth, then centrifuged for 15 min at 26,000 X g (Sorvall RC5B, SS34 rotor). The resulting mitochondrial pellet is resuspended in 50 ml Smith Sucrose solution and homogenized with two passes (600 rpm, Talboys Model 102 motorized stirrer, Talboys Engineering Corp., Thorofare, N.J) of a Teflon-glass Potter-Elvejhem homogenizer (clearance 0.006 in). Total volume is brought to ~140 ml and the suspension is centrifuged for 30 min at 12,000 X g (Sorvall). The mitochondrial pellet is resuspended in a small volume (less than 10 ml) Smith Sucrose buffer and the protein determined (BCA Protein Assay, Pierce). The beef heart mitochondria (BHM) are stored at a concentration >10 mg/ml at −80°C.
c. Preparation of submitochondrial particles (SMP)
The SMP preparation protocol is adapted from methods described by Thierbach [46] and Pallotti [45]. Briefly, the beef heart mitochondrial preparation (BHM) is thawed and suspended to 10 mg/ml protein, sonicated on ice 10 times, with 30 sec cooling intervals, for 15 sec each (continuous, maximum output with a microprobe; Model W-225R, Ultrasonics, Inc. Plainview, N.Y.). The sonicated suspension is centrifuged at 12,000x g for 10 minutes (Sorvall RC5B, SS34 rotor), the pellet disgarded, and the supernatant collected and centrifuged at >100,000 x g for 45 minutes ( Beckman Optima XL 70, 70ti rotor). The SMP pellet is resuspended in Smith Sucrose buffer, the protein determined (BCA Protein Assay, Pierce),then aliquoted at >10mg/ml and stored at −80°C.
d. Assay for Complex I NADH:Ubiquinone Oxidoreductase Activity in Beef Heart Submitochondrial Particles
Mitochondrial Complex I activity in beef heart SMP was determined by monitoring the rate of absorbance decrease at 340 nm due to NADH (ε=6.81 mM−1 cm−1) [47] oxidation by Coenzyme Q1 [48], with 425 nm as the reference wavelength. The rate of absorbance decrease at 620 nm representing 2,6 Dichlorophenolindophenol (DCIP, ε=19.1 mM−1 cm−1) reduction by Coenzyme Q1H2served as an additional measurement of activity [49–51]. The assay is performed basically as described by Birch-Machin [50] in a 1 cm temperature-controlled cuvette (30°C), stirring, in a final volume of 2.05ml using the Hewlett-Packard Model 8453 Diode-array Spectrophotometer in Kinetics mode.
Assay Buffer contains 0.025M KPO4 ,pH 7.2, 5 mM MgCl2, 2.5 mg/ml bovine serum albumin (BSA, fatty acid free), 50μM Coenzyme Q1, 70μM 2,6 Dichlorophenolindophenol (DCIP), Antimycin A (2μg/ml), 2mM potassium cyanide (KCN). Rotenone (2μg/ml) is added to the Assay Buffer for the rotenone control. Two ml of Assay Buffer, 10μl DMSO (Dimethyl sulphoxide-DMSO control) or 10μl of inhibitor in DMSO, and mitochondria (30–50μg of SMP protein, activated by freeze-thawing two times) is added to the cuvette and incubated for two minutes. The addition of 0.13mM NADH initiates the complex I reaction, and absorbance readings continue for three minutes. Since the assay is performed with saturating concentrations of NADH , CoQ1, and DCIP, the enzyme velocity is constant and product formation linear with time. Specific activity is reported as μmoles/min/mg SMP based on the rates (Au/sec) of either NADH oxidation or DCIP reduction obtained with the Kinetics Program of the Hewlett Packard Spectrophotometer. Total activity of the DMSO control minus the activity in the presence of rotenone constitutes 100% rotenone-sensitive NADH:ubiquinone oxidoreductase activity. Dose response curves of the five best complex I inhibitors were obtained using dilutions at final concentrations of 20μM to 1nM in the complex I assay described above and IC50's were determined by non-linear regression analysis (GraphPad Prizm).
e. Polarographic Assay (Oxygen Electrode)
Digitonin is added to 2ml (20x106 cells in “Attardi” Buffer A [52] -- 10mM MgCl2, 250 mM Sucrose, 20 mM HEPES, pH 7.1) at .005% (1μl of 10% solution to two mls). The mixture is gently mixed for ~15sec, then cells are suspended to 10 ml with Buffer A (without digitonin [53]), then centrifuged 800xg, 5min (T40 rotor, Jouan centrifuge). Cell pellets are resuspended in 2 ml Buffer A (aerated and pre-equilibrated to 37°C), and 1ml of the suspension is added to each of two Hansatech Oxygraph (Hansatech Instruments Ltd.) oxygen electrode water-jacketed chambers connected to a circulating water bath at 37°C with integral constant magnetic stirring. The chamber top is inserted and all additions are made with Hamilton syringes through the slotted top. ADP is added at 0.5mM, along with 2mM KPi, pH 7.1. The NADH substrates for Complex I, 5mM malate (pH 7.1) and 10mM glutamate are added and oxygen consumption is recorded. Drugs are added (1μl of 1mM stock in DMSO) and immediate interference with oxygen consumption occurs with inhibition of Complex I. Succinate addition (5mM) results in the resumption of oxygen consumption which indicates that complex III is still active. Addition of Antimycin A inhibits complex III activity and oxygen consumption (and oxidative phosphorylation) is stopped. The change in rate of oxygen consumption is recorded with a custom Windows® data acquisition and instrument control program provided by Hansatech, and rates of oxygen consumption after the addition of each reagent are computed using this software.
f. Preparation of Inhibitor solutions
Each candidate inhibitor (1–2mg) was weighed using a Cahn microbalance and DMSO was added to yield a 20mM solution. Immediately prior to the initial testing of all ten compounds, the 20mM concentrates were diluted into a series of stock solutions so that a 10μl aliquot would be diluted to 0.5, 1.0 or 5μM final concentration in the 2.0ml assay buffer . Those compounds which exhibited significant inhibitory activity at 0.5μM final concentration were re-tested over a range of at least eight inhibitor concentrations (final concentrations ranging from 5μM to 1nM) in order to determine their IC50's For all of these compounds the stock solution in 100%DMSO was diluted 1:200 when added to the assay buffer.
3. RESULTS
Complex I in bovine SMP’s was measured using a coupled assay involving NADH, hydrophilic Coenzyme Q1 as the electron acceptor for NADH oxidation, and DCIP [26,49]. In the assay, electrons were passed from NADH to CoQ1, generating CoQ1H2 which then proceeded to reduce DCIP , changing its color from the blue (Oxidized) form to the colorless (Reduced) form (Figure 1.) The Coenzyme Q1 was chosen as the initial exogenous electron acceptor for NADH because it is rotenone sensitive in aqueous buffer solutions, yields one of the lowest rotenone insensitive rates of the less hydrophobic coenzyme Q analogs, and generates a high enzymatic rate [12,49,54]. DCIP was added to accelerate the rates and increase specificity for NADH dehydrogenase [55–57] and would also facilitate the development of a plate assay for use in a high-throughput screen, should that prove advantageous [49]. CoQ1H2 was prevented from passing its electrons to complex III of the electron transport chain by the addition of antimycin A, an inhibitor of complex III. Complex IV was also inhibited by the inclusion of KCN in the reaction buffer.
Figure 1.
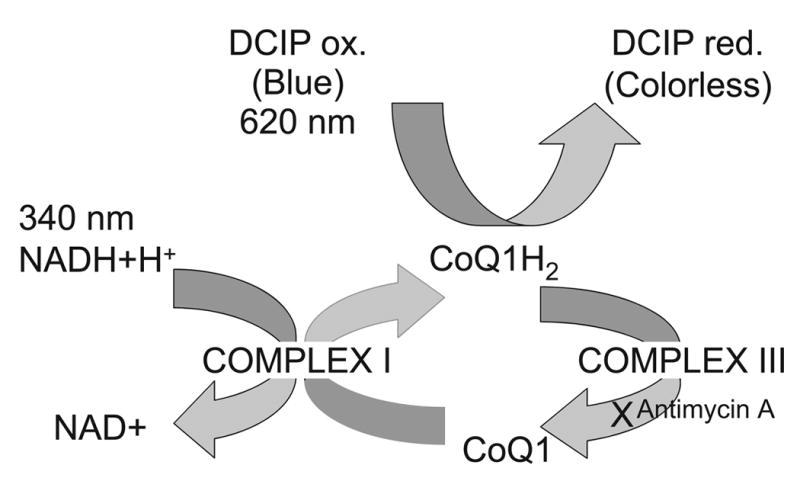
The electron transport mechanism in the bovine sub-mitochondrial particles (SMP) complex I assay (as in Jewess et al [49]). In a buffer enriched with excess NADH, Coenzyme Q1 and DCIP, SMP containing bovine complex I enzyme oxidize NADH to NAD+ and reduce Coenzyme Q1 to Coenzyme Q1H2, resulting in a decline of NADH absorbance at 340nm. Coenzyme Q1H2 reduces DCIP, resulting in a decrease in absorbance at 620nm. The decrease in absorbance at 340nm, and also at 620nm continues until all exogenously added NADH is oxidized. Antimycin A (an inhibitor of complex III) prevents Coenzyme Q1H2 from passing reducing components to complex III, and KCN (an inhibitor of complex IV) prevents cytochrome oxidase activity of complex IV.
The reaction occurred under zero-order steady state conditions with respect to coenzyme Q1 with rates initially recorded as AU (Absorbance Units)/sec). Complex I (NADH: CoQ oxidoreductase) activity was initiated by the addition of NADH and enzyme rates monitored simultaneously following NADH oxidation at 340nm and DCIP reduction at 620nm, with a slight delay between the start of NADH oxidation and that of DCIP reduction. Rates are expressed as specific activities (μmoles/min/mg SMP protein) for NADH oxidation and DCIP reduction both in the presence or absence of rotenone or inhibitor. The maximal control rate for complex I activity in the absence of rotenone varied between 1.03 and 1.203 μmolesNADH/min/mg SMP and agrees well with published rates for NADH oxidation with CoQ1 (0.965μmol/min/mg SMP [48]). The slightly higher rate we recorded may be an acceleration in NADH oxidase due to the inclusion of DCIP as the final electron acceptor. In the presence of rotenone, the rate of complex I activity decreased to ~0.167 μmolesNADH/min/mg. A small amount of apparent enzyme activity was expected even with rotenone due to background NADH oxidation (e.g. by cytochrome b5 reductase), and was termed the ”rotenone-insensitive” activity. The activity obtained from the DMSO control when no inhibitor was present minus the “rotenone-insensitive” activity is termed the 100% “rotenone-sensitive” activity. The specific activities at three inhibitor concentrations (0.5,1 and 5μM) was divided by the “rotenone-sensitive” specific activity and presented as “% Control” in Figure 2. Some of the agents tested inhibited all enzyme activity, and this complete inhibition is shown by deflection below the 0% line. Five (Figure 3) of the ten compounds tested demonstrated significant inhibition of complex I activity and were chosen for further dose-response analysis. Four of these five most potent compounds inhibited the control more than 70% at the lowest concentration of inhibitor. Subsequent dose-response analysis was determined for NADH oxidation (data not shown) and DCIP reduction (Figure 4) with the IC50's for the five inhibitors determined for both dose-response curves. For increased clarity the graph shows only the DCIP set of dose response curves, but the IC50 for each inhibitor reported on the graph represents the average of computations for both NADH oxidation and DCIP reduction.
Figure 2.
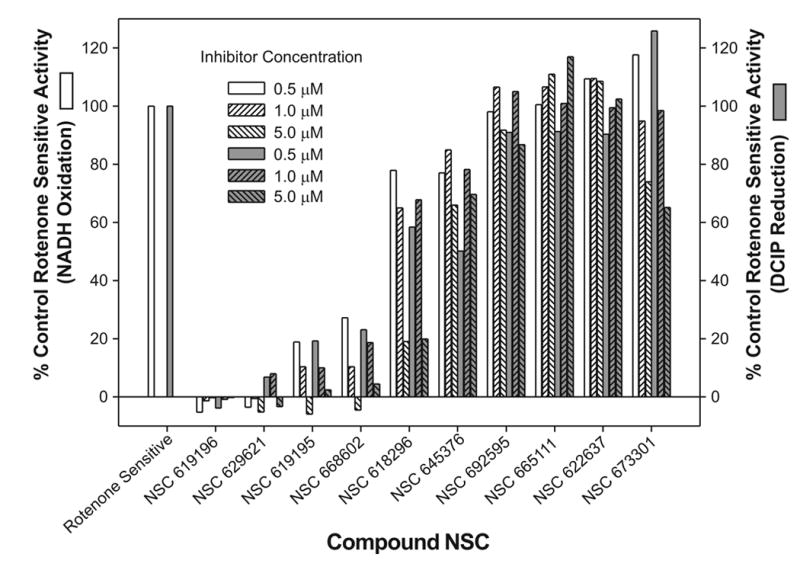
The % Control complex I activity as monitored by NADH oxidation (white columns) and DCIP reduction (grey columns) for each of the ten compounds tested at concentrations of 0.5, 1.0 and 5.0μM. Each compound was incubated with the SMP for at least two minutes before the addition of NADH. The specific activity of the complex I enzyme in the presence of the three concentrations of compounds was compared to total rotenone sensitive activity (DMSO control-Rotenone control).
Figure 3.
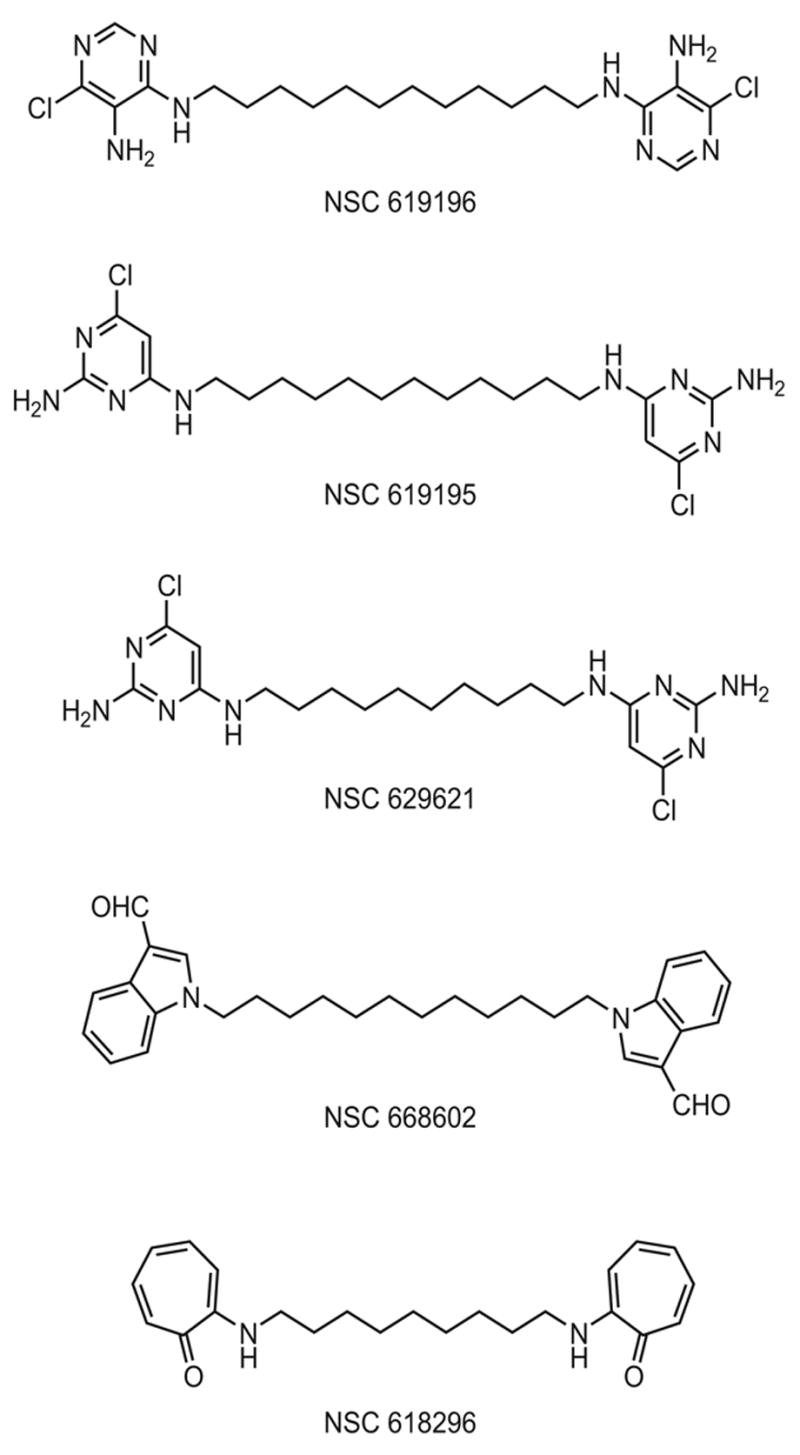
Structures of the five most potent inhibitors. NSC 619196, N4-(12-((5-amino-6-chloro-4-pyrimidinyl)amino)dodecyl)-6-chloro-4,5-pyrimidinediamine; NSC 619195,N4-(12-((2-amino-6-chloro-4-pyrimidinyl)amino)dodecyl)-6-chloro-2,4-pyrimidinediamine; NSC 629621, N4-(10-((2-amino-6-chloro-4-pyrimidinyl)amino)decyl)-6-chloro-2,4-pyrimidinediamine; NSC 668602, 1-(12-(3-formyl-1H-indol-1-yl)dodecyl)-1H-indole-3-carbaldehyde; and NSC 618296,2-((9-((7-oxo-1,3,5-cycloheptatrien-1-yl)amino)nonyl)amino)-2,4,6-cycloheptatrien-1-one.
Figure 4.
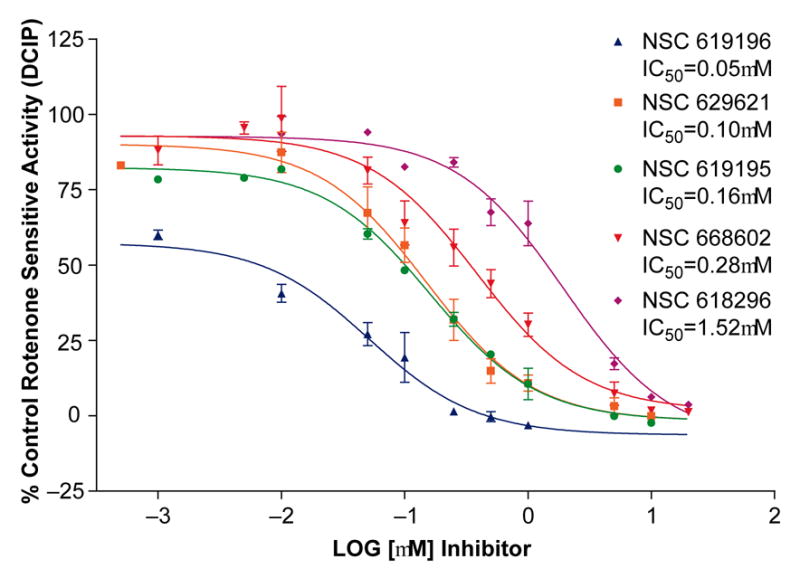
Dose response curves for the five strongest inhibitors discovered by the survey in Figure 1. Inhibitor concentrations for NSC 619196, 629621, 619195 and 668602, and 618296 ranged from an initial concentration of 20μM to 1nM final concentration in the complex I assay. Inhibitors were incubated with the SMP for at least two minutes prior to addition of NADH to initiate the assay , then NADH oxidation and DCIP reduction was monitored for several minutes and slopes at each concentration of inhibitor were used to calculate specific activities. The IC50's obtained by the dose -response curves for each compound were obtained by non-linear regression analysis, one-site competition, using GraphPad Prizm. The specific activity of complex I enzyme in the presence of the serial dilutions of the compounds was divided by maximal (total) rotenone sensitive specific activity (DMSO control DCIP activity-Rotenone control DCIP activity) to obtain calculations for % Control (n=3).
The cytotoxicity profiles toward the NCI 60-cell line cancer screen revealed that the five inhibitors were particularly cytotoxic to leukemia cells, especially the K562 cell line. We decided to determine if the ten original compounds would show a pattern of inhibition similar to that with bovine heart SMP’s once Complex I inhibition was measured by effects on oxygen consumption in K562 cells. Complex I inhibitors should decrease oxygen consumption and oxidative phosphorylation through the mitochondrial electron transport system when NADH generating substrates, such as malate or glutamate, are added to the cellular milieu after the plasma membrane is disrupted by digitonin permeabilization. The digitonin-permeabilized K-562 cells consumed oxygen at the rate (avg) of ~22nmol/ml/min for 1x107 K562 cells upon addition of malate, glutamate. When rotenone, as control, and the ten compounds were added (in individual experiments at a concentration of 1μM), oxygen consumption became severely limited only by rotenone and the five compounds we had determined to be complex I inhibitors, e.g. NSC 619196 (see Table I). The other five non-inhibitors (e.g. NSC 622637) had no effect on oxygen consumption. Succinate addition produces FADH from the TCA cycle which enters the electron transport system at complex II; when added to the cells, oxygen consumption in all cases resumed (avg ~32 nmol/ml/min), demonstrating that neither complex II nor complex III (nor any latter portion of the ETS) was inhibited. Although it has often been shown that complex I inhibitors may also inhibit complex III ( [58]), these results indicate that the inhibitors we discovered are exclusively inhibiting complex I of the ETS. Dose-response curves for the five inhibitors also confirmed that NSC 619196 was the most potent, and that the other four were in similar rank order as that determined with bovine heart SMP’s (Figure 5).
Table I. Effect on the Rate of Oxygen Consumption (nmol/ml/min) (% of Maximal Rate) In K562 Leukemic Cells of Compounds Tested for Complex I Inhibition.
This table reports oxygen electrode data based upon the specific activity (nmol/ml/min) of the rates of oxygen consumption determined when NADH producing substrates (malate, glutamate) are added to the chamber of digitonin-permeabilized K562 cells, and the effect on the slopes when the Complex I inhibitors (and non-inhibitors) are added. The five inhibitors severely limit oxygen consumption, and the non-inhibitors have no effect. Succinate, when added, enters the ETC via complex II, and thus maximal rates of oxygen consumption resume. As expected, Antimycin A eliminates oxygen consumption as it is a powerful inhibitor of Complex III. This data shows that the complex I inhibitors are inhibiting complex I exclusively. If they were inhibitors of complex II or III, the addition of succinate would not have stimulated the resumption of oxygen consumption through the electron transport system.
| Oxygen Consumption as % of Malate, Glutamate Rate
|
|||||
|---|---|---|---|---|---|
| Add Complex I | Name or NSC# of Compound now added to Chamber | Compound at 1 μM | Compound at 3 μM | Add Complex II | + Antimycin A Oxygen Consumption at % Succinate Rate |
| Substrates; Malate, Glutamate : All Chambers with Cells are at Maximal Oxygen Consumption, i.e. 100%=22.03 nmol/ml/min (AVG) | Rotenone | 6.5% | Substrate; Succinate, Return of All Chambers with Cells to Maximal Oxygen Consumption, i.e. 100%=31.6 nmol/ml/min (AVG) | 1.1% | |
| 619196 | 2.3% | <0 | |||
| 619195 | 4.3% | <0 | |||
| 629621 | 0.7% | <0 | |||
| 668602 | 54.7% | 10.5% | 0.83% | ||
| 618296 | 26.6% | 17.7% | 0.27% | ||
| 673301 | 95.3% | <0 | |||
| 665111 | 92.5% | <0 | |||
| 622637 | 100% | <0 | |||
| 692595 | 93.6% | <0 | |||
| 645376 | 97.8% | 3.5% | |||
Figure 5.
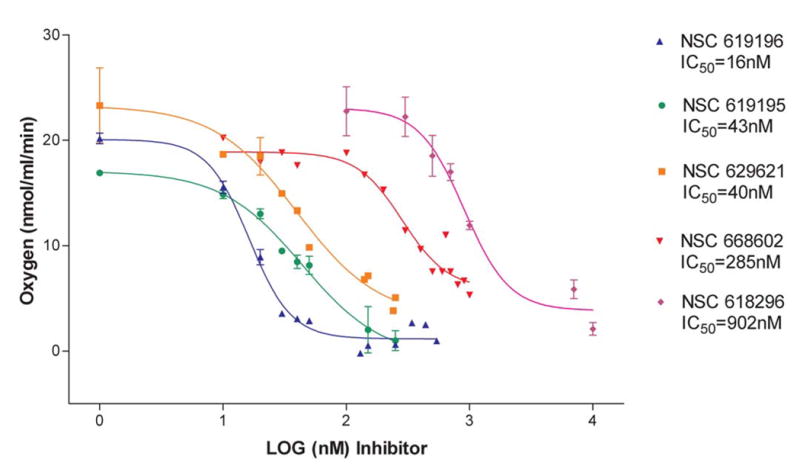
Dose response curves for Complex I inhibition of K562 Cells by the five strongest inhibitors confirmed by Table I. Dose response curves are based upon the change in rate of oxygen consumption when increasing concentrations (range=1nM to 1mM) of inhibitors of Complex I are added, after 1x107 cells have been permeabilized with digitonin and energized with Complex I substrates. The IC50's obtained by the dose -response curves for each compound were obtained by non-linear regression analysis, one-site competition, using GraphPad Prizm. n=2–4 separate experiments for each concentration.
Under normal circumstances in the cell, the main source of reactive oxygen species is the mitochondrial electron transport system when single electrons pass to oxygen from reduced substrates entering the chain at complex I and III [59]. Mitochondrial complex I inhibitors can stimulate increased production of reactive oxygen species [15], which may signal mitochondrial permeability transition and/or apoptosis. Preliminary experiments with NSC 619196, using rotenone and the acetogenin Bullatacin as controls, indicated that reactive oxygen species (H2O2) increased in the presence of all three compounds (data not shown).
Regarding structure, the five tested compounds that showed the most significant inhibition of Complex I, NSC 619196, NSC 619195, NSC 629621, NSC 668602 and NSC 618296 are barbell-shaped, possessing aromatic cyclic and heterocyclic end groups separated by 9, 10 or 12 saturated carbon spacers or “handles” (Figure 3). Three of the most potent inhibitors displayed pyrimidinediamine headgroups (NSC’s 619196, 619195 and 629621). NSC 619195 and NSC 629621, which possess spacer chain lengths of 12C (dodecyl) and 10C (decyl), respectively, have identical headgroups, very similar IC50 ‘s (629621=0.10μM and 619195=0.16μM), and a Tanimoto index of 0.99 [60,61]. The most potent compound, NSC 619196 (IC50 of ~50nM), also has a 12C spacer and head groups which differ from NSC 619195 and 629621 only in the relative placement of Cl- and NH2, an alteration in substitutions which lowers the Tanimoto index between NSC 619195 and NSC 619196 to 0.87. Furthermore, although the forth most potent compound, NSC 668602 ( IC50 0.28μM) possesses a 12C spacer, its head group, an indole-3-carboxaldehyde [62,63] markedly differs from the pyrimidinediamines. A fifth compound, NSC 618296, whose IC50 is 1.52μM differs from all the others in possessing a cycloheptatrienone head group with a 9 carbon spacer.
4. DISCUSSION
The DTP self-organized maps (SOM’s) were constructed not only to organize large collections of drug cytotoxicity patterns observed toward the panel of ~80-cancer cell lines, but to simplify the process of finding new chemotherapeutic agents whose mechanism of action is unknown and co-located in loci which contain compounds with known molecular mechanism. Within a cluster may be found structural analogs and/or diverse structures which nevertheless demonstrate similar cytotoxic patterns, inferring that even structurally dissimilar compounds may be directed toward the same molecular target. Considering that known complex I inhibitors encompass a large variety of chemical families [26], compounds of markedly variant structure but unknown mechanism which sort to loci of known inhibitors of complex I may also share the ability to inhibit mitochondrial complex I activity.
In this report we examined a group of ten structurally varied compounds whose mechanism of action is unknown but can be found in the same SOM map loci as known complex I inhibitors. Of the ten tested, we describe the discovery of five new complex I inhibitors by utilizing an assay with bovine sub-mitochondrial particles (SMPs), and confirmed their activity in cancer cells by measuring interruption of oxygen consumption in the K562 leukemic cell line. All five compounds display certain structural similarities, i.e. they are all “barbell” shaped with cyclic or heterocyclic ring moieties separated by a saturated carbon (alkyl) spacer. These new compounds share attributes of many known inhibitors of complex I which mimic coenzyme Q (“Q”), usually with a cyclic “head” corresponding to the ubiquinone ring and a hydrophobic “tail” [26]. Compounds with similarity to “Q” have been shown to be Type I/A (quinone antagonists) inhibitors according to the classification cited by Okun, and include the annonaceous acetogenins and the aminopyrimidines [30]. Three of the more effective new inhibitors have pyrimidinediamine cyclic head groups (NSC’s 619195, 619196, and 629621) which bear some structural similarity to the aminopyrimidines such as the AE F117233 4(cis-4-tert-butylcyclohexylamino)5-chloro-6-ethylpyrimidine), a robust Type I/A complex I inhibitor [30]. The most potent among the new inhibitors identified in this study (NSC 619196) was included within the same SOM cluster (k20.15) as the acetogenins bullatacin, NSC 615484, asimicin,NSC 609700, and annonacin, NSC 606194, lipophilic compounds which contain one or more tetrahydrofuran (THF) rings, two alkyl chains, and a γ-lactone ring [64–66]. It is notable that each of the barbell shaped compounds we identified as inhibitors has an alkyl spacer between the ring moieties which possibly contributes to potency: three have 12C, one has 10C, and the least potent has 9C. The γ-lactone ring of certain acetogenins has been described as interacting directly with the complex I binding site [67] and experiments with synthesized acetogenins indicated that the γ-lactone and the THF rings act in a cooperative manner on the enzyme, but both functional units work efficiently only if separated by an alkyl spacer of ~10–13 carbons [68,69]. It would appear that as acetogenins partition into lipid membranes, the alkyl spacer apparently is necessary to place the two ring moieties in optimal spatial position for enzyme inhibition. A similar function would be presumed for the alkyl spacer in the more potent of the new compounds we have identified.
Recently it has been shown that the γ-lactone ring of acetogenins may be substituted with a ubiquinone ring and still retain complex I inhibitory activity [70], indicating that there is broad specificity or loose recognition of inhibitor ring moieties, possibly attributed to the “cavity -like structure” of the enzyme’s ubiquinone binding domains [69,71]. In addition, evidence for at least two, perhaps three Q binding sites has been advanced, making it possible for inhibitors from structurally diverse chemical classes to interact specifically with different Q binding sites [71–73]. The compounds we have identified are likely to be categorized as Type I/A inhibitors in the classification by Okun [30], bearing resemblance at least in part to the acetogenins and aminopyrimidines.
Novel complex I inhibitors have now been identified in loci known to contain other compounds acknowledged to be potent complex I inhibitors. In addition to identifying these new inhibitors which may serve to promote apoptosis, especially in chronic myeloid leukemic K562 cells which have demonstrated resistance to apoptosis [74,75], the present study has validated the hypothesis (in vitro recapitulates in silico) that the computational procedures used to reorganize the NCI drug-screening data can in fact identify clusters of compounds with similar cellular reactivity, thus significantly narrowing the search for novel agents which share common mechanisms of action.
Acknowledgments
6. ACKNOWLEDGEMENTS
The authors are grateful to Dr. Ronald Felsted and Dr.Gary Fiskum for helpful discussions.
ABBREVIATIONS
- NCI
(National Cancer Institute)
- DTP
(Developmental Therapeutics Program)
- SOM
(Self-Organized Maps of the Developmental Therapeutics Program of the National Cancer Institute)
- ETS
(Mitochondrial Electron Transport System)
- MOM
(Mitochondrial Outer Membrane)
- IMS
(Mitochondrial Intermembrane Space)
- ROS
(Reactive Oxygen Species)
- CoQ or “Q”
(Coenzyme Q)
- CoQ1
(Coenzyme Q1)
- MPP+
(1-methyl-4-phenylpyridinium ion)
- bis THF alkane
(Tetrahydrofuran)
- SMP
(bovine heart Sub-Mitochondrial Particles)
- BHM
(Bovine Heart Mitochondria)
- DCIP
(2,6-Dichlorophenolindophenol)
- DMSO
(Dimethyl sulphoxide)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferreira CG, Epping M, Kruyt FAE, Giaccone G. Apoptosis: Target of cancer therapy. Clin Cancer Res. 2002;8:2024–2034. [PubMed] [Google Scholar]
- 2.Makin G. Targeting apoptosis in cancer chemotherapy. Expert Opin Ther Targets. 2002;6:73–84. doi: 10.1517/14728222.6.1.73. [DOI] [PubMed] [Google Scholar]
- 3.Kaufman SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 4.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 5.Yano T. The energy-transducing NADH: quinone oxidoreductase, complex I. Mol Aspects Med. 2002;23:345–368. doi: 10.1016/s0098-2997(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 6.Ricci J, Muñoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of Complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Y, Jones DJ. Superoxide in apoptosis. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 9.Genova ML, Ventura B, Giuliano G, Bovina C, Formiggini G, Castelli GP, et al. The site of production of superoxide radical in mitochondrial complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001;505:364–368. doi: 10.1016/s0014-5793(01)02850-2. [DOI] [PubMed] [Google Scholar]
- 10.Ricci J, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol. 2003;160:65–75. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricci J, Waterhouse N, Green DR. Mitochondrial functions during cell death, a complex(I–V) dilemma. Cell Death Differ. 2003;10:488–492. doi: 10.1038/sj.cdd.4401225. [DOI] [PubMed] [Google Scholar]
- 12.Degli Esposti M, Ngo A, Ghelli A, Benelli B, Carelli V, McLennan H, et al. The interaction of Q analogs, particularly hydroxydecyl benzoquinone (Idebenone), with the respiratory complexes of heart mitochondria. Arch Biochem Biophys. 1996;330:395–400. doi: 10.1006/abbi.1996.0267. [DOI] [PubMed] [Google Scholar]
- 13.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 14.Clayton R, Clark JB, Sharpe M. Cytochrome c release from rat brain mitochondria is proportional to the mitochondrial functional deficit: implications for apoptosis and neurodegenerative disease. J Neurochem. 2005;92:840–849. doi: 10.1111/j.1471-4159.2004.02918.x. [DOI] [PubMed] [Google Scholar]
- 15.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 16.Brill LB, Bennett JPJ. Dependence on electron transport chain function and intracellular signaling of genomic responses in SH-SY5Y cells to the mitochondrial neurotoxin MPP+ Exp Neurol. 2003;181:25–38. doi: 10.1016/s0014-4886(02)00045-6. [DOI] [PubMed] [Google Scholar]
- 17.Yuan SF, Chang H, Chen H, Yeh Y, Kao Y, Lin K, et al. Annonacin, a mono-tetrahydrofuran acetogenin, arrests cancer cells at the G1 phase and causes cytotoxicity in a Bax- and caspase-3-related pathway. Life Sci. 2003;72:2853–2861. doi: 10.1016/s0024-3205(03)00190-5. [DOI] [PubMed] [Google Scholar]
- 18.Zhu X, Liu Z, Xie B, Li Z, Feng G, Xie H, et al. Involvement of caspase-3 activation in squamocin-induced apoptosis in leukemia cell line HL-60. Life Sci. 2002;70:1259–1269. doi: 10.1016/s0024-3205(01)01501-6. [DOI] [PubMed] [Google Scholar]
- 19.Chih H, Chiu H, Tang K, Chang F, Wu Y. Bullatacin, a potent antitumor annonaceous acetogenin, inhibits proliferation of human hepatocarcinoma cell line 2.2.15 by apoptosis induction. Life Sci. 2001;69:1321–1331. doi: 10.1016/s0024-3205(01)01209-7. [DOI] [PubMed] [Google Scholar]
- 20.Kallio A, Zheng A, Dahllund J, Heiskanen KM, Härkönen P. Role of mitochondria in tamoxifen-induced rapid death of MCF-7 breast cancer cells. Apoptosis. 2005 doi: 10.1007/s10495-005-2137-z. 3 October, 2005. E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 21.Moreira PI, Custódio J, Moreno A, Oliveira CR, Santos MS. Tamoxifen and estradiol interact with the flavin mononucleotide site of complex I leading to mitochondrial failure. J Biol Chem. 2006 doi: 10.1074/jbc.M510249200. ;e-pub. [DOI] [PubMed] [Google Scholar]
- 22.Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol Cell Proteomics. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Grigorieff N. Three-dimensional structure of bovine NADH:ubiquinone oxidoreductase (complex I) at 22 Å in ice. J Mol Biol. 1998;277:1033–1046. doi: 10.1006/jmbi.1998.1668. [DOI] [PubMed] [Google Scholar]
- 24.Yagi T, Matsuno-Yagi A. The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry. 2003;42:2266–2274. doi: 10.1021/bi027158b. [DOI] [PubMed] [Google Scholar]
- 25.Tormo JR, Gonzalez MC, Cortes D, Estornell E. Kinetic characterization of mitochondrial Complex I inhibitors using annonaceous acetogenins. Arch Biochem Biophys. 1999;369:119–126. doi: 10.1006/abbi.1999.1343. [DOI] [PubMed] [Google Scholar]
- 26.Degli Esposti M. Inhibitors of NADH-ubiquuinone reductase: an overview. Biochim Biophys Acta. 1998;1364:222–235. doi: 10.1016/s0005-2728(98)00029-2. [DOI] [PubMed] [Google Scholar]
- 27.Miyoshi H. Probing the ubiquinone reduction site in bovine mitochondrial complex I using a series of synthetic ubiquinones and inhibitors. J Bioenerg Biomembr. 2001;33:223–231. doi: 10.1023/a:1010735019982. [DOI] [PubMed] [Google Scholar]
- 28.Degli Esposti M, Ghelli A. The mechanism of proton and electron transport in mitochondrial complex I. Biochim Biophys Acta. 1994;1187:116–120. doi: 10.1016/0005-2728(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 29.Friedrich T, Van Heek P, Leif H, Ohnishi T, Forche E, Kunze B, et al. Two binding sites of inhibitors in NADH:ubiquinone oxidoreductase (complex I) Eur J Biochem. 1994;219:691–698. doi: 10.1111/j.1432-1033.1994.tb19985.x. [DOI] [PubMed] [Google Scholar]
- 30.Okun JG, Lümmen P, Brandt U. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase) J Biol Chem. 1999;274:2625–2630. doi: 10.1074/jbc.274.5.2625. [DOI] [PubMed] [Google Scholar]
- 31.Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Mining the National Cancer Institute's tumor-screening database: identification of compounds with similar cellular activities. J Med Chem. 2002;45:818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]
- 32.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, et al. Feasibility of a high-flux anticancer drug screen using s diverse panel of cultured human tumor cell lines. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 33.Monks A, Scudiero DA, Johnson GS, Paull KD, Sausville EA. The NCI anti-cancer drug screen: a smart screen to identify effectors of novel targets. Anticancer Drug Des. 1997;12:533–541. [PubMed] [Google Scholar]
- 34.Boyd MR. The NCI In Vitro Anticancer Drug Discovery Screen. In: Teicher BA, editor. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials and Approval. Vol. 1995. Totowa, NJ: Humana Press; pp. 23–41. [Google Scholar]
- 35.Shoemaker RH, Scudiero D, Melillo G, Currens MJ, Monks AP, Rabow AA, et al. Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr Top Med Chem. 2002;2:229–246. doi: 10.2174/1568026023394317. [DOI] [PubMed] [Google Scholar]
- 36.Monga M, Sausville EA. Developmental therapeutics program at the NCI: molecular target and drug discovery process. Leukemia. 2002;16:520–526. doi: 10.1038/sj.leu.2402464. [DOI] [PubMed] [Google Scholar]
- 37.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 38.Shi LM, Fan Y, Myers TG, O'Connor PM, Paull KD, Friend SH, et al. Mining the NCI anticancer drug discovery databases: genetic function approximation for the QSAR study of anticancer ellipticine analogues. J Chem Inf Comput Sci. 1998;38:189–199. doi: 10.1021/ci970085w. [DOI] [PubMed] [Google Scholar]
- 39.Shi LM, Fan Y, Lee JK, Waltham M, Andrews DT, Scherf U, et al. Mining and visualizing large anticancer drug discovery databases. J Chem Inf Comput Sci. 2000;40:367–379. doi: 10.1021/ci990087b. [DOI] [PubMed] [Google Scholar]
- 40.Blower PE, Yang C, Fligner MA, Verducci JS, Richman S, Weinstein JN. Pharmacogenomic analysis: correlating molecular substructure classes with microarray gene expression data. Pharmacogenomics J. 2002;2:259–271. doi: 10.1038/sj.tpj.6500116. [DOI] [PubMed] [Google Scholar]
- 41.Wallqvist A, Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Establishing connections between microarray expression data and chemotherapeutic cancer pharmacology. Mol Cancer Ther. 2002;1:311–320. [PubMed] [Google Scholar]
- 42.Wallqvist A, Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Linking the growth inhibition response from the National Cancer Institute's anticancer screen to gene expression levels and other molecular target data. Bioinformatics. 2003;19:2212–2224. doi: 10.1093/bioinformatics/btg302. [DOI] [PubMed] [Google Scholar]
- 43.Kohonen T. Self-Organizing Maps. Berlin, Germany: Springer Verlag; 1995. [Google Scholar]
- 44.Smith AL. Preparation, properties, and conditions for assay of mitochondria: slaughterhouse material, small-scale. Methods Enzymol. 1967;10:81–86. [Google Scholar]
- 45.Pallotti F, Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Meth Cell Biol. 2001;65:1–35. doi: 10.1016/s0091-679x(01)65002-7. [DOI] [PubMed] [Google Scholar]
- 46.Thierbach G, Reichenbach H. Myxothiazol, a new inhibitor of the cytochrome b-c1 segment of the respiratory chain. Biochim Biophys Acta. 1981;638:282–289. doi: 10.1016/0005-2728(81)90238-3. [DOI] [PubMed] [Google Scholar]
- 47.Santos DL, Moreno AJM, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicol Appl Pharmacol. 2002;185:218–227. doi: 10.1006/taap.2002.9532. [DOI] [PubMed] [Google Scholar]
- 48.Estornell E, Fato R, Pallotti F, Lenax G. Assay conditions for the mitochondrial NADH:coenzyme Q oxidoreductase. FEBS Lett. 1993;332:127–131. doi: 10.1016/0014-5793(93)80498-j. [DOI] [PubMed] [Google Scholar]
- 49.Jewess PJ, Devonshire AL. Kinetic microplate-based assays for inhibitors of mitochondrial NADH:ubiquinone oxidoreductase (Complex I) and succinate:cytochrome c oxidoreductase. Anal Biochem. 1999;272:56–63. doi: 10.1006/abio.1999.4168. [DOI] [PubMed] [Google Scholar]
- 50.Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Meth Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- 51.Lenaz G, Fato R, Baracca A, Genova ML. Mitochondrial quinone reductases: Complex I. Methods Enzymol. 2004;382:3–20. doi: 10.1016/S0076-6879(04)82001-9. [DOI] [PubMed] [Google Scholar]
- 52.Hofhaus G, Shakeley RM, Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- 53.Chretien D, Bénit P, Chol M, Lebon S, Rötig A, Munnich A, et al. Assay of mitochondrial respiratory chain complex I in human lymphocytes and cultured skin fibroblasts. Biochem Biophys Res Commun. 2003;301:222–224. doi: 10.1016/s0006-291x(02)03016-4. [DOI] [PubMed] [Google Scholar]
- 54.Nakashima Y, Shinzawa-Itoh K, Watanabe K, Naoki K, Hano N, Yoshikawa S. Steady-state kinetics of NADH:coenzyme Q oxidoreductase isolated from bovine heart mitochondria. J Bioenerg Biomembr. 2002;34:11–19. doi: 10.1023/a:1013862502185. [DOI] [PubMed] [Google Scholar]
- 55.Andreani A, Rambaldi M, Leoni A, Locatelli A, Ghelli A, Ratta M, et al. Thienylimidazo[2,1-b]thiazoles as inhibitors of mitochondrial NADH dehyrogenase. J Med Chem. 1995;38:1090–1097. doi: 10.1021/jm00007a006. [DOI] [PubMed] [Google Scholar]
- 56.Andreani A, Rambaldi M, Locatelli A, Leoni A, Ghelli A, Degli Esposti M. Thienylvinylindoles as inhibitors of mitochondrial NADH dehydrogenase. Pharm Acta Helv. 1994;69:15–20. doi: 10.1016/0031-6865(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 57.Majander A, Huoponen K, Savontaus M, Nikoskelainen E, Wikström M. Electron transfer properties of NADH:ubiquinone reductase in the ND1/3450 and the ND4/11778 mutations of the Leber hereditary optic neuroretinopathy (LHON) FEBS Lett. 1991;292:289–292. doi: 10.1016/0014-5793(91)80886-8. [DOI] [PubMed] [Google Scholar]
- 58.Degli Esposti M, Ghelli A, Crimi M, Estornell E, Fato R, Lenaz G. Complex I and Complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem Biophys Res Commun. 1993;190:1090–1096. doi: 10.1006/bbrc.1993.1161. [DOI] [PubMed] [Google Scholar]
- 59.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (Complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 60.Willett P, Barnard JM, Downs GM. Chemical similarity searching. J Chem Inf Comput Sci. 1998;38:983–996. [Google Scholar]
- 61.Martin YC, Kofron JL, Traphagen LM. Do structurally similar molecules have similar biological activity? J Med Chem. 2002;45:4350–4358. doi: 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- 62.Anderton MJ, Manson MM, Verschoyle RD, Gescher A, Lamb JH, Farmer PB, et al. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clin Cancer Res. 2004;10:5233–5241. doi: 10.1158/1078-0432.CCR-04-0163. [DOI] [PubMed] [Google Scholar]
- 63.Staub RE, Feng C, Onisko B, Bailey GS, Firestone GL, Bjeldanes LF. Fate of Indole-3-carbinol in cultured human breast tumor cells. Chem Res Toxicol. 2002;15:101–109. doi: 10.1021/tx010056m. [DOI] [PubMed] [Google Scholar]
- 64.Miyoshi H, Ohshima M, Shimada H, Akagi T, Iwamura H, McLaughlin JL. Essential structural factors of annonaceous acetogenins as potent inhibitors of mitochondrial complx I. Biochim Biophys Acta. 1998;1365:443–452. doi: 10.1016/s0005-2728(98)00097-8. [DOI] [PubMed] [Google Scholar]
- 65.Alali FQ, Liu X, McLaughlin JL. Annonaceous acetogenins: recent progress. J Nat Prod. 1999;62:504–540. doi: 10.1021/np980406d. [DOI] [PubMed] [Google Scholar]
- 66.González-Coloma A, Guadaño A, de Inés C, Martínez-Díaz R, Cortes D. Selective action of acetogenin mitochondrial Complex I inhibitors. Z Naturforsch. 2002;57c:1028–1034. doi: 10.1515/znc-2002-11-1213. [DOI] [PubMed] [Google Scholar]
- 67.Shimada H, Grutzner JB, Kozlowski JF, McLaughlin JL. Membrane conformations and their relation to cytotoxicity of asimicin and its analogues. Biochemistry. 1998;37:854–866. doi: 10.1021/bi972348l. [DOI] [PubMed] [Google Scholar]
- 68.Motoyama T, Yabunaka H, Miyoshi H. Essential structural factors of acetogenins, potent inhibitors of mitochondrial Complex I. Bioorg Med Chem. 2002;12:2089–2092. doi: 10.1016/s0960-894x(02)00374-8. [DOI] [PubMed] [Google Scholar]
- 69.Kuwabara K, Takada M, Iwata J, Tatsumoto K, Sakamoto K, Iwamura H, et al. Design syntheses and mitochondrial complex I inhibitory activity of novel acetogenin mimics. Eur J Biochem. 2000;267:2538–2546. doi: 10.1046/j.1432-1327.2000.01263.x. [DOI] [PubMed] [Google Scholar]
- 70.Yabunaka HY, Abe M, Kenmochi A, Hamada T, Nishioka T, Miyoshi H. Synthesis and inhibitory activity of ubiquinone-acetogenin hybrid inhibitor with bovine mitochondrial Complex I. Bioorg Med Chem Lett. 2003;13:2385–2388. doi: 10.1016/s0960-894x(03)00439-6. [DOI] [PubMed] [Google Scholar]
- 71.Tormo JR, Estornell E. New evidence for the multiplicity of ubiquinone- and inhibitor-binding sites in the mitochondrial complex I. Arch Biochem Biophys. 2000;381:241–246. doi: 10.1006/abbi.2000.1969. [DOI] [PubMed] [Google Scholar]
- 72.Esposti MD, Ghelli A. Ubiquinone and inhibitor sites in complex I: one, two, or three? Biochem Soc Trans. 1999;27:606–609. doi: 10.1042/bst0270606. [DOI] [PubMed] [Google Scholar]
- 73.Ohshima M, Miyoshi H, Sakamoto K, Takegami K, Iwata J, Kuwabara K, et al. Characterization of the ubiquinone reduction site of mitochondrial Complex I using bulky synthetic ubiquinones. Biochemistry. 1998;37:6436–6445. doi: 10.1021/bi9800202. [DOI] [PubMed] [Google Scholar]
- 74.McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- 75.Cao D, Qiao B, Ge Z, Yuan Y. Comparison of burst of reactive oxygen species and activation of caspase-3 in apoptosis of K562 and HL-60 cells induced by docetaxel. Cancer Lett. 2004;214:103–113. doi: 10.1016/j.canlet.2004.03.047. [DOI] [PubMed] [Google Scholar]