

Abstract
A possible origin of novel coding sequences is the removal of stop codons, leading to the inclusion of 3′ UTRs within genes. We classified changes in the position of stop codons in closely related Saccharomyces species and in a mouse/rat comparison as either additions to or subtractions from coding regions. In both cases, the position of stop codons is highly labile, with more subtractions than additions found. The subtraction bias may be balanced by the input of new coding regions through gene duplication. Saccharomyces shows less stop codon lability than rodents, probably due to greater selective constraint. A higher proportion of 3′ UTR incorporation events preserve frame in Saccharomyces. This higher proportion is consistent with the action of the [PSI+] prion as an evolutionary capacitor to facilitate 3′ UTR incorporation in yeast.
Keywords: evolution of novelty, evolutionary innovation, alternative splicing, genetic assimilation, gene length
Introduction
The origin of new protein-coding sequences is of great interest to understanding evolutionary innovation. New protein-coding sequence can result from gene duplication (Ohno 1970; Lynch and Conery 2000), from rearrangements between genes (Long and Langley 1993; Yun et al. 1999; Powell et al. 2000; Leong et al. 2003; Conant and Wagner 2005), from the insertion of new nucleotide sequences (Claverie and Ogata 2003), or from the inclusion of nucleotide sequences that were formerly non-coding (Levine et al. 2006).
One major route by which non-coding regions are believed to become coding is via alternative splicing (Modrek and Lee 2003). Alternative splicing is associated with an increased rate of exon creation and loss (Modrek and Lee 2003). 25 instances of the addition of novel sequence have been implicated in alternative splicing, most likely through intron inclusion, while 48 cases were found in which alternative splicing led to loss of sequence, usually through exon skipping (Kondrashov and Koonin 2003). Alternative splicing is also the most likely mechanism by which transposable elements are incorporated into coding sequences (Nekrutenko and Li 2001)
Just as novel sequences can be incorporated into coding regions via intron inclusion, the addition of new exons, or the expansion of exon boundaries, they might also be incorporated via shifts in the position of the start (Lynch, Scofield, and Hong 2005) and stop codons. In yeast, the [PSI+] prion might facilitate the incorporation of sequences beyond stop codons. [PSI+] is an epigenetically inherited aggregate of the Sup35 protein (Paushkin et al. 1996), which is involved in translation termination (Stansfield et al. 1995; Zhouravleva et al. 1995). When [PSI+] appears, translation termination is impaired, and phenotypic variation is revealed as a consequence of the expression of sequences beyond stop codons (True and Lindquist 2000; True, Berlin, and Lindquist 2004; Wilson et al. 2005). Variation is “revealed” rather than created, since the effect of [PSI+] depends strongly on the genetic background (True and Lindquist 2000). [PSI+]-dependent phenotypes could be a consequence of the resurrection of pseudogenes that are normally disabled by premature stop codons (Harrison et al. 2002), the merging of two ORFs through readthrough translation (Harrison et al. 2002; Namy et al. 2003), or the addition of new coding sequence by the addition of 3′ UTR to a single ORF (Namy, Duchateau-Nguyen, and Rousset 2002). Some are also the result of nonstop mRNA decay (Wilson et al. 2005). Surprisingly, revealed variation need not be pathological, and can increase growth rates under a range of stressful conditions (True and Lindquist 2000).
As an epigenetically inherited protein aggregate, [PSI+] can easily be lost after some generations (Cox, Tuite, and Mundy 1980). This returns the lineage to its normal [psi−] state and restores translation fidelity. If a subset of revealed phenotypic variation is adaptive, it will have lost its dependence on [PSI+] by this time (True, Berlin, and Lindquist 2004). The exact molecular mechanism of this genetic assimilation has not been elucidated since it occurs so rapidly (True, Berlin, and Lindquist 2004), but it may, for example, involve one or more point mutations in stop codons, leading to the incorporation of 3′ UTR into the coding region. The revelation and genetic assimilation of [PSI+]-mediated traits is believed to increase phenotypic evolvability in yeast (True and Lindquist 2000; Masel and Bergman 2003). The ability to form the [PSI+] prion is not restricted to Saccharomyces cerevisiae, but has instead been conserved over long evolutionary time periods (Chernoff et al. 2000; Kushnirov et al. 2000; Santoso et al. 2000; Nakayashiki et al. 2001).
Here we examine changes in the position of stop codons between closely related yeast species. If [PSI+] mediates the incorporation of 3′ UTR into coding regions, this leads to two predictions. Firstly, we should find evidence for a large number of incorporation events during the evolution of closely related yeast species. Secondly, frame-preserving incorporation events should be more common in yeast than in other species such as mammals for which no [PSI+]-like mechanism is known.
Materials and Methods
Yeast data
Genome sequences of S. cerevisiae, S. mikatae, S. bayanus and S. paradoxus were downloaded from the Saccharomyces Genome Database (Balakrishnan et al. 2005), together with ORF annotation using a release that was current as of 7/21/2005. The latter three genomes were originally sequenced by Kellis et al. (2003), but also include a number of substantial updates since first publication. For all four genomes, annotated ORFs marked dubious by SGD, lacking 3′ UTR sequence, or not ending in a stop codon were excluded. Additionally, since a minimum of 3 species is needed to identify the location of an event, only orthologs present in 3 or more species were used. In the event that SGD had annotated more than one ortholog in a given species, whichever one aligned best to S. cerevisiae or S. paradoxus was used. These exclusions reduced the total number of sequences from 24727 to 18490, organized into 4882 orthologous groups of 3 or 4 species each.
In order to check for genes for which the gene tree and species tree might not match (due, for example, to paralogy and lineage sorting), aligned sequences were fed into PAUP* (Swofford 2003) and groups of orthologous ORFs were deemed suspect if the species tree (Rokas et al. 2003) returned p<0.05 according to the Shimodaira-Hasegawa test (Shimodaira and Hasegawa 1999). However, only one gene gave a significant result of p=0.02, and this one gene had no variation in stop codon position.
Alignments were performed on nucleic acids using POA (Lee, Grasso, and Sharlow 2002) with default options. The included scoring matrix used +4 for all matches, −2 for a mismatch, a gap open penalty of 15 and a gap extension penalty of 2. When sections of alignments needed to be scored during analysis, the same scoring matrix and penalties were used, and scores were normalized for length.
To locate potential stop codon change events, the 3′ UTR of each gene was concatenated to the end of its ORF, and the orthologous group of sequences was aligned. The stop codons and trailing 3′ UTR were then extracted. If stop codon position was inconsistent across species, the aligned sequences between the first and last stop codon positions were extracted from each species and scored using the matrix specified above. Because short, unrelated sequences sometimes align well due to chance alone, multiple protocols were tested on randomly sampled sequences, to minimize the frequency of false positive alignments. Longer sequences were less likely to align well by chance, but more likely to contain a repetitive or other sequence element that aligns well for a reason. We obtained optimal results by using all sequence between the two stop codon positions in question, and additionally padding out in the 3′ direction until a minimum of 18 bp was used. Setting a score threshold of 0.2 using the parameters described above, this resulted in rejection of 97.7% of aligned unrelated 3′ UTR sequences during testing.
Classification of events as additions or subtractions was performed using published phylogeny (Rokas et al. 2003) and maximum parsimony, as shown in Figure 1. Classification was implemented by noting the rank order of lengths (in bp and ignoring gaps) beyond the earliest terminating member for each ortholog family. Differences of less than 9 base pairs were ignored when ranking sequence lengths. The rank order data were then compared to a set of all observed combinations and classified accordingly.
Figure 1.
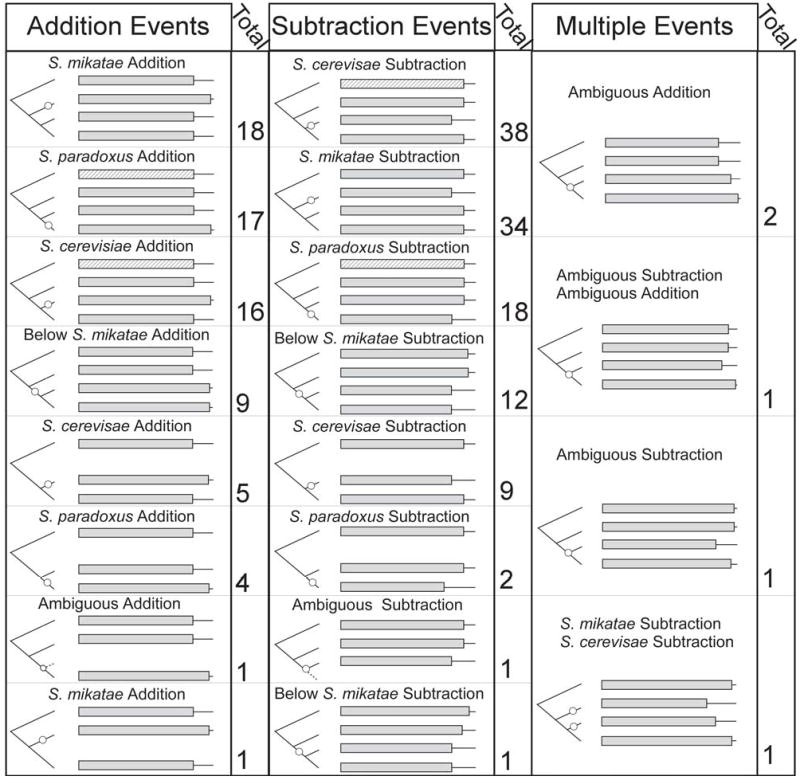
A list of all observed stop codon shifts that could be classified as additions or subtractions. The phylogenetic tree contains S. bayanus, S. mikatae, S. cerevisiae and S. paradoxus respectively from top to bottom. Gene lengths shown in stripes can take any value without changing the classification, while gene lengths left blank represent an ortholog that was not present or not identified in one species. Many additional configurations are possible, however we present only those observed. The genes corresponding to each classification are listed in Supplementary Table 1.
Finally, the classified results were cross checked against a list of known introns in S. cerevisiae. Introns are not annotated in Saccharomyces species other than S. cerevisiae, and several questionable stop codon annotations were identified that most likely correspond to a false stop codon located in an intron, rather than to a genuine change in stop codon position. All genes that showed both stop codon lability and contained an intron in S. cerevisiae (Grate et al. 2000) were verified by hand, and a total of 7 stop codon shifts were rejected.
Mammalian data
The procedure developed in yeast was repeated on the human, mouse and rat genomes provided by the UCSC Genome Browser database. The databases used were the May 2004 release of the human genome, the March 2005 mouse genome, and the June 2003 rat genome. Orthologs were identified using data for all three mammal species provided by the Mouse Genome Database (Eppig et al. 2005). For each gene, we extracted the exon containing the annotated stop codon, and, when necessary, the subsequent exon. In order to ensure that exons were orthologous between genes, sequences downloaded in the previous step were aligned and scored as before in yeast. Exons scoring less than 0.2 were rejected.
Classification was conducted as in yeast, however with only 3 species, there are only two possible addition configurations and two possible subtraction configurations.
Frame-preserving vs. non-frame-preserving addition events
The 43 addition events on the S. cerevisiae and S. paradoxus branches, and the 67 addition events on the mouse and rat branches were analyzed by hand. If the stop codon was disabled by a point mutation or other event that did not disrupt the reading frame of the new stop codon, the gene was put into one category. If the new stop codon was in a different frame, the gene was put into a second category. Finally, 2 genes in yeast and 12 in mammals were rejected from this portion of the analysis because frame identity was obscured by poor conservation or because a change in splicing was suspected such that the region around the ancestral stop codon had likely been spliced out.
Error Rates in Yeast
Sequencing errors, rather than true evolutionary change, may lead to apparent changes in stop codon location. In order to assess the prevalence of sequencing errors incorrectly altering stop codon annotation, the data sequenced by Kellis et al. (2003) was compared to an independent sequencing effort by Cliften et al. (2003) of S. mikatae and S. bayanus at 2x coverage. Although the Cliften data is at lower coverage, it is of sufficiently high quality to provide an effective check for a data set that is expected to be enriched for errors (see below).
In order to exclude the possibility of differing annotation, a BLAST search for each ORF in the Kellis data was performed against the Cliften data. The stop codon location annotated by SGD was then compared to the aligned sequence in the Cliften data. Synonymous differences were ignored under the assumption that they may represent either polymorphisms or neutral evolution between the strains sequenced by the two groups. Additionally, the 48 bp immediately 5′ of the stop codon were examined for frameshifts and the sequence was flagged as a mismatch if the annotated codon was out of frame, or rejected entirely if insufficient base pairs were available in the alignment to determine the reading frame.
False positive subtraction events are expected to be more common than false positive addition events because a sequencing error can easily create a subtraction by inserting a stop codon into any position along an ORF. However, to create an addition, a sequencing error must occur within the stop codon, or else a frameshift must occur in the right location to knock the real stop codon out of frame, but not introduce a new premature stop codon from another frame. This biased sequencing error rate parallels the mutational bias towards subtractions.
Events occurring at S. mikatae were broken down into additions and subtractions in order to test the extent to which sequencing errors favor subtractions. These genes were then checked against the Cliften data to locate stop codon events that were not supported by both datasets. In S. mikatae 1 of 20 additions and 9 of 44 subtractions were not supported by a comparison to the Cliften data. This allows us to reject the null hypothesis that the proportion of errors of each type is equal in favor of the alternative hypothesis of subtraction-biased errors with p < 0.05. Subtraction errors are also more common in absolute terms, with p < 0.02.
Just 67/3969=1.68% of stop codon positions in all available S. mikatae genes showed disagreement between the two data sets, with the majority likely to be sequencing errors in the Cliften data, due to its lower coverage. This illustrates the extent to which our search for stop codon shifts enriches for errors, since the disagreement rate in gene undergoing stop codon shifts was (1+9)/(20+44)=15%. For this reason, when a stop codon shift is not validated by the Cliften data, we are able to assume that this is due to enrichment for errors in the Kellis data rather than an error the Cliften data used to verify it.
Statistical analysis
Confidence intervals on proportions were calculated as described by Clopper and Pearson (1934) and implemented at http://statpages.org/confint.xls by John Pezzullo. Differences between proportions were tested using a G-test of independence performed on contingency tables (Sokal and Rohlf 1995).
Results
Changes in the location of stop codons can correspond either to the addition of new coding regions during evolution, or to the subtraction of old coding regions. Additions represent the destruction of the terminal stop codon and the expansion of an ORF into the 3′ UTR. Subtractions represent the creation of a new stop codon prior to the terminal stop codon, causing translation to terminate prematurely. Here we use the terms “addition” and “subtraction” rather than “insertion” and “deletion”, since the events we consider need not involve the physical insertion or deletion of nucleotide sequence.
Both additions and subtractions were detected by aligning orthologous genes in related species to find those that differ in the location of their stop codon, and confirming that the additional 3′ coding regions align well with 3′ UTRs in the appropriate region, as described in Materials and Methods. Addition events can be distinguished from subtractions according to maximum parsimony, as shown in Figure 1, by referring to the known phylogeny of Saccharomyces (Rokas et al. 2003). Some changes in stop codon location cannot be classified as either additions or subtractions, either because they occurred on the ancestral branch, or because multiple events lead to more than one equally parsimonious way to interpret the data.
The results of the classification process are presented in Figure 2A. Perhaps most striking is the high degree of stop codon lability in this closely related yeast clade. Out of 4882 ortholog families with data suitable for analysis, 371 displayed both a change in stop codon position of at least 9 nucleotides and strong sequence conservation between the two stop codon locations. As described in Materials and Methods, we confirmed that this high degree of lability was not a consequence of sequencing errors by crosschecking results generated with one data set (Kellis et al. 2003) against an independent sequencing effort (Cliften et al. 2003). Using this approach, all genes with addition or subtraction events on the S. mikatae branch were found in the Cliften data. Through this independent verification, 1 addition out of 20 and 9 subtractions out of 44 were found to be sequencing errors, and were removed from the analysis.
Figure 2.
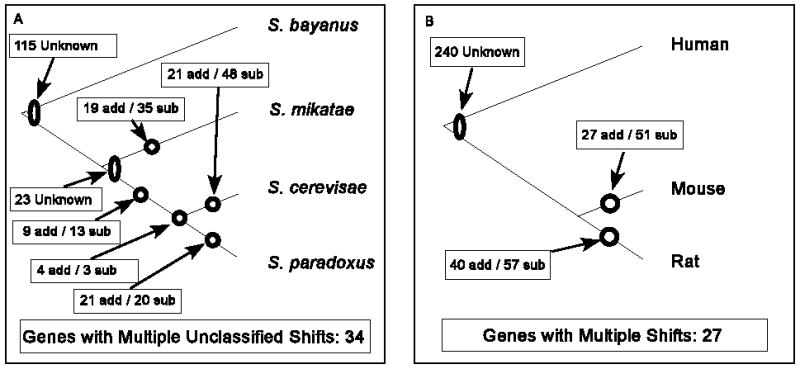
Summary of addition and subtraction events in Saccharomyces (A), and in mammals (B). Nodes on a single branch of a phylogenetic tree represent events that can be fully resolved. Nodes spanning multiple cannot be resolved to a specific branch, but may still be classifiable as additions or subtractions. There were no significant differences between the ratios of additions/subtractions in the different branches of the yeast phylogeny or between mouse and rat.
Protein length is stable over the long term, possibly as a tradeoff between the metabolic costs vs. increased stability of longer proteins (Wang, Hsieh, and Li 2005). Since there is no evidence for a change in the total number of genes within the Saccharomyces lineages considered here, we expect a balance between all forms of addition to coding sequences and all forms of subtraction from coding sequences. Additions can take the form of insertions of nucleotide sequence, as well as incorporation of untranslated regions as studied here. Subtractions can take the form of deletions of nucleotide sequence, as well as the reversion of coding sequences to an untranslated state studied here.
If gene duplication is sufficiently common, it could create a bias towards deletions and/or subtractions to maintain the long term stability of protein length. The fate of most duplicates is relaxed selection leading to partial or complete degeneration (Ohno 1970), typically through a series of small deletions (Kellis, Birren, and Lander 2004). Under relaxed selection, there is a strong mutational bias towards premature termination rather than stop codon removal. Premature stop codons play a major role in pseudogenation, and intermediate stages of either ongoing or partially reversed degeneration may be detectable as an elevated number of subtraction events relative to additions. This was observed in the yeast data: the ratio of additions/(additions+subtractions) = 74/(74+119) = 0.383 with a 95% confidence interval of 0.31–0.46. The confidence interval needed to be expanded to 99.9% confidence before it included 0.5.
Note that subtraction events are more likely than additions to result from a sequencing error rather than a real evolutionary change (see Materials and Methods). We estimated the likely effect of this bias on our results as follows. Given the small number of errors in S. mikatae, together with the higher quality of the S. cerevisiae data, we assume that S. cerevisiae is essentially free from errors. Stop codon changes on the common branch between S. mikatae and the divergence of S. paradoxus and S. cerevisiae are also highly unlikely to be errors, given that only a very narrow class of errors could create an event on this branch. Assuming that S. paradoxus has at most the same number of false positives as S. mikatae (which we believe to exaggerate the errors, given subjective experience with the quality of the data from the different species), we estimate one more addition and 9 more subtractions to be false. Ignoring the error associated with this estimate, this would give the proportion of additions as 73/(73+110) = 0.40 with a 95% confidence interval of 0.33–0.47, and so a significant subtraction bias is still observed. This confidence interval needed to be expanded to 99.3% confidence before it included 0.5.
To test whether the high level of stop codon lability might be a consequence of [PSI+] activity, we repeated the same analysis in mammals, using human, mouse, and rat sequences. The results are summarized in Figure 2B. Out of 5180 sets of orthologs suitable for analysis, we found evidence of 67 addition events, 108 subtraction events, and 267 events whose phylogeny could not be resolved. There was no significant difference in the proportion of additions/subtractions between yeast and mammals. The 95% confidence interval on the ratio of additions/(additions+subtractions) in rodents is 0.31–0.46, and needed to be expanded to 99.78% confidence before it included 0.5. The high proportion of unresolved events can be attributed to the much greater evolutionary distance between humans and rodents than between mice and rats.
The bias towards subtractions becomes still stronger when we consider the number of amino acids affected by stop codon changes rather than merely the number of events changing stop codon location. The average lengths of additions and subtractions in Saccharomyces branches other than the error-prone S. paradoxus branch are 11 and 18 codons respectively, and the average lengths on the mouse branch (mouse has better quality sequence than rat) are 35 and 43 codons. In Figure 3 we see that the longer mean of subtractions can be entirely accounted for by a small number of very long subtractions, which are possibly part of the process of pseudogenation. For example, when the 1 yeast addition and 11 yeast subtractions longer then 42 codons are ignored, then the average length for both additions and subtractions is 9 codons. Since the Saccharomyces branches analyzed are close to error-free, these occasional long events are most likely real rather than a consequence of sequencing errors.
Figure 3.
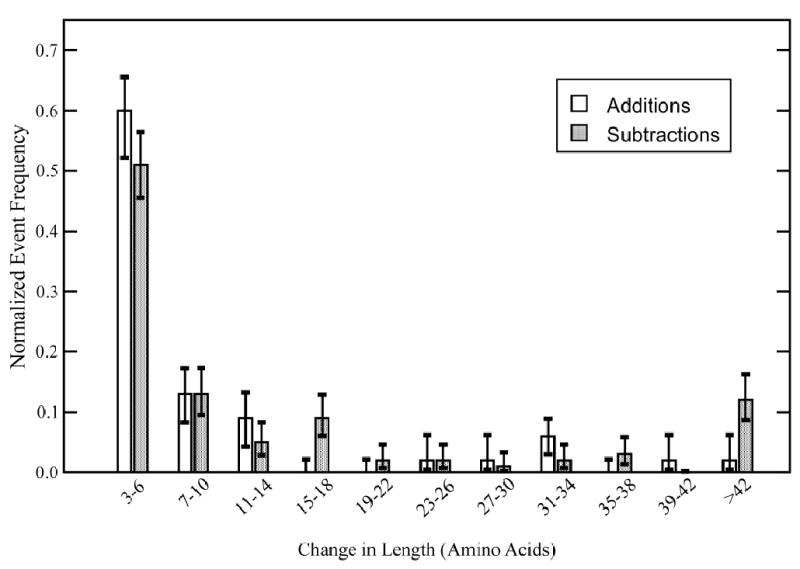
Length distributions of additions and subtractions in all Saccharomyces branches except S. paradoxus. Standard errors were calculated as 78.2% confidence intervals as described in the Methods.
It is possible that additions simply increase the length of the C-terminal tail, but do not damage domains, wherease subtractions may truncate a conserved domain and therefore destroy protein function. Using S. cerevisiae domain information from the SGD domain browser, we found that 13/55 subtractions not on a lineage leading to S. cerevisiae came within 3 amino acids of a domain and hence were likely to disrupt it, whereas 9/33 additions on a lineage leading to S. cerevisiae came within 3 amino acids of a domain. Despite the larger size of a small category of subtraction events, there is no significant difference between these proportions, showing that addition events are able to create or extend protein domains.
The S. cerevisiae vs. S. paradoxus comparison exhibits a similar evolutionary divergence to mouse vs. rat (see more details below), so we now concentrate on these two pairs of species. On the S. cerevisiae vs. S. paradoxus branches alone, 43/4710=0.9% of the genes underwent an addition, while 69/4710=1.5% of the genes underwent a subtraction. 34 genes were observed to have undergone a sequence of multiple stop codon events that could not be resolved, contributing additional stop codon variation. This high degree of lability compares to only 1–5 gene duplication events occurring during this time (Gao and Innan 2004). Given the average lengths of additions and subtractions, orthologous genes have on average 0.1 amino acids added through 3′ UTR incorporation, 0.3 amino acids lost through nonsense mutations, and, given an average gene length of 486 amino acids, have 0.1–0.5 amino acids added through gene duplication. These figures, perhaps coincidently, yield approximately neutral balance before insertions, deletions and other events are considered. Note that most eukaryotes show a mutational bias towards deletions rather than insertions (Gregory 2004), although the existence and strength of this effect is not known for Saccharomyces (Byrnes, Morris, and Li 2006).
In comparison, on the mouse/rat branches, using humans as an outgroup, 67/5180=1.3% of the genes have an addition while 108/5180=2.0% of the genes have a subtraction. Given the average lengths of additions and subtractions on the mouse lineage, orthologous genes on the mouse/rat branches have on average 0.45 amino acids added through 3′ UTR incorporation and 0.90 amino acids lost through nonsense mutations. As another point of reference, the rate of newly evolved exons on the mouse branch alone has been estimated 2695/79098=3%, although many of these new exons are minor forms with low expression levels (Wang et al. 2005).
To see whether the apparently higher stop codon lability in rodents was due to increased divergence between this species pair, we compared a set of 406 genes for which orthologs existed in all species considered. Genes were selected by performing a BLAST search of each S. cerevisiae gene against each rat gene, and accepted only if a hit covered >80% of the mammal gene with an expect value of less than 10−20. In this comparable gene set, identity at 4-fold degenerate sites was 75% in yeast and 83% in rodents, showing that the higher rodent lability occurred despite lower neutral divergence. This is most likely due to relaxed selective constraint in rodents relative to yeast due to their smaller effective population size. In agreement with stronger selective constraint in yeast, the same set of genes had amino acid identity of 95% in both yeast and rodents. Note that this matched data set is biased towards conserved genes; taking all genes, the respective identities for Saccharomyces and rodents are 73.3% and 82.6% for 4-fold degenerate sites and 88.7% and 87.9% for amino acids. The same patterns are observed using either set of genes.
If changes in stop codon position, are under the same selective constraint as other changes to the amino acid sequence, then we would expect equal stop codon lability in yeast and rodents, consistent with equal percent identity in amino acid sequence. Changes in stop codon position typically affect multiple stop codons at a time, and may therefore be under greater selective constraint. This greater selective constraint might explain the higher stop codon lability in rodents than yeast.
Addition events can occur in one of two ways. Firstly, a point mutation could destroy the stop codon, leading to an extension to the next in-frame stop codon in what was previously 3′ UTR. Deletions that include the stop codon and whose sizes are multiples of three can have a similar effect. Secondly, an insertion or deletion that is not a multiple of three could create a frameshift. When such an indel occurs near the end of a gene, the next stop codon in the new frame could lie beyond the ancestral stop codon, creating an addition event. Only the first category of additions mimics the effect of [PSI+]-mediated readthrough translation, and so only this category of additions should be promoted by the evolutionary capacitance activity of [PSI+].
The S. cerevisiae and S. paradoxus branches have 19 of the first category of additions (all of them point mutations in the stop codon), 20 of the second in which frame is not preserved, and one intermediate event in which frame appears to change twice, resulting in an addition with 3 codons in frame and 4 out of frame. The rat and mouse branches have 5 of the first category of additions (4 point mutations and 1 3bp deletion precisely removing a stop codon) and 50 of the second. Two events in yeast and 12 in rodents were unclassifiable. Three of the rodent unclassifiable events resulted from small changes in splicing: most splicing changes were already excluded by our 3′UTR alignment criteria. The proportion of classifiable addition events compatible with [PSI+]-mediated activity is significantly higher in yeast (p<0.00002). This strong result seems unlikely to be explained by different mutational biases.
The majority (13/19 = 68%) of yeast addition events in the first category jumped to a new stop codon still retained in the 3′ UTR of the ortholog. The remaining 6 events may also have been jumps to the next in-frame stop codon, but this history has been masked by subsequent mutations. The 68% preservation rate is compatible with the 73% rate of conservation under neutral evolution measured at 4-fold degenerate sites.
Our results show that the high levels of stop codon lability and 3′ UTR incorporation are not restricted to yeast and are in fact higher in rodents. Nevertheless, the subset of 3′ UTR incorporation events that have a form consistent with [PSI+]-mediated activity are twice as frequent in yeast than in rodents. This provides evidence for the action of [PSI+] as an evolutionary capacitor in natural populations of yeast.
Discussion
High levels of stop codon lability were found, relative to other large changes at the molecular level. There was a bias towards subtractions rather than additions, likely due to ongoing gene duplication and degeneration as well as to sequencing errors. Despite this bias towards subtractions, frequent incorporation of 3′ UTR into coding regions still occurred, potentially supplying a significant source of novelty at the molecular level. Pathological effects of C-terminal extensions are known (Weatherall and Clegg 1979), making the high level of stop codon lability seen here somewhat surprising. In some cases, however, the addition of substantial C-terminal tails to proteins can have surprisingly little effect, and may in fact increase protein stability (Matsuura et al. 1999; Chow et al. 2003). Previous work suggests that gene length tends to remain constant over evolutionary time, perhaps as tradeoff between protein stability and metabolic efficiency (Zhang 2000; Wang, Hsieh, and Li 2005; Xu et al. 2006). Our results suggest that this process may be highly dynamic over shorter timescales.
Although the overall ratio of additions to subtractions was the same in mouse vs. rat as in Saccharomyces, there was an elevated rate of frame-preserving additions in yeast, consistent with a role for [PSI+] as an evolutionary capacitor. The number of potentially [PSI+]-mediated events is modest, with only 19 genes identified as potentially involved, above a control background of 5 genes in the absence of [PSI+] in species with similar evolutionary divergence.
The role of [PSI+] as a source of novelty at the phenotypic level may, however, be greater than this. The mechanistic basis by which [PSI+]-mediated phenotypes are genetically assimilated is not known (True, Berlin, and Lindquist 2004). The disappearance of stop codons is one obvious example, but one well-supported alternative is the acquisition of mutations that change the susceptibility of mRNAs to nonsense-mediated decay (True, Berlin, and Lindquist 2004). There is evidence that [PSI+]-mediated traits may be complex phenotypes dependent on changes at multiple loci (True, Berlin, and Lindquist 2004). Genetic assimilation can involve single or multiple mutations at entirely different loci from those responsible for the initial condition-dependent phenotype (Stern 1958; Bateman 1959; Dworkin et al. 2003; Masel 2004). Genetic complexity combined with the rapidity of genetic assimilation make it difficult to elucidate in a direct manner the molecular basis of genetic assimilation (True, Berlin, and Lindquist 2004). Our more indirect approach show that at least in some cases, genetic assimilation may take place in a straightforward manner through mutational loss of one or more stop codons.
Supplementary Material
Supplementary Tables are available giving the identity of all ORFs showing changes in stop codon position, as well as the 19 yeast ORFs showing addition events compatible with [PSI+]-mediated evolutionary capacitance.
Supplementary Material
Acknowledgments
We thank Aron Talenfeld and Joel Wertheim for help with the PAUP* analysis, Oliver King, Michael Nachman, Roy Parker and Heather True for helpful discussions, and two anonymous reviewers for helpful suggestions. Financial support came from BIO5 Institute at the University of Arizona and National Institutes of Health grant GM076041. M.G. and A.H. were also supported by the Interdisciplinary UBRP program funded by the National Institutes of Health (R25 GM072733).
References
- Balakrishnan R, Christie KR, Costanzo MC, et al. [(accessed 21 July 2005)];2005 (23 co-authors) Saccharomyces Genome Database. ftp://ftp.yeastgenome.org/yeast/
- Bateman KG. The genetic assimilation of the dumpy phenocopy. J Genet. 1959;56:341–351. [Google Scholar]
- Byrnes JK, Morris GP, Li W-H. Reorganization of Adjacent Gene Relationships in Yeast Genomes by Whole-Genome Duplication and Gene Deletion. Mol Biol Evol. 2006;23:1136–1143. doi: 10.1093/molbev/msj121. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol. 2000;35:865–876. doi: 10.1046/j.1365-2958.2000.01761.x. [DOI] [PubMed] [Google Scholar]
- Chow CC, Chow C, Raghunathan V, Huppert TJ, Kimball EB, Cavagnero S. Chain length dependence of apomyoglobin folding: Structural evolution from misfolded sheets to native helices. Biochemistry. 2003;42:7090–7099. doi: 10.1021/bi0273056. [DOI] [PubMed] [Google Scholar]
- Claverie JM, Ogata H. The insertion of palindromic repeats in the evolution of proteins. Trends Biochem Sci. 2003;28:75–80. doi: 10.1016/S0968-0004(02)00036-1. [DOI] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26:404–413. [Google Scholar]
- Conant GC, Wagner A. The rarity of gene shuffling in conserved genes. Genome Biol. 2005;6 doi: 10.1186/gb-2005-6-6-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B, Tuite M, Mundy C. Reversion from suppression to nonsuppression in SUQ5 [psi+] strains of yeast: the classification of mutations. Genetics. 1980;95:589–609. doi: 10.1093/genetics/95.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin I, Palsson A, Birdsall K, Gibson G. Evidence that Egfr contributes to cryptic genetic variation for photoreceptor determination in natural populations of Drosophila melanogaster. Curr Biol. 2003;13:1888–1893. doi: 10.1016/j.cub.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Eppig JT, Bult CJ, Kadin JA, Richardson JE, Blake JA. Mouse Genome Database (MGD) [(accessed 9/16/2005)];2005 doi: 10.1093/nar/29.1.91. http://www.informatics.jax.org. [DOI] [PMC free article] [PubMed]
- Gao L-z, Innan H. Very low gene duplication rate in the yeast genome. Science. 2004;306:1367–1370. doi: 10.1126/science.1102033. [DOI] [PubMed] [Google Scholar]
- Grate L, Telerski A, Clark T, Centers R, Ares M. Ares lab Yeast Intron Database. [(accessed Aug 1, 2005)];2000 http://www.cse.ucsc.edu/research/compbio/yeast_introns/currentDB/Extras.html.
- Gregory TR. Insertion-deletion biases and the evolution of genome size. Gene. 2004;324:15–34. doi: 10.1016/j.gene.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Harrison P, Kumar A, Lan N, Echols N, Snyder M, Gerstein M. A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J Mol Biol. 2002;316:409–419. doi: 10.1006/jmbi.2001.5343. [DOI] [PubMed] [Google Scholar]
- Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Kondrashov FA, Koonin EV. Evolution of alternative splicing: deletions, insertions and origin of functional parts of proteins from intron sequences. Trends Genet. 2003;19:115–119. doi: 10.1016/S0168-9525(02)00029-X. [DOI] [PubMed] [Google Scholar]
- Kushnirov VV, Kochneva-Pervukhova N, Chechenova MB, Frolova NS, Ter-Avanesyan MD. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000;19:324–331. doi: 10.1093/emboj/19.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18:452–464. doi: 10.1093/bioinformatics/18.3.452. [DOI] [PubMed] [Google Scholar]
- Leong SR, Chang JCC, Ong R, Dawes G, Stemmer WPC, Punnonen J. Optimized expression and specific activity of IL-12 by directed molecular evolution. Proc Natl Acad Sci U S A. 2003;100:1163–1168. doi: 10.1073/pnas.0237327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 2006;103:9935–9939. doi: 10.1073/pnas.0509809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MY, Langley CH. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science. 1993;260:91–95. doi: 10.1126/science.7682012. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Lynch M, Scofield DG, Hong X. The evolution of transcription-initiation sites. Mol Biol Evol. 2005;22:1137–1146. doi: 10.1093/molbev/msi100. [DOI] [PubMed] [Google Scholar]
- Masel J. Genetic assimilation can occur in the absence of selection for the assimilating phenotype, suggesting a role for the canalization heuristic. J Evol Biol. 2004;17:1106–1110. doi: 10.1111/j.1420-9101.2004.00739.x. [DOI] [PubMed] [Google Scholar]
- Masel J, Bergman A. The evolution of the evolvability properties of the yeast prion [PSI+] Evolution. 2003;57:1498–1512. doi: 10.1111/j.0014-3820.2003.tb00358.x. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Miyai K, Trakulnaleamsai S, Yomo T, Shima Y, Miki S, Yamamoto K, Urabe I. Evolutionary molecular engineering by random elongation mutagenesis. Nat Biotechnol. 1999;17:58–61. doi: 10.1038/5232. [DOI] [PubMed] [Google Scholar]
- Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nature Genet. 2003;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell. 2001;7:1121–1130. doi: 10.1016/s1097-2765(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Namy O, Duchateau-Nguyen G, Hatin I, Hermann-Le Denmat S, Termier M, Rousset JP. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:2289–2296. doi: 10.1093/nar/gkg330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Duchateau-Nguyen G, Rousset JP. Translational readthrough of the PDE2 stop codon modulates cAMP levels in Saccharomyces cerevisiae. Mol Microbiol. 2002;43:641–652. doi: 10.1046/j.1365-2958.2002.02770.x. [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Li WHS. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001;17:619–621. doi: 10.1016/s0168-9525(01)02445-3. [DOI] [PubMed] [Google Scholar]
- Ohno S. Evolution by Gene Duplication. Springer-Verlag; Heidelberg: 1970. [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Powell SK, Kaloss MA, Pinkstaff A, McKee R, Burimski I, Pensiero M, Otto E, Stemmer WPC, Soong NW. Breeding of retroviruses by DNA shuffling for improved stability and processing yields. Nat Biotechnol. 2000;18:1279–1282. doi: 10.1038/82391. [DOI] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–288. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol BiolEvol. 1999;16:1114–1116. [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry. W.H. Freeman and Company; New York: 1995. pp. 724–739. [Google Scholar]
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. The products of the sup45 (eRF1) and sup35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern C. Selection for subthreshold differences and the origin of pseudoexogenous adaptations. Am Nat. 1958;92:313–316. [Google Scholar]
- Swofford DL. Phylogenetic Analysis Using Parsimony (*and other methods) Sinauer Associates; Sunderland, Massachusetts: 2003. PAUP*. [Google Scholar]
- True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature. 2004;431:184–187. doi: 10.1038/nature02885. [DOI] [PubMed] [Google Scholar]
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- Wang DY, Hsieh M, Li WH. General tendency for conservation of protein length across eukaryotic kingdoms. Mol Biol Evol. 2005;22:142–147. doi: 10.1093/molbev/msh263. [DOI] [PubMed] [Google Scholar]
- Wang W, Zheng H, Yang S, et al. Origin and evolution of new exons in rodents. Genome Res. 2005;15:1258–1264. doi: 10.1101/gr.3929705. (17 co-authors) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall DJ, Clegg JB. Recent developments in the molecular genetics of human hemoglobin. Cell. 1979;16:467–479. doi: 10.1016/0092-8674(79)90022-9. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Meaux S, Parker R, van Hoof A. Genetic interactions between [PSI+] and nonstop mRNA decay affect phenotypic variation. Proc Natl Acad Sci U S A. 2005;102:10244–10249. doi: 10.1073/pnas.0504557102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Chen H, Hu XH, Zhang RM, Zhang Z, Luo ZW. Average gene length is highly conserved in prokaryotes and eukaryotes and diverges only between the two kingdoms. Mol Biol Evol. 2006;23:1107–1108. doi: 10.1093/molbev/msk019. [DOI] [PubMed] [Google Scholar]
- Yun SH, Berbee ML, Yoder OC, Turgeon BG. Evolution of the fungal self-fertile reproductive life style from self-sterile ancestors. Proc Natl Acad Sci U S A. 1999;96:5592–5597. doi: 10.1073/pnas.96.10.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ. Protein-length distributions for the three domains of life. Trends Genet. 2000;16:107–109. doi: 10.1016/s0168-9525(99)01922-8. [DOI] [PubMed] [Google Scholar]
- Zhouravleva G, Frolova L, Legoff X, Leguellec R, Inge-Vechtomov S, Kisselev L, Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide-chain release factors, eRF1 and eRF3. EMBO J. 1995;14:4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.