

Summary
Lambda integrase (Int) forms higher-order protein–DNA complexes necessary for site-specific recombination. The carboxy-terminal domain of Int (75–356) is responsible for catalysis at specific core-type binding sites whereas the amino-terminal domain (1–70) is responsible for cooperative arm-type DNA binding. Alanine scanning mutagenesis of residues 64–70, within full-length integrase, has revealed differential effects on cooperative arm binding interactions that are required for integrative and excisive recombination. Interestingly, while these residues are required for cooperative arm-type binding on both P′1,2 and P′2,3 substrates, cooperative binding at the arm-type sites P′2,3 was more severely compromised than binding at arm-type sites P′1,2 for L64A. Concomitantly, L64A had a much stronger effect on integrative than on excisive recombination. The arm-binding properties of Int appear to be intrinsic to the amino-terminal domain because the phenotype of L64A was the same in an amino-terminal fragment (Int 1–75) as it was in the full-length protein.
Introduction
The λ Int recombinase catalyses the integration and excision of viral DNA into and out of the chromosome of its Escherichia coli host (Campbell, 1962; Nash, 1974). The product of recombination between viral attP (POP′) and bacterial attB (BOB′) sites is an integrated prophage bounded by hybrid att sites, attL (BOP′) and attR (POB′), which are themselves substrates for excisive recombination (Fig. 1). The strand nicking and joining events take place at the core sites within a synaptic higher-order structure that brings together two core regions bound by an Int tetramer. Recombination proceeds by means of an ordered pair of transesterification reactions that first generate and then resolve a Holliday junction recombination intermediate (Fig. 1; Hsu and Landy, 1984; Nunes-Düby et al., 1987; Kitts and Nash, 1988a,b). This mechanism comprises the hallmark for the tyrosine recombinase family.
Fig. 1.
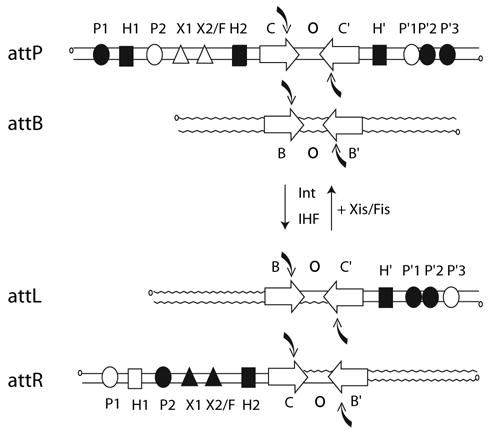
Location and occupancy of protein-binding sites during integrative and excisive recombination. Integrative recombination between the phage attP and bacterial attB sites requires the phage-encoded Int protein and the host encoded IHF to generate the attL and attR prophage sites. Excisive recombination between attL and attR additionally requires the phage-encoded Xis protein and is assisted by the host-encoded Fis protein. Protein binding sites in the arms of attP, attL and attR that are occupied during integration and excision are shown as filled symbols. Open symbols represent unoccupied sites. Four core-type sites (C, C′, B and B′) flank two 7 bp overlap regions (O) as inverted repeats (inverted open arrows) and encompass each of the DNA-cleavage sites (curved arrows). There are five arm-type Int-binding sites (circles): two single sites in the P arm (P1 and P2) and three adjacent sites in the P′ arm (P′1, P′2 and P′3). IHF has three binding sites, H1, H2 and H′ (squares), and Xis has two binding sites, X1 and X2 (triangles), one of which (X2) overlaps with the single Fis-binding site (F; not illustrated).
For the heterobivalent recombinases, such as λ Int, there is an overlay of additional complexity that is to be found in the phage DNA sequences (P and P′) flanking the core region in attP and distributed to each of the prophage att sites (BOP′ and POB′). Encoded within these DNA sequences are the five arm-type Int-binding sites and the six sites for the accessory DNA-bending proteins (Fig. 1). Two overlapping subsets of this ensemble are used for integrative and excisive recombination (Thompson et al., 1987; Numrych et al., 1990). This complexity facilitates the unique directional control of recombination that is characteristic of λ Int and related heterobivalent recombinases.
The λ Int protein can be cleaved by limited proteolysis into two fragments: a small amino-terminal fragment (1–64) and a large carboxy-terminal fragment (65–356). The carboxy-terminal domain is responsible for binding to core-type DNA (Tirumalai et al., 1998), and contains the catalytic residues (Kwon et al., 1997; Tirumalai et al., 1997). The amino-terminus has been shown to bind to arm-type DNA sites (Moitoso de Vargas et al., 1988; Kim et al., 1990; Sarkar et al., 2002; Wojciak et al., 2002), to bind cooperatively to Xis protein (Sarkar et al., 2002; Swalla et al., 2003; Warren et al., 2003) and to act as a modulator of recombinase functions (Sarkar et al., 2001).
The minimal arm-type DNA binding domain is Met-1 to Leu-64 (Sarkar et al., 2002). However, unlike fragments ending at Ser-70 and Thr-75, this fragment failed to show cooperativity when binding to a P′1,2 substrate (Sarkar et al., 2002). These results suggest that the residues between Leu-64 and Ser-70 mediate protein–protein interactions, although this conclusion is compromised by the inability to rule out salutary ‘end effects’; for example, a simple lengthening of the protein by five amino acids might have restored proper folding and/or context to some of the more proximal residues of the amino-terminal domain which were then able to promote protein–protein interactions. In this report we demonstrate that residues 64–70, within the context of full-length recombinogenic Int protein, are important for the cooperativity in binding to arm-type sites. These interactions are required for both cooperative P′1,2 and P′2,3 binding with the effect on P′2,3 being much greater than on P′1,2. These interactions also appear to be required for recombination because the mutant L64A was also defective for both integrative and excisive recombination. The defect in integrative recombination was greater than that for excisive recombination and correlates well with the arm-binding data. These data suggest that the nature of protein–protein interactions mediated by the amino-terminal domain differs between the two sets of arm-binding sites and, concomitantly, between the two forms of recombination.
Results and discussion
Residues 64–70 are required for cooperativity in arm-type DNA binding
To ascertain which residues in the 64–70 region are involved in cooperative arm-binding interactions in the context of full-length Int, we performed Alanine Scanning Mutagenesis and constructed a panel of five single-point mutants (L64A, R67A, I68A, N69A, S70A). The alanine at position 66 was not changed and the mutant at position 65 was not isolated. Each of the five mutant proteins, as well as wild-type Int, were examined for their ability to bind to neighbouring arm-type DNA sites. These experiments were carried out on a double-stranded, 50 bp radiolabelled oligonucleotide substrate containing the adjacent P′1,2 arm-type sites. All proteins yield DNA complexes with retarded electrophoretic mobilities relative to unbound DNA (P′1,2) that are consistent with singly (1x)-and doubly (2x)-bound protein complexes (Fig. 2A). However, for any given protein concentration the percentage of doubly bound species relative to total substrate bound is greater for wild-type Int than for any of the mutants (Fig. 2B).
Fig. 2.
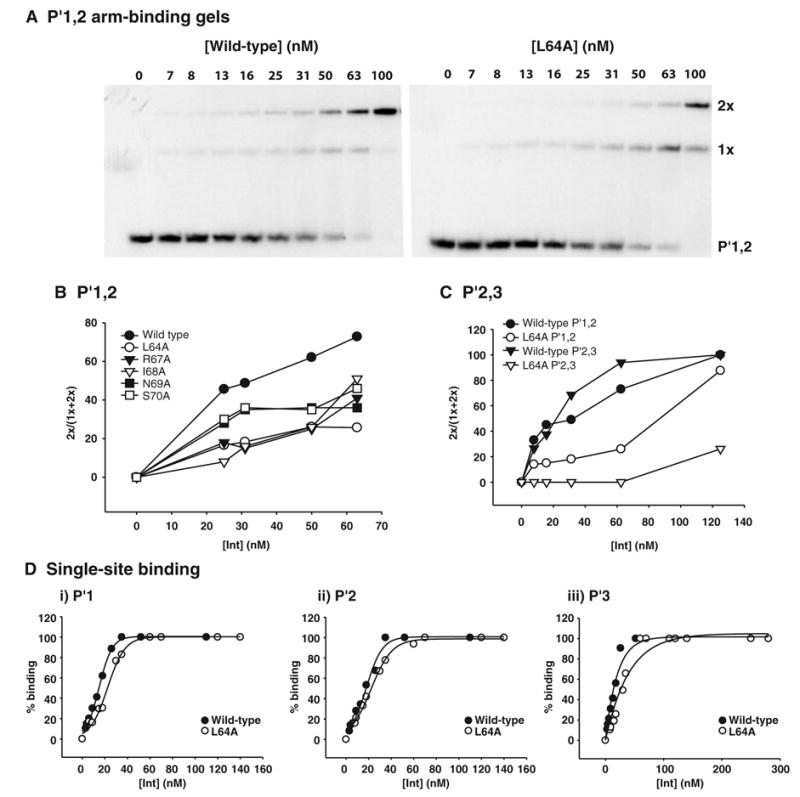
Residues within the 64–70 region are required for cooperative arm-binding. Full-length Int, various mutant proteins were tested in gel shift assays for their abilities to bind to the following arm-type DNA substrates; (A,B) 50 bp P′1,2, (C) 50 bp P′2,3 double arm-type sites and (D) 40 bp P′1, P′2 and P′3 single arm-type sites. The indicated concentrations of proteins were mixed with 50 nM and 10 nM radiolabelled DNA substrate for the single and double arm-type substrates, respectively, and incubated at 19°C for 30min. Reactions were analysed by electrophoresis on native 8% polyacrylamide gels which were subsequently dried and visualized by autoradiography. The intensity of the bands was measured as described in Experimental procedures. For the double and single arm-type sites the ratio of the doubly bound species to the total amount of substrate bound and the total amount of substrate bound has been calculated, respectively, and plotted as a function of protein concentration. Results are representative of at least three independent experiments.
Because L64A has one of the strongest phenotypes on P′1,2, it was also tested on the adjacent P′2,3 sites. With this substrate the difference between mutant and wild-type Int is even greater (Fig. 2C). Because wild-type Int binds more cooperatively to P′2,3 than to P′1,2 (this study; Sarkar et al., 2002), any mutant, such as L64A, that decreases cooperative arm-type binding might be expected to have a stronger effect on P′2,3 than on P′1,2 binding. The binding of L64A to the single sites P′1, P′2 and P′3 was comparable to wild-type Int (Fig. 2D). Taken together these results demonstrate that residues within the region 64–70 are required for cooperative arm-binding by full-length Int which correlates well with the results obtained with amino-terminal fragments (Sarkar et al., 2002).
The cooperative arm-binding defect of L64A is independent of the carboxy-terminus
There are protein–protein interactions between the amino- and carboxy-terminal domains in full-length proteins that affect carboxy-terminal functions (Sarkar et al., 2001). In particular, C65 is known to possess enhanced cleavage activity relative to wild-type, suggesting that the amino-terminal domain partially inhibits carboxy-terminal function in full-length proteins. To ascertain whether the L64A phenotype was due to a loss of interactions between the amino- and carboxy-terminal domains, we characterized L64A in the context of Int 1–75 (N1–75), a carboxy-terminus-truncated form of Int (Sarkar et al., 2002). These experiments were carried out on a double-stranded, 50 bp radiolabelled oligonucleotide substrate containing the two adjacent P′1,2 arm-type sites. The wild-type amino-terminal fragment Int 1–75 yields predominantly doubly bound (2x) protein complexes and only very small amounts of singly bound (1x) complexes (Fig. 3A). In contrast, the mutant Int 1–75L64A yields one complex with an electrophorectic mobility corresponding to the singly bound species. The fact that the L64A mutation has the same phenotype in both the amino-terminal fragment and full-length Int indicates that the deficiency in cooperative arm binding of full-length L64A is not due to the loss of an interaction between L64 and the carboxy-terminal domain.
Fig. 3. The defect in cooperative arm-binding of L64A is independent of the carboxy-terminus.
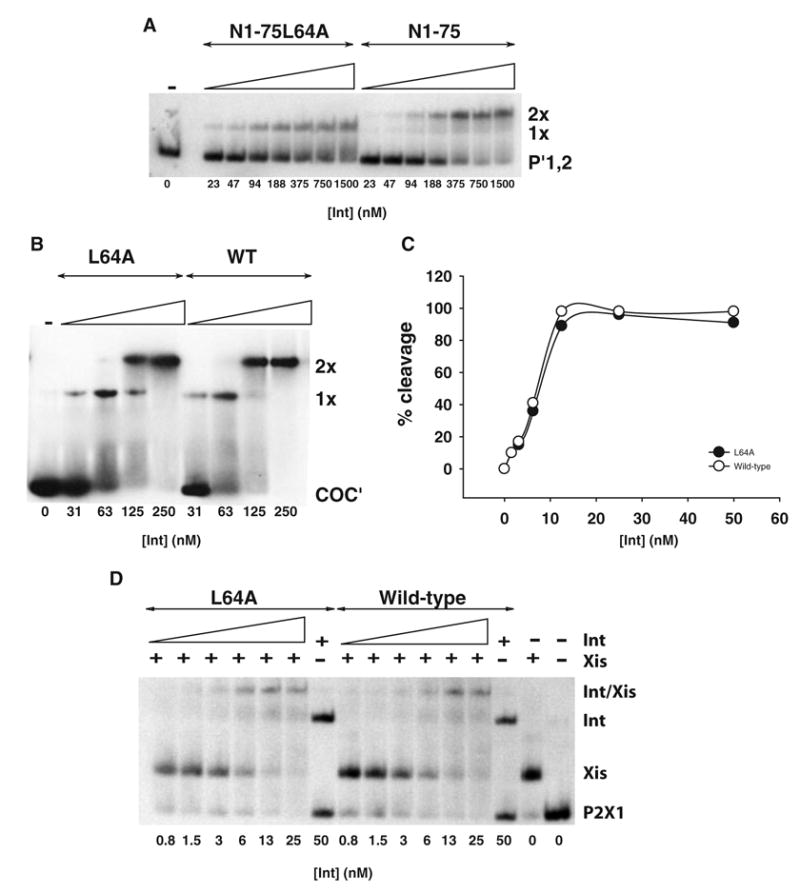
A. Int 1–75 (N1–75) and Int 1–75L64A (N1–75L64A) proteins were tested in a gel-shift assay for their abilities to bind to arm-type DNA sites. A total of 200 nM radiolabelled 50 bp DNA substrate containing the two adjacent P′1,2 sites was incubated with the indicated concentrations of proteins at 19°C for 30min.
B. Full-length wild-type and L64A proteins were tested in a gel shift assay for their abilities to bind to core-type substrate. In total, 100 nM radiolabelled 35 bp COC′ substrate was incubated with the indicated concentrations of protein at 19°C for 30min.
C. Full-length wild-type and L64A were tested for their abilities to cleave a nicked COC′ suicide substrate. The indicated concentrations of protein were mixed with 5 nM radiolabelled DNA substrate and incubated at 25°C for 30min. The percent cleavage of the nicked COC′ suicide substrate is plotted as a function of protein concentration.
D. Full-length wild-type and L64A proteins were tested in a gel-shift assay for their abilities to bind to a 40 bp duplex containing the P2X1 binding sites. The substrate DNA (13 nM) was preincubated at 19°C for 10 min with Xis (200 nM). Wild-type and L64A proteins (25 nM) were then added to the first tube followed by five serial two-fold dilutions and the reactions incubated at 19°C for 30min. Results are representative of at least two independent experiments.
To confirm the absence of a functional interaction between L64 and the carboxy-terminal domain, we tested the carboxy-terminal functions of the full-length wild-type and L64A mutant proteins. Using a double-stranded, 35 bp radiolabelled oligonucleotide substrate containing the two adjacent core-type sites, COC′, L64A and wild-type Int were found to bind comparably (Fig. 3B). Using a double-stranded, 31 bp radiolabelled nicked COC′ suicide substrate (Sarkar et al., 2002), the two proteins were also found to form covalent complexes with equal efficiency (Fig. 3C).
Taken together, these results suggest that the carboxy-terminal functions of full-length L64A are equivalent to those of wild-type Int and that the arm-binding properties of the amino-terminal domain are not influenced by the carboxy-terminal domain. Although there does not seem to be a functional interaction between L64 and the carboxy-terminal domain, interactions involving the carboxy-terminal domain may play a role in optimizing arm-type binding in full-length Int because the L64A defect is more severe in the context of the amino-terminal fragment than in the full-length protein (Figs 2A and 3A). This could reflect interactions between the core-binding and/or catalytic domains of two adjacent Int protomers.
Residue L64 is not required for Int-Xis-DNA ternary complex formation
To determine whether L64 was required for complex formation with DNA and Xis, we tested the ability of full-length wild-type and L64A Int proteins to form a ternary complex on a 40 bp duplex containing the adjacent binding sites P2 (Int) and X1 (Xis). When this DNA substrate is incubated with Xis or Int alone, single retarded bands are observed with relative mobilities consistent with the difference in size between the two proteins (Fig. 3D). Presence of both Int and Xis yields a third band that migrates more slowly than with Int or Xis alone (Fig. 3D). Wild-type Int and L64A form this complex with comparable efficiencies. This result suggests that L64 is dispensable when complexes between Int and Xis are formed.
Two distinct modes of amino-terminus-mediated protein–protein interactions
To directly test for a role of the amino-terminal domain of full-length Int in protein–protein interactions, we devised a coprecipitation assay based on Int fusions to a chitin-binding domain (CBD). Amino-terminal fragments of Int were expressed as CBD-fusion products and assayed for their ability to pull down either full-length Int or an amino-terminal truncation (C75). The presence of the CBD domain alone or the CBD–Int fusion was confirmed by immunoblotting with rabbit anti-CBD (Fig. 4B, lanes 2–4; 6–8). The CBD–Int fusions were also detected when immunoblotting performed with rabbit anti-Int (Fig. 4A, lanes 3,4,7 and 8). The CBD-binding domain alone fails to pull down Int (Fig. 4A, lane 2) although a low level of non-specific binding to C75 is observed (Fig. 4A, lane 6). The CBD–Int 1–75 fusion pulls down full-length Int (Fig. 4A, lane 4) but is unable to coprecipitate C75 any better than the CBD alone (Fig. 4A, lanes 6 and 8). These results suggest that the amino-terminal 75 residues of Int (fused to the CBD) are interacting with the first 74 residues of the full-length protein.
Fig. 4.
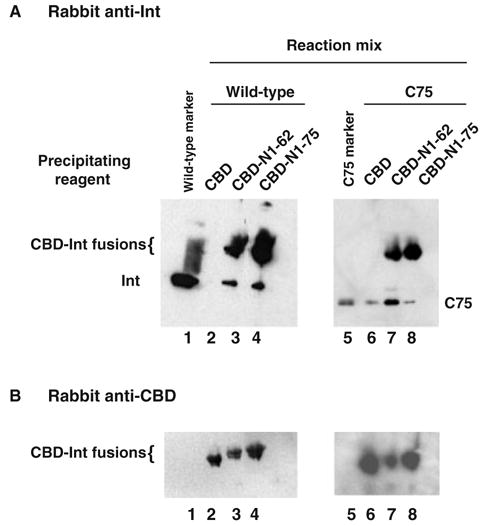
Two-distinct binding sites reside within the amino-terminus of Int. The chitin-binding domain (CBD) and the CBD–Int amino-terminal fusions (CBD–Int 1–62 and CBD–Int 1–75) were used to affinity precipitate full-length Int or C75, an amino-terminus-truncated Int as described in Experimental procedures. Briefly, reaction mixtures containing the appropriate Int protein, full-length wild-type or C75, were incubated with either the CBD alone or a CBD–Int fusion. The reactions were incubated at 4°C for 2.5 h and the CBD complexes precipitated via the addition of chitin. The precipitates were subsequently washed and loaded onto denaturing 12% SDS-PAGE gels. The proteins in the gel were subsequently transferred to PVDF membrane and analysed via Western blotting with (A) polyclonal rabbit anti-Int and (B) polyclonal rabbit antichitin binding domain antibodies. Results are representative of at least three independent experiments.
The CBD–Int 1–62 fusion brings down both full-length Int (Fig. 4A, lane 3) and C75 (Fig. 4A, lane 7). Previously published results demonstrated that the amino-terminal domain (Int 1–64) stimulates cleavage activity of C65 in trans (Sarkar et al., 2001) and it is possible that the functional interaction between Int 1–64 and C65 involves the same interface as the one responsible for the coprecipitation reported here between CBD–Int 1–62 and C75 (Fig. 4A, lane 7). Interestingly, the epitope within Int 1–62 that interacts with the carboxy-terminal domain of Int appears to be masked in the context of Int 1–75 because the CBD–Int 1–75 fusion is unable to bring down C75 (Fig. 4A, lane 8). Although the ability of Int residues 1–62 and 1–64 to interact with C75 and C65, respectively (this study, Sarkar et al., 2001), is only known to manifest itself in truncated forms of Int, the data suggest that the amino-terminal domain has the capacity to form at least two distinct surfaces for protein–protein interactions. Depending on their context, such differences may play a role in distinguishing between integrative- and excisive-competent recombinogenic complexes.
L64 and R67 are required for recombination
To determine whether the amino-terminal arm-binding interactions mediated by the region 64–70 are required for recombination, linear attL or attB substrates were recombined with supercoiled attR or attP substrates, respectively, with full-length wild-type Int and two of the alanine mutants, L64A and R67A. Whereas L64A and R67A are reduced relative to wild-type Int for both types of recombination, the effect of these mutations is much greater on integrative than on excisive recombination (Fig. 5A and B). Mutational inactivation of individual arm-type sites has shown that within the small cluster of three sites on the P′ arm, only P′1 and P′2 are required for excisive recombination, whereas P′2 and P′3 are primarily involved in integrative recombination (Bauer et al., 1986; Numrych et al., 1990). The stronger effect of L64A on P′2,3 than on P′1,2 binding and on integrative versus excisive recombination is consistent with these studies. We have shown that the differential effect of these mutants on integrative versus excisive recombination is not due to a general reduction in overall Int activity (Fig. 3B and C), as observed for a rare class of mutants isolated by Enquist et al. (Enquist and Weisberg, 1977; Enquist et al., 1979), and secondary effects on other residues in Int seem unlikely because mutations of two different residues produce the same phenotype.
Fig. 5.
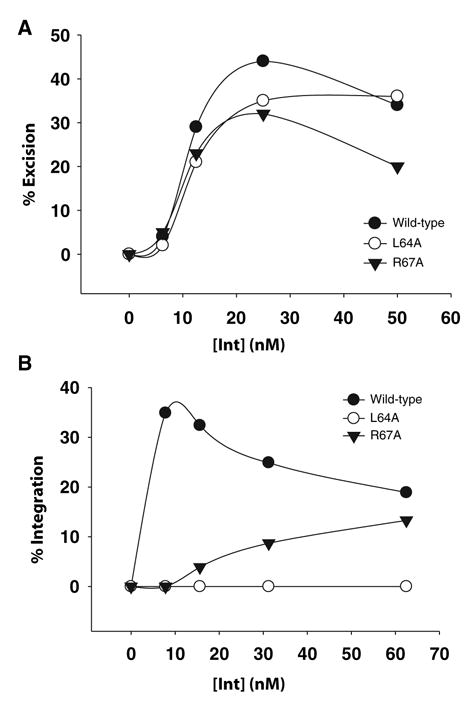
L64 is required for both excisive and integrative recombination. Recombination reactions were carried out as described in the Experimental procedures. Briefly, reaction mixtures containing the appropriate substrates (A) supercoiled attR and linear attL and (B) supercoiled attP and linear attB were incubated with full-length wild-type, L64A, or R67A Int. The reactions were allowed to proceed for 1 h at 25°C before being analysed by agarose gel electrophoresis. The percent recombination is plotted as a function of protein concentration. Results are representative of two independent experiments.
Other studies have also examined amino-terminus-mediated protein–protein interactions necessary for recombination. Although cross-linking studies have implicated C25 of the amino-terminal domain in protein–protein interactions (Jessop et al., 2000) and its modification by N-ethylmaleimide (NEM) abolishes recombination (Tirumalai et al., 1996), C25 is dispensable for recombination, as judged by mutagenesis (Tirumalai et al., 1997), and is suggested to be near the DNA-binding surface by NMR studies (Wojciak et al., 2002). A mutant, R42L, isolated in the amino-terminal domain of HK022 Int, which is 86% identical to λ Int for the first 64 amino acids, has been shown to increase binding to P′1,2,3 10-fold more than to P′1, as if it had enhanced protein–protein interactions (Cheng et al., 2000). This is consistent with the NMR structure of the amino-terminal domain of λ Int in which the position of residue R42 is near a solvent-exposed hydrophobic surface that might be involved in protein–protein interactions. In addition, substitutions at Int position 47 also affect amino-terminus-mediated protein–protein interactions (Swalla et al., 2003; Warren et al., 2003). These studies demonstrate that E47 is required for both Int–Int and Int–Xis interactions and show that this residue is essential for excisive recombination and contributes to the efficiency of the integrative reaction.
Conclusion
The results reported here identify a site of protein–protein interaction within the amino-terminus of full-length Int which is necessary for cooperative arm-binding interactions required for recombination. This site has a differential importance in integrative and excisive recombination. We further suggest that for any given site of protein–protein interaction within the amino-terminus its function may be dependent on its context.
Experimental procedures
Oligonucleotides and mutagenesis
The single-point mutations were cloned into the full-length (pRT101) and the amino-terminal Int 1–75 (pDS203) Int expression vectors using the Quikchange site-directed mutagenesis kit (Stratagene) with the following SDS-PAGE-purified oligonucleotides (Operon Technologies, Alameda, CA):
L64A 5′-GATTCTCGCTGTCGCAGGCTTGTGTTTGTG-3′;
R67A 5′-GGAATTATCACTGTTGATCGCCGCTGTCAGAGGCTTGTG-3′;
I68A 5′-GGAATTATCACTGTTCGCTCTCGCTGTCAG-3′;
N69A 5′-GGAATTATCACTCGCGATTCTCGCTGTCAG-3′;
S70A 5′-CGTAACGGAATTATCCGCGTTGATTCTCGC-3′ and their complementary strands.
Sequences of the top strands for each of the double-stranded substrates used in this work are as follows with Int binding sites noted in bold caps:
40 bp P′1 5′-CATTGCTCAACGAACAGGTCACTATTCATTGATTTCATGG-3′;
40 bp P′2 5′-CGCTCAACGAACTATCAGTCAAAATAAATTGATTTCATGG-3′;
40 bp P′3 5′-CATTGCTCAACGATAAAATCATTATTTGATTTCATACTGG-3′;
50 bp P′1,2 5′-CATTGCTCAACGAACAGGTCACTATCAGTCAAAATTTGATTTCATACCAT-3′;
50 bp P′2,3 5′-CATTGCGCAACGAACCAGTCAAAATAAAATCATTATTTGATTTCATACCT;
40 bp P′2 × 1 5′-TAGGATTCATAGTGACTGCATATGTTGTGTTTTCGAGATG-3′;
35 bp COC′ 5′-GTATTGCCAGCTTTATTCAACAAAGTTGGAGCAGT-3′;
31 bp nicked COC′ 5′-TCGAGCAGCTTTTTT( )ATATTAAGTTGGAATT-3′ (the empty parentheses indicate a nick). The bottom strand of the nicked COC′ substrate was unnicked.
SDS-PAGE-purified substrate oligonucleotides were 5′ end-labelled with 32P by the use of phage T4 polynucleotide kinase (Sambrook Mol Cloning) and were separated from free label by using a Biospin P-30 chromatography column (Bio-Rad). Oligos were annealed with unlabelled complementary partner strands in 10 mM Tris-HCl pH 7.5 containing 50 mM NaCl.
Protein expression and purification
Proteins were produced in E. coli BL21(DE3)pRIL from expression plasmids under the control of the T7 promoter. Full-length Int proteins, amino-terminal fragments, and integration host factor (IHF) were expressed and purified as described previously (Nash et al., 1987; Chong et al., 1998; Tirumalai et al., 1998). The Met-Gly-(His)10-(Ser)2-Gly-His-Ile-Glu-Gly-Arg-His-tagged Xis protein was purified as described previously (Warren et al., 2003) with one modification. After extensive washing with 10 column volumes (100 ml) of potassium phosphate buffer containing 0.5 M NaCl, Xis was stepwise eluted off the column using potassium phosphate buffers containing 0.8, 1.2, 1.6 and 2.2 M sodium chloride. The majority of Xis eluted off the column in the 1.2 M fraction. The concentrations of all proteins were estimated by the dye-binding method using bicinchoninic acid (Pierce) (Smith et al., 1985; Wiechelman et al., 1988). The purity of all the proteins was > 90% as judged by Coomassie Blue staining of overloaded SDS-polyacrylamide gels.
Gel retardation and Cleavage assays. Binding of Int proteins to double-stranded P′1,2, P′2,3, COC′ and P2 × 1 DNA substrates was carried out as previously described (Sarkar et al., 2001; Sarkar et al., 2002) in 10 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5% glycerol, 0.5 mg ml−1 BSA, 1 mM EDTA and 2.5 mM DTT. In arm-binding experiments, 20 ng μ−1l sheared herring sperm DNA was also present. In all cases the proteins were mixed with DNA and incubated for 30 min at 19°C. The reactions were analysed by electrophoresis (100 V, 4–6 h) through native, non-denaturing 8% (w/v) polyacrylamide gels.
DNA cleavage by Int protein was assayed using a ‘top-strand’-nicked COC′ suicide substrate (Pargellis et al., 1988; Tekle et al., 2002). The cleavage reactions were performed at 25°C for 30min in reaction mixtures containing 10 mM Tris (pH 8.0), 50 mM NaCl, 1 mM EDTA, 0.5 mg ml−1 BSA, 2.5 mM DTT and 5 nM end-labelled substrate at indicated amounts of protein. The reactions were terminated by adding 0.2% SDS and analysed by electrophoresis on a 8% (w/v) SDS-polyacrylamide gel. The gels were dried, visualized by autoradiography and scanned for phosphor image analysis (Fuji film). The bands corresponding to free DNA or protein–DNA complex were quantified with MacBAS-2500 image analysis software. All quantification was performed within the linear response range.
Affinity-precipitation reaction
The CBD and the CBD–Int amino-terminal fusions (CBD–Int 1–62 and CBD–Int 1–75) were purified as described (Chong et al., 1998) with one modification. The DTT-mediated cleavage step was omitted and the proteins were recovered bound to chitin beads. The affinity precipitation reactions were carried out in Tris buffer (10 mM Tris (pH 8.0), 0.5 mg ml−1 BSA, 5% glycerol, 1 mm EDTA (pH 8.0), 50 mM NaCl) containing 10 nM full-length Int or the carboxy-terminal fragment C75 (Sarkar et al., 2002). One hundred microlitre of the appropriate CBD fusion protein were added to 900 μl of Tris buffer containing either full-length Int or C75 and incubated with gentle mixing at 4°C for 2.5 h. The CBD complexes were pelleted by the addition of chitin followed by centrifugation (4000 r.p.m., 4°C, 2 min). The complexes were subsequently washed three times with 1 ml of Tris buffer minus Int. After the final wash, the beads were resuspended in 50 μl of 2x SDS-PAGE loading solution, boiled, briefly centrifuged and the protein extract transferred to a clean eppendorf tube. The extracts were then analysed via SDS-PAGE and Western analysis. The proteins were electrotransferred to polyvinylidene difluoride (PVDF) membranes (U.S. Biochemicals) which were blocked with 5% BSA and 0.1% Tween 20 for at least 1 h. Blotting was performed according to standard protocols using polyclonal rabbit anti-Int, polyclonal rabbit antichitin-binding domain (S6654S, NEB), and horseradish peroxidase-conjugated goat antirabbit immunoglobulin (NA934, Amersham). Blots were visualized with ECL from Amersham (RPN 2106) and exposed to Kodak XAR-5 film.
Recombination assays
Recombination reactions were carried out as described previously (Nunes-Düby et al., 1987) in Tris recombination buffer (0.03 pmol assay−1 linear attL or attB, 0.02 pmol assay−1 supercoiled attR or attP, 5 mM EDTA pH 8, 6 mM Spermidine, 25 mM Tris (pH 8), 0.5 mg ml−1 BSA, 2.5 mM DTT, 30 mM NaCl and 2 U assay−1 IHF. One unit being defined as the minimum amount of protein giving maximum amount of product under standard conditions (Nunes-Düby et al., 1995). For excisive recombination reactions the buffer was further supplemented with 60 nM Xis. In all experiments, Int proteins were mixed with the DNA in the assay mix and incubated for 1 h at 25°C. The reactions were stopped by the addition of SDS containing loading solution and analysed by electrophoresis (100 V, 4 h) through 1.2% agarose gels in Trisacetate-EDTA buffer. The gels were stained with 0.5 ng ml−1 Ethidium Bromide solution and quantified with the Gel Doc-It system from UVP according to the manufacturers instructions.
Acknowledgments
We thank Joan Boyles for manuscript preparation; Christine Lank for technical assistance; Simone Nunes-Düby and members of the Ellenberger and Landy laboratories for advice and helpful discussions. This work was supported by National Institutes of Health Grants GM62723 and GM33928 to A.L.
References
- Bauer CE, Hesse SD, Gumport RI, Gardner JF. Mutational analysis of integrase arm-type binding sites of bacteriophage lambda. J Mol Biol. 1986;192:513–527. doi: 10.1016/0022-2836(86)90273-1. [DOI] [PubMed] [Google Scholar]
- Campbell AM. Episomes. In: Caspari EW, Thoday JM, editors. Advances in Genetics. New York: Academic Press; 1962. pp. 101–145. [Google Scholar]
- Cheng Q, Swalla BM, Beck M, Alcaraz R, Jr, Gumport RI, Gardner JF. Specificity determinants for bacteriophage Hong Kong 022 integrase: analysis of mutants with relaxed core-binding specificities. Mol Microbiol. 2000;36:424–436. doi: 10.1046/j.1365-2958.2000.01860.x. [DOI] [PubMed] [Google Scholar]
- Chong S, Montello GE, Zhang A, Cantor EJ, Liao W, Xu MQ, Benner J. Utilizing the C-terminal cleavage activity of a protein splicing element to purify recombinant proteins in a single chromatographic step. Nucl Acids Res. 1998;22:5109–5115. doi: 10.1093/nar/26.22.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist L, Weisberg R. Flexibility in attachment site recognition by λ integrase. In: Bukhari A, Shaprio J, Adhya S, editors. DNA Insertion Elements, Plasmids and Episomes. New York: Cold Spring Harbor Press; 1977. pp. 343–348. [Google Scholar]
- Enquist LW, Kikuchi A, Weisberg RA. The role of 1 integrase in integration and excision. Cold Spring Harb Symp. 1979;43:1115–1120. doi: 10.1101/sqb.1979.043.01.124. [DOI] [PubMed] [Google Scholar]
- Hsu PL, Landy A. Resolution of synthetic att-site Holliday structures by the integrase protein of bacteriophage λ. Nature. 1984;311:721–726. doi: 10.1038/311721a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop L, Bankhead T, Wong D, Segall AM. The amino terminus of bacteriophage λ integrase is involved in protein–protein interactions during recombination. J Bacteriol. 2000;182:1024–1034. doi: 10.1128/jb.182.4.1024-1034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Moitoso de Vargas L, Nunes-Düby SE, Landy A. Mapping of a higher order protein-DNA complex: two kinds of long–range interactions in λ attL. Cell. 1990;63:773–781. doi: 10.1016/0092-8674(90)90143-3. [DOI] [PubMed] [Google Scholar]
- Kitts PA, Nash HA. An intermediate in the phage λ site-specific recombination reaction is revealed by phosphorothioate substitution in DNA. Nucl Acids Res. 1988a;16:6839–6856. doi: 10.1093/nar/16.14.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitts PA, Nash HA. Bacteriophage λ site-specific recombination proceeds with a defined order of strand-exchanges. J Mol Biol. 1988b;204:95–108. doi: 10.1016/0022-2836(88)90602-x. [DOI] [PubMed] [Google Scholar]
- Kwon HJ, Tirumalai RS, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the λ integrase catalytic core. Science. 1997;276:126–131. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitoso de Vargas L, Pargellis CA, Hasan NM, Bushman EW, Landy A. Autonomous DNA binding domains of λ integrase recognize different sequence families. Cell. 1988;54:923–929. doi: 10.1016/0092-8674(88)90107-9. [DOI] [PubMed] [Google Scholar]
- Nash HA. Purification of bactriophage λ Int protein. Nature. 1974;247:543–545. doi: 10.1038/247543a0. [DOI] [PubMed] [Google Scholar]
- Nash HA, Robertson CA, Flamm E, Weisberg RA, Miller HI. Overproduction of Escherichia coli integration host factor, a protein with nonidentical subunits. J Bacteriol. 1987;169:4124–4127. doi: 10.1128/jb.169.9.4124-4127.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numrych TE, Gumport RI, Gardner JF. A comparison of the effects of single-base and triple-base changes in the integrase arm-type binding sites on the site-specific recombination of bacteriophage lambda. Nucl Acids Res. 1990;18:3953–3959. doi: 10.1093/nar/18.13.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Düby SE, Matsumoto L, Landy A. Site-specific recombination intermediates trapped with suicide substrates. Cell. 1987;50:779–788. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- Nunes-Düby SE, Smith-Mungo LI, Landy A. Single base-pair precision and structural rigidity in a small IHF-induced DNA loop. J Mol Biol. 1995;253:228–242. doi: 10.1006/jmbi.1995.0548. [DOI] [PubMed] [Google Scholar]
- Pargellis CA, Nunes-Düby SE, Moitoso de Vargas L, Landy A. Suicide recombination substrates yield covalent λ integrase-DNA complexes and lead to identification of the active site tyrosine. J Biol Chem. 1988;263:7678–7685. [PubMed] [Google Scholar]
- Sarkar D, Azaro MA, Aihara H, Papagiannis C, Tirumalai RS, Nunes-Düby SE, et al. Differential affinity and cooperativity functions of the amino-terminal 70 residues of λ integrase. J Mol Biol. 2002;324:775–789. doi: 10.1016/s0022-2836(02)01199-3. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Radman-Livaja M, Landy A. The small DNA binding domain of λ Int is a context-sensitive modulator of recombinase functions. EMBO J. 2001;20:1203–1212. doi: 10.1093/emboj/20.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Swalla BM, Cho EH, Gumport RI, Gardner JF. The molecular basis of cooperative DNA binding between lambda integrase and excisionase. Mol Microbiol. 2003;50:89–99. doi: 10.1046/j.1365-2958.2003.03687.x. [DOI] [PubMed] [Google Scholar]
- Tekle M, Warren DJ, Biswas T, Ellenberger T, Landy A, Nunes-Düby SE. Attenuating functions of the carboxyl-terminus of λ integrase. J Mol Biol. 2002;324:649–665. doi: 10.1016/s0022-2836(02)01108-7. [DOI] [PubMed] [Google Scholar]
- Thompson JF, Moitoso de Vargas L, Skinner SE, Landy A. Protein–protein interactions in a higher-order structure direct lambda site-specific recombination. J Mol Biol. 1987;195:481–493. doi: 10.1016/0022-2836(87)90177-x. [DOI] [PubMed] [Google Scholar]
- Tirumalai RS, Healey E, Landy A. The catalytic domain of λ site-specific recombinase. Proc Natl Acad Sci USA. 1997;94:6104–6109. doi: 10.1073/pnas.94.12.6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumalai RS, Kwon H, Cardente E, Ellenberger T, Landy A. The recognition of core-type DNA sites by λ Integrase. J Mol Biol. 1998;279:513–527. doi: 10.1006/jmbi.1998.1786. [DOI] [PubMed] [Google Scholar]
- Tirumalai RS, Pargellis CA, Landy A. Identification and characterization of the NEM-sensitive site in lambda integrase. J Biol Chem. 1996;271:29599–29604. doi: 10.1074/jbc.271.47.29599. [DOI] [PubMed] [Google Scholar]
- Warren D, Sam M, Manley K, Sarkar D, Lee SY, Abbani M, et al. Identification of the λ integrase surface that interacts with the Xis accessory protein reveals a residue that is also critical for homomeric dimer formation. Proc Natl Acad Sci USA. 2003;100:8176–8181. doi: 10.1073/pnas.1033041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechelman KJ, Braun RD, Fitzpatrick JD. Investigation of the bicinchoninic acid protein assay: identification of the groups responsible for color formation. Anal Biochem. 1988;175:231–237. doi: 10.1016/0003-2697(88)90383-1. [DOI] [PubMed] [Google Scholar]
- Wojciak JM, Sarkar D, Landy A, Clubb RT. Arm-site binding by the lambda integrase protein: solution structure and functional characterization of its amino-terminal domain. Proc Natl Acad Sci USA. 2002;99:3434–3439. doi: 10.1073/pnas.052017999. [DOI] [PMC free article] [PubMed] [Google Scholar]