

Abstract
High mobility group box 1 (HMGB1) can be actively secreted by macrophages/monocytes in response to exogenous and endogenous inflammatory stimuli (such as bacterial endotoxin, TNF-α, IL-1, and IFN-γ) or passively released by necrotic cells and mediates innate and adaptive inflammatory responses to infection and injury. Here, we demonstrated that a reactive oxygen species, hydrogen peroxide (H2O2), induces active and passive HMGB1 release from macrophage and monocyte cultures in a time- and dose-dependent manner. At nontoxic doses (e.g., 0.0125–0.125 mM), H2O2 induced HMGB1 cytoplasmic translocation and active release within 3–24 h. At higher concentrations (e.g., 0.25 mM), however, H2O2 exhibited cytotoxicity to macrophage and monocyte cell cultures and consequently, triggered active and passive HMGB1 release. In addition, H2O2 stimulated potential interaction of HMGB1 with a nuclear export factor, CRM1, in macrophage/monocyte cultures. Inhibitors specific for the JNK (SP600125) and MEK (PD98059), but not p38 MAPK (SB203580), abrogated H2O2-induced, active HMGB1 release. Together, these data establish an important role for oxidative stress in inducing active HMGB1 release, potentially through a MAPK-and CRM1-dependent mechanism.
INTRODUCTION
Oxidative stress is caused by an excessive accumulation of reactive oxygen species (ROS) and has been implicated in the pathogenesis of a growing number of inflammatory diseases, including arthritis, ischemia/reperfusion (I/R) injury, stroke, and sepsis. Excessive production of ROS can lead to damage of macromolecules (such as lipids, proteins, nucleic acids, or carbohydrates), thereby affecting normal cellular functions [1]. MAPKs can be activated by a variety of stimuli, including growth factors, hormones, and ROS, and are involved in a number of physiological (e.g., cell proliferation and differentiation) [2] and pathological (oxidative stress/damage) processes [3, 4]. However, the molecular mechanism by which ROS causes oxidative stress/damage is poorly characterized.
A ubiquitous nuclear protein, high-mobility group box 1 protein (HMGB1), has been established recently as a crucial mediator of lethal systemic inflammatory diseases [5-7]. Residing predominantly in the nucleus of quiescent macophages/monocytes, HMGB1 can be secreted actively by macrophages/monocytes in response to exogenous and endogenous inflammatory stimuli such as endotoxin, CpG DNA, TNF-α, IL-1, and IFN-γ [6, 8]. In addition, HMGB1 can be released passively by necrotic cells [9]. Once released, extracellular HMGB1 mediates a wide range of biological responses in diverse cell types and tissues. In vitro, extracellular HMGB1 can activate macrophages, monocytes [10], and promote dendritic cell (DC) maturation [11-13]. In vivo, HMGB1 cause acute lung inflammation, epithelial-cell barrier leaking, and even death [14, 15]. Moreover, increased levels of HMGB1 are found in patients with sepsis and other major inflammatory diseases including rheumatoid arthritis [5, 16].
However, the potential role of oxidative stress in the regulation of HMGB1 release has not been investigated previously. These studies were therefore undertaken to determine the effects of hydrogen peroxide (H2O2)-induced oxidative stress/damage on the potential active or passive HMGB1 release in macrophage and monocyte cultures. Here, we demonstrated that H2O2 stimulates macrophages/monocytes to actively release HMGB1, potentially through a MAPK- and CRM1-dependent mechanism.
MATERIALS AND METHODS
Cell culture and treatment
Murine macrophage-like RAW 264.7 cells were obtained from the Shanghai Type Culture Collection (China) and cultured in RPMI-1640 medium (Life Technologies, Gaithersburg, MD) supplemented with 10% heat-inactivated FBS, 2 mM glutamine, and an antibiotic-antimycotic mix in a humidified incubator with 5% CO2 and 95% air. Human PBMCs were isolated from healthy donor blood by Ficoll density gradient centrifugation and cultured in RPMI-1640 medium/10% heat-inactivated human serum/2 mM glutamine overnight. Nonadherent cells were subsequently removed, and adherent monocyte-enriched cultures were stimulated with H2O2 at various concentrations.
H2O2 was diluted in PBS and further diluted in culture medium. The specific MAPK inhibitors PD98059 (Promega, Madison, WI), SB203580 (Promega), and SP600125 (KangChen Biotechnology, China) were dissolved in DMSO. Cells were pretreated for 30 min with or without MAPK inhibitor prior to the addition of H2O2.
Cell viability assay
Cells were plated at a density of 104 cells/well on 96-well plates in 100 μl RPMI. Cell viability was evaluated using the conventional MTT reduction assays, where cells of each microwell were incubated with 20 μl 0.5% MTT for 2 h at 37°C, and the reaction was stopped by adding 150 μl DMSO. The amount of MTT formazan product was determined by measuring absorbance using a micro-plate reader (Bio-Rad, Hercules, CA) at a test wavelength of 570 nm and a reference wavelength of 630 nm.
Preparation of cellular extracts
At indicated time-points after the treatment, cells were harvested and washed twice with cold PBS; nuclear and cytoplasmic extracts were prepared according to the method of Schreiber et al. [17]. Briefly, the cell pellet was resuspended in 400 μl cold buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF). The cells were allowed to swell on ice for 15 min, after which 25 μl of a 10% solution of Nonidet P-40 (NP-40) was added, and the tube was vortexed vigorously for 10 s. The homogenate was centrifuged for 30 s, and the nuclear pellet was resuspended in 50 μl ice-cold buffer B (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF). After vigorously rocking at 4°C for 15 min on a shaking platform, the nuclear extract was centrifuged for 5 min in a microfuge at 4°C, and the supernatant was frozen in aliquots at −80°C. The protein content of the different fractions was determined by a Bradford method.
Western blotting analysis
After various treatments, cell-conditioned medium was harvested and filtered through Millex-GP (Millipore, Bedford, MA) to remove cell debris and macromolecular complexes. Samples were then concentrated 40-fold with Amicon Ultra-4-10000 NMWL (Millipore) following the manufacturer's instructions. Proteins in the whole-cell lysate, subcellular fractions, or concentrated cell culture supernatants were resolved on 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride membrane. After blocking the membrane at room temperature for 6 h, the membrane was incubated for 2 h with various primary antibodies specific for HMGB1, CRM1, proliferating cell nuclear antigen (PCNA; BD Biosciences, San Jose, CA), β-actin, and GAPDH (KangChen Biotechnology), respectively. After incubation with peroxidase-conjugated secondary antibodies for 1 h at 25°C, the signals were visualized by 3,3′-diaminobenzidine detection (Boster Biotech, China) according to the manufacturer's instruction.
Immunocytochemical analysis
Cells were cultured on glass coverslips and fixed in 4% formaldehyde for 30 min at room temperature prior to detergent extraction with 0.1% Triton X-100, 10 min at 4°C. Coverslips were saturated with PBS containing 2% BSA for 1 h at room temperature and processed for immunofluorescence with anti-HMGB1 antibody (BD Biosciences), followed by Cy3-conjugated Ig (Sigma Chemical Co., St. Louis, MO) and Hoechst 33258 (Sigma Chemical Co.). Between all incubation steps, cells were washed three times for 3 min with PBS containing 0.2% BSA. Coverslips were mounted on slides using Movio (Sigma Chemical Co.). Fluorescence signals were analyzed using a fluorescence microscope (Nikon, Japan).
Coimmunoprecipitation analysis
Nuclear extracts were lysed at 4°C in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, protease inhibitor cocktail). Lysates were cleared by centrifugation at 12,000 g for 10 min and then incubated for 2 h or overnight at 4°C with 5 μg/mL of the appropriate antibody and protein A or G agarose/sepharose beads (Amersham Biosciences, Sweden). Immune complexes were washed extensively with PBS, and proteins were eluted by boiling in 2× SDS sample buffer. Proteins were assayed by Western blotting as above.
Statistical analysis
Significance of differences between groups was determined by two-tailed Student's t-test or Fisher's LSD test, as indicated. P < 0.05 was considered significant.
RESULTS
H2O2 induces HMGB1 release in RAW 264.7 macrophages and human PBMCs
The effects of H2O2 on cell viability were first determined by MTT reduction assay (Fig. 1A). At lower (0.0125–0.125 mM) concentrations, H2O2 was not toxic to macrophage cultures. At higher (0.25–1.0 mM) concentrations, however, H2O2 exhibited a dose-dependent cytotoxicity to macrophage cultures.
Fig. 1.
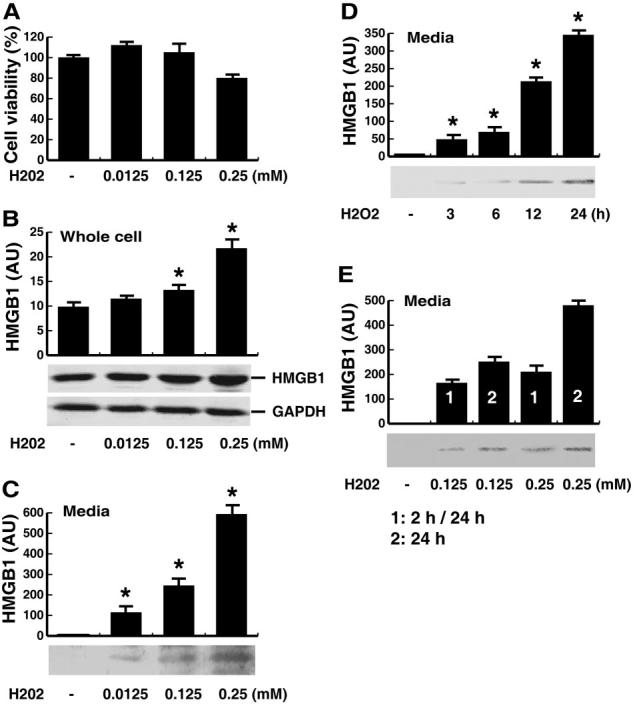
Effects of H2O2 on the release of HMGB1 in macrophage cultures. (A) RAW 264.7 macrophages were stimulated with H2O2 at indicated doses for 24 h, and cell viability was determined by MTT assay and expressed as mean ± sem of four experiments in duplicate. In parallel experiments, HMGB1 levels in the whole-cell lysate (B) or culture medium (C–E) were determined by the relative optical intensity [in arbitrary units (AU)] of the immunoreactive bands on Western blots and expressed as mean ±sem of three experiments in duplicate. *, Statistically significant versus control group; P < 0.05. (D) Levels of HMGB1 in the culture medium at various time-points following stimulation with H2O2 at a nontoxic dose (0.125 mM). (E) Levels of HMGB1 in the culture medium at 24 h after the onset of a brief (for 2 h, after which H2O2 was washed out; marked as “1” or “2 h/24 h”) or a persistent (for 24 h; marked as “2” or “24 h”) exposure to H2O2 at indicated concentrations.
To determine if H2O2 induces active HMGB1 release in the absence of cell death, macrophage cell cultures and cell-conditioned medium were collected separately and assayed for HMGB1 levels by Western blotting analysis. At nontoxic doses (e.g., 0.0125–0.125 mM), H2O2 increased cellular HMGB1 protein levels slightly (Fig. 1B) and induced active HMGB1 release within 3–24 h (Fig. 1, C and D). At higher concentrations (e.g., 0.25 mM), however, H2O2 exhibited cytotoxicity to macrophage cell cultures (Fig. 1A) and consequently, triggered a more pronounced, robust HMGB1 release, possibly as a result of active and passive HMGB1 release (Fig. 1C). The inducible nature of HMGB1 release was reproduced in cultures of primary human PBMCs, which actively released HMGB1 within 3 h after stimulation with H2O2 at nontoxic concentrations (0.125 mM, data not shown).
To gain insight into the mechanisms underlying H2O2-mediated HMGB1 release, H2O2 was washed out after a brief (2-h) exposure, and HMGB1 levels in the culture medium were determined 24 h after the onset of H2O2 stimulation. A brief exposure of macrophage cultures to H2O2 at doses nontoxic (0.125 mM) or otherwise low toxic (0.25 mM if exposed persistently) did not reduce cell viability significantly (data not shown) but triggered a marked HMGB1 release (Fig. 1E).
H2O2 induces the translocation of HMGB1 in RAW 264.7 macrophages and human PBMCs
To determine if H2O2 induces HMGB1 cytoplasmic translocation, macrophage and monocyte cultures were immunostained with HMGB1-specific antibodies. In agreement with previous reports [18-21], HMGB1 was noted predominantly in the nucleus of quiescent macrophages and monocytes (Fig. 2A). At 12 h after H2O2 treatment, HMGB1 staining was observed in nuclear and cytoplasmic regions of macrophage and monocyte cultures (Fig. 2A). To confirm HMGB1 cytoplasmic translocation, cytoplasmic and nuclear fractions of macrophage or monocyte cultures were isolated and immunoblotted with antibodies specific for HMGB1, PCNA (a nuclear protein), or β-actin (a cytoplasmic protein), respectively. Consistently, levels of HMGB1 in the cytoplasmic fractions were increased dramatically after treatment with H2O2 at a nontoxic concentration (0.125 mM; Fig. 2B).
Fig. 2.
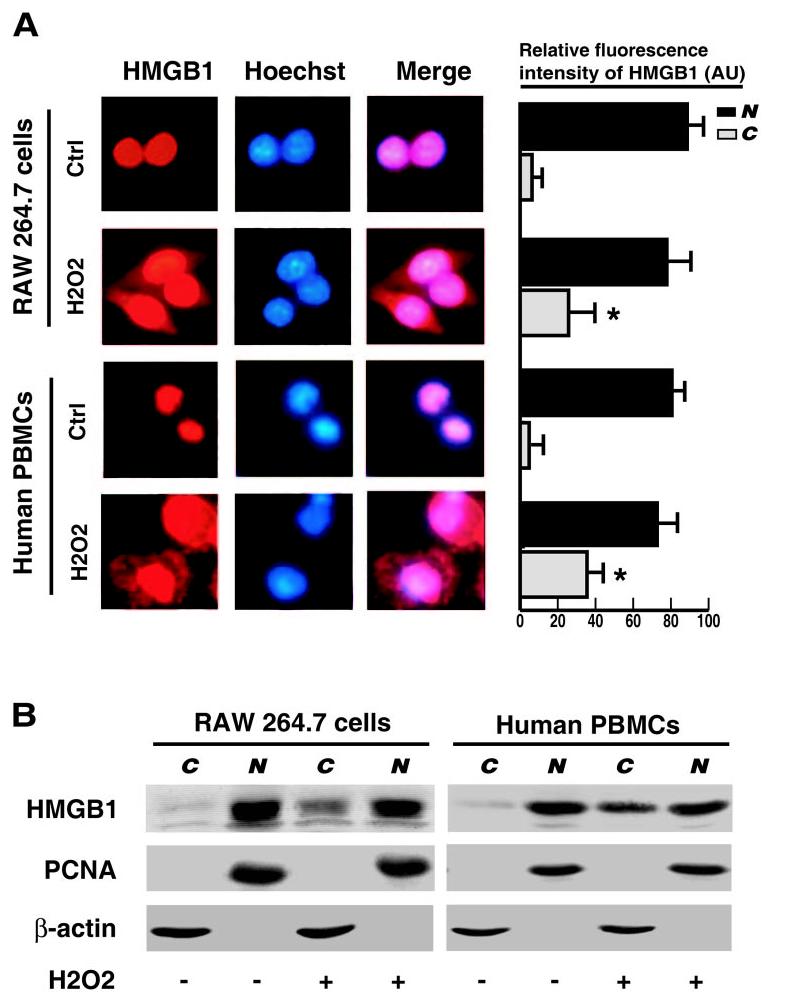
Effects of H2O2 on HMGB1 cytoplasmic translocation in macrophage and monocyte cultures. RAW 264.7 macrophages and human PBMCs were stimulated with H2O2 at a nontoxic concentration (0.125 mM) and monitored for HMGB1 cytoplasmic translocation by immunocytochemistry (A) or cell fractionation/Western blot (B) at 12 h poststimulation (A). The relative fluorescence intensity in the nuclear (“N”) or cytoplasmic (“C”) regions of multiple representative cells was determined using the ImageProPlus software and expressed as mean ± sem (in arbitrary units) of three independent experiments. Red, HMGB1; blue, nuclei; pink, Merge (original magnification, ×400). (B) Following cell fractionation, HMGB1 content in the cytoplasmic (“C”) or nuclear (“N”) fraction was determined by Western blotting analysis. Equal loading of samples was confirmed by Western blotting analysis of each fraction with antibodies specific for a nuclear (PCNA, BD Biosciences) or cytoplasmic (β-actin, KangChen Biotechnology) protein. Blots are representative of three independent experiments with similar results.
H2O2 increased interaction of HMGB1 with a nuclear export factor, CRM1, in macrophage and monocyte cultures
The nuclear pore complex (NPC) is a 125-MDa nuclear envelope complex, which mediates nucleocytoplasmic shuttling of RNA and protein. NPC is comprised of more than 30 different proteins, including Ran/TC4, importins, and exportins. CRM1 is a widely expressed importin involved in the maintenance of chromatin structure and export of nuclear proteins. To determine the potential involvement of CRM1 in H2O2-mediated HMGB1 cytoplasmic translocation, we first determined whether H2O2 affects CRM1 levels in macrophage and monocyte cultures. Following H2O2 stimulation, the relative CRM1 content (as normalized by a cytoplasmic housekeeping protein, GAPDH) was increased slightly in macrophage and monocyte cultures (Fig. 3A), implicating a possibility that H2O2 may induce CRM1 cytoplasmic translocation. To examine potential CRM1-HMGB1 interaction, nuclear proteins were immunoprecipitated with CRM1- or HMGB1-specific antibodies, and the precipitated protein complexes were subsequently assayed for HMGB1 or CRM1 content by Western blotting analysis. In quiescent macrophage or monocyte cultures, a small amount of HMGB1 was coimmunoprecipitated with CRM1 protein, indicating a poor interaction between HMGB1 and CRM1. After H2O2 exposure, however, significant, higher amounts of HMGB1 were coimmunoprecipitated with CRM1 (Fig. 3B), indicating an increase in HMGB1-CRM1 interaction in H2O2-stimulated macrophage/monocyte cultures. In light of the potential role of CRM1 in LPS-induced HMGB1 release [22], it is plausible that CRM1 may be involved in H2O2-mediated HMGB1 cytoplasmic translocation in macrophage and monocyte cultures.
Fig. 3.
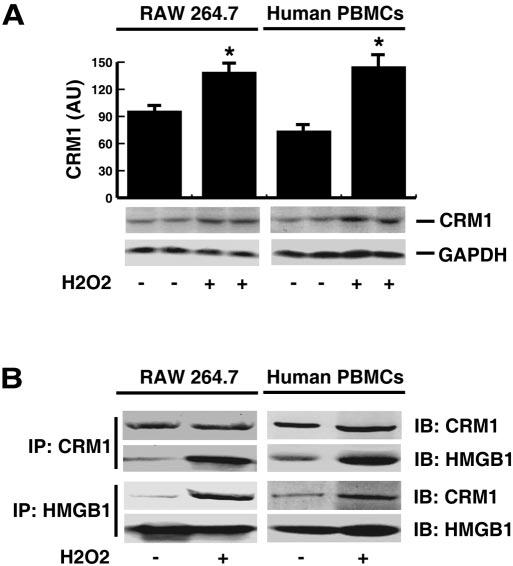
Effects of H2O2 on CRM1 expression and interaction with HMGB1 in macrophage and monocytes cultures. (A) RAW 264.7 macrophages and human PBMCs were stimulated with H2O2 at a nontoxic concentration (0.125 mM) for 24 h, and whole-cell lysate assayed CRM1 content by Western blotting analysis. Shown in the bar graph was the relative optical intensity (in arbitrary units) of the CRM1-immunoreactive band. A cytoplasmic housekeeping protein, GAPDH, was used as a loading control. In parallel experiments, nuclear fractions were isolated and immunoprecipitated (“IP”) with CRM1- or HMGB1-specific antibodies. The precipitated proteins were subsequently immunoblotted (“IB”) with CRM1- or HMGB1-specific antibodies. Blot shown is representative of three experiments with similar results.
SP600125 and PD98059 inhibit H2O2-induced HMGB1 release and translocation in RAW 264.7 macrophages and human PBMCs
In response to various stimuli, the p38, JNK, and ERK MAPKs are phosphorylated quickly. Indeed, H2O2 effectively induced phosphorylation of p38, JNK, and ERK MAPK in macrophages and monocyte cultures (data not shown). To determine the potential involvement of various MAPK pathways in H2O2-induced HMGB1 release, we determined whether specific inhibitors for p38 (SB203580), JNK (SP600125), or MEK (PD98059) affect H2O2-induced HMGB1 release. Pretreatment of macrophage cell cultures with inhibitors for JNK and MEK, but not p38 MAPK, dose-dependently attenuated H2O2-induced HMGB1 release (Fig. 4A). Consistently, pretreatment with JNK inhibitor dramatically reduced cytoplasmic HMGB1 levels in H2O2-stimulated macrophages (Fig. 4B), suggesting that JNK inhibitor attenuates H2O2-induced HMGB1 release through interfering with its cytoplasmic translocation.
Fig. 4.
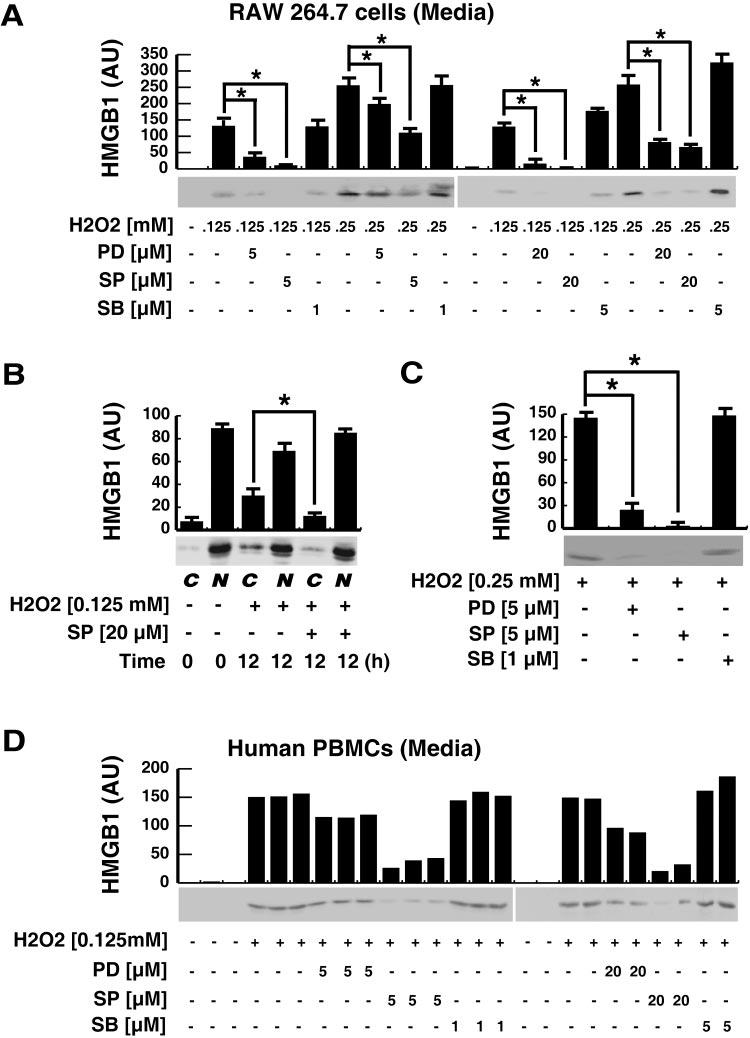
Effects of MAPK inhibitors on H2O2-induced HMGB1 translocation and release in macrophage and monocyte cultures. (A, B) RAW 264.7 macrophages were pretreated with various MAPK inhibitors [SB203580 (SB), SP600125 (SP), PD98059 (PD)] at indicated concentrations for 30 min before stimulation with H2O2 at indicated doses. At 12 h post-H2O2 stimulation, levels of HMGB1 in the culture medium (A) or cellular cytoplasmic or nuclear fractions (B) were determined by Western blotting analysis, and the relative optical intensity of HMGB1-immunoreactive band (in arbitrary units) was expressed as mean ± sem of three experiments in duplicate. *, P < 0.05. (C) RAW 264.7 macrophages were pretreated with various MAPK inhibitors for 30 min before exposure to 0.25 mM H2O2. At 2 h post-H2O2 stimulation, H2O2 was washed out, and HMGB1 levels in the culture medium were determined at 24 h after the onset of H2O2 exposure. The relative optical intensity of HMGB1-immunoreactive band (in arbitrary units) was expressed as mean ± sem of three experiments in duplicate. *, P < 0.05. (D) Human PBMCs (n = 5 per group) were similarly pretreated with various MAPK inhibitors (SB203580, SP600125, PD98059), at indicated concentrations for 30 min before addition of H2O2 (0.125 mM). At 6 h post-H2O2 stimulation, the levels of HMGB1 in the culture medium were assayed by Western blotting analysis and expressed as the relative optical intensity of a HMGB1-immunoreactive band (in arbitrary units). Blots are representative of three independent experiments with similar results.
As mentioned above, a brief exposure of macrophage cultures to H2O2 at a dose (0.25 mM) otherwise slightly toxic if exposed persistently (for 24 h) did not reduce cell viability significantly (data not shown) but triggered a marked HMGB1 release (Figs. 1E and 4C). This HMGB1 release was almost completely abrogated by specific inhibitors for JNK and MEK, but not p38 MAPK (Fig. 4C), when they were used at well-documented, nontoxic concentrations [23-27].
To confirm the role of MAPKs in H2O2-induced HMGB1 release, we examined the effects of various MAPK inhibitors on H2O2-induced HMGB1 release in human monocyte cultures. Similarly, specific inhibitors for the JNK and MEK, but not p38 MAPK, significantly attenuated HMGB1 release induced by H2O2 at nontoxic doses (Fig. 4D). Taken together, these data support a potential role for JNK and ERK, but not p38 MAPK, in H2O2-induced HMGB1 release in macrophage and monocyte cultures.
DISCUSSION
HMGB1, a 30-kDa nuclear protein, widely studied as a transcription factor and growth factor, has been identified recently as a cytokine mediator of lethal systemic inflammation (e.g., endotoxemia and sepsis), arthritis, and local inflammation (e.g., hepatic injury after I/R and LPS-induced acute lung injury) [5, 14, 16, 28, 29]. In response to exogenous or endogenous inflammatory stimuli (such as bacterial endotoxin, LPS, TNF-α, IL-1, or IFN-γ), monocytes/macrophages and pituicytes actively release HMGB1 [5, 18, 19, 30]. In addition, HMGB1 is released passively from necrotic or damaged cells [9] and triggers an inflammatory response to injury. Extracellular HMGB1 can activate macrophages, monocytes, DC, and T cells and mediates a rigorous innate and adaptive inflammatory response to infection and injury [11-13, 31].
Although various HMGB1-inducing, inflammatory stimuli (such as LPS, TNF-α, and IFN-γ) can also stimulate macrophages and monocytes to produce oxidative-free radicals, the potential role of oxidative stress (such as H2O2) in the regulation of HMGB1 release was unknown previously. Consistent with the notion that LPS and hypoxia up-regulate HMGB1 expression in vivo [29, 32], we found that H2O2, at non- to low toxic doses (e.g., 0.0125–0.25 mM), similarly increases cellular HMGB1 expression levels in macrophage cultures. It is currently unknown whether up-regulation of HMGB1 expression influences its subsequent release in response to oxidative stress.
It is more important that we demonstrated that oxidative stress could effectively stimulate macrophage/monocyte cell cultures to actively secrete HMGB1 at nontoxic doses (e.g., 0.0125–0.125 mM). Consistently, H2O2 induces dramatic cytoplasmic translocation of HMGB1 in macrophage and monocyte cell cultures. At higher concentrations (e.g., 0.25 mM), however, H2O2 exhibited a dose-dependent cytotoxicity and consequently, triggered a more robust release of HMGB1 as a possible result of active secretion from activated cells and passive leakage from injured cells. It is notable that a brief exposure with H2O2 was enough to induce active HMGB1 release, implicating a potential involvement of early signaling molecules (such as MAPKs) in the regulation of H2O2-induced HMGB1 release.
Various MAPKs play important roles in physiological and pathological events, including cell-cycle regulation, proliferation, apoptosis, and inflammation [2, 33]. We observed that specific inhibitors for JNK (SP600125) [23] and MEK (PD98059) [34], but not for p38 MAPK (SB203580) [35], almost completely abrogated H2O2-induced, active HMGB1 release. The suppression of H2O2-induced HMGB1 release by JNK and MEK inhibitors was not a result of their cell toxicity, as at concentrations effectively abrogating H2O2-induced HMGB1 release, these inhibitors did not affect cell viability of macrophage cultures. Taken together, our experimental results support a potential role for JNK and ERK (but not p38) MAPKs in the regulation of H2O2-induced, HMGB1 active release.
The mechanisms underlying regulation of active HMGB1 release are complex and still remain elusive. For instance, early studies indicate that HMGB1 is secreted by macro-phages/monocytes via a nonclassical, vesicle-mediated, secre-tory pathway [22, 36], but the identity of the HMGB1-containing vesicles is not yet fully characterized. Although ERK MAPK is not important in the regulation of endotoxin- or IFN-γ-mediated HMGB1 release [18, 20], it may be important in H2O2-induced HMGB1 translocation and release. It is thus plausible that macrophages/monocytes use distinct signaling pathways to trigger HMGB1 release in response to different inflammatory stimuli.
The CRM1 protein is a leucine-rich nuclear export signal receptor involved in export of nuclear proteins [37] such as HMGB1 [22]. In the present study, we demonstrated that H2O2-induced HMGB1 release is accompanied by an increase in CRM1-HMGB1 interaction. In light of the important role of CRM1 in export of nuclear RNA and proteins [38], it will be important to determine whether oxidative stress induces active HMGB1 release through a CRM1-dependent mechanism in future studies.
In summary, we demonstrated here that oxidative stress such as H2O2 induces active and/or passive HMGB1 release from macrophage and monocyte cultures in a time- and dose-dependent manner. In view of the observations that specific inhibitors for JNK and ERK (but not p38) abrogated H2O2-induced HMGB1 release and that H2O2 enhanced CRM1-HMGB1 interaction significantly, we propose that oxidative stress induces active HMGB1 release in a MAPK and CRM1-dependent mechanism. Thus, it may be important to explore the therapeutic potential of many antioxidant agents in the treatment of various inflammatory diseases.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Sciences Foundation of China (30500485, 30330280, and 30371382), the Major National Basic Research Program of China (G2000056908), and the Innovative Program of Central South University for Post-Graduate Research (2005-75239). It was supported in part by grants from the National Institute of General Medical Sciences (NIGMS; to H. W.). We thank Dr. Giddabasappa Anand (College of Optometry, University of Houston, TX) for critical reading of the manuscript, Meidong Liu for help in preparing reagents, and Mr. Haibo Chen (Center Laboratory, Dalian Medical University) for help in fluorescence intensity assay.
REFERENCES
- 1.Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr. Pharm. Des. 2004;10:1611–1626. doi: 10.2174/1381612043384664. [DOI] [PubMed] [Google Scholar]
- 2.Pouyssegur J. Signal transduction. An arresting start for MAPK. Science. 2000;290:1515–1518. doi: 10.1126/science.290.5496.1515. [DOI] [PubMed] [Google Scholar]
- 3.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 4.Handley ME, Thakker M, Pollara G, Chain BM, Katz DR. JNK activation limits dendritic cell maturation in response to reactive oxygen species by the induction of apoptosis. Free Radic. Biol. Med. 2005;38:1637–1652. doi: 10.1016/j.freeradbiomed.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 6.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 7.Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J. Leukoc. Biol. 2005;78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 8.Jiang W, Li J, Gallowitsch-Puerta M, Tracey KJ, Pisetsky DS. The effects of CpG DNA on HMGB1 release by murine macrophage cell lines. J. Leukoc. Biol. 2005;78:930–936. doi: 10.1189/jlb.0405208. [DOI] [PubMed] [Google Scholar]
- 9.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 10.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Muller S, Iannacone M, Traversari C, Bianchi ME, Manfredi AA. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, Tracey KJ, Chiorazzi N. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 2004;173:307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 13.Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur. J. Immunol. 2005;35:2184–2190. doi: 10.1002/eji.200526066. [DOI] [PubMed] [Google Scholar]
- 14.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 15.Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, Inoue K, Yamada S, Ijiri K, Matsunaga S, Nakajima T, Komiya S, Maruyama I. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–981. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 17.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, Susarla S, Czura C, Mitchell RA, Chen G, Sama AE, Tracey KJ, Wang H. IFN-γ induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J. Immunol. 2003;170:3890–3897. doi: 10.4049/jimmunol.170.7.3890. [DOI] [PubMed] [Google Scholar]
- 19.Tang D, Shi Y, Jang L, Wang K, Xiao W, Xiao X. Heat shock response inhibits release of high mobility group box 1 protein induced by endotoxin in murine macrophages. Shock. 2005;23:434–440. doi: 10.1097/01.shk.0000159556.95285.df. [DOI] [PubMed] [Google Scholar]
- 20.Chen G, Li J, Ochani M, Rendon-Mitchell B, Qiang X, Susarla S, Ulloa L, Yang H, Fan S, Goyert SM, Wang P, Tracey KJ, Sama AE, Wang H. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J. Leukoc. Biol. 2004;76:994–1001. doi: 10.1189/jlb.0404242. [DOI] [PubMed] [Google Scholar]
- 21.Chen G, Li J, Qiang X, Czura CJ, Ochani M, Ochani K, Ulloa L, Yang H, Tracey KJ, Wang P, Sama AE, Wang H. Suppression of HMGB1 release by stearoyl lysophosphatidylcholine: an additional mechanism for its therapeutic effects in experimental sepsis. J. Lipid Res. 2005;46:623–627. doi: 10.1194/jlr.C400018-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyper-acetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morita Y, Naka T, Kawazoe Y, Fujimoto M, Narazaki M, Nakagawa R, Fukuyama H, Nagata S, Kishimoto T. Signal transducers and activators of transcription (STAT)-induced STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1 (SOCS-1) suppresses tumor necrosis factor α-induced cell death in fibroblasts. Proc. Natl. Acad. Sci. USA. 2000;97:5405–5410. doi: 10.1073/pnas.090084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shyu KG, Chao YM, Wang BW, Kuan P. Regulation of discoidin domain receptor 2 by cyclic mechanical stretch in cultured rat vascular smooth muscle cells. Hypertension. 2005;46:614–621. doi: 10.1161/01.HYP.0000175811.79863.e2. [DOI] [PubMed] [Google Scholar]
- 27.Sim S, Yong TS, Park SJ, Im KI, Kong Y, Ryu JS, Min DY, Shin MH. NADPH oxidase-derived reactive oxygen species-mediated activation of ERK1/2 is required for apoptosis of human neutrophils induced by Entamoeba histolytica. J. Immunol. 2005;174:4279–4288. doi: 10.4049/jimmunol.174.7.4279. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Yang H, Tracey KJ. Extracellular role of HMGB1 in inflammation and sepsis. J. Intern. Med. 2004;255:320–331. doi: 10.1111/j.1365-2796.2003.01302.x. [DOI] [PubMed] [Google Scholar]
- 29.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Vishnubhakat JM, Bloom O, Zhang M, Ombrellino M, Sama A, Tracey KJ. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 1999;126:389–392. [PubMed] [Google Scholar]
- 31.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 32.Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor α, interleukin-1β, -6, and high-mobility-group protein-1 in skeletal muscle. Shock. 2003;19:538–546. doi: 10.1097/01.shk.0000055237.25446.80. [DOI] [PubMed] [Google Scholar]
- 33.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 34.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 35.Wilson KP, McCaffrey PG, Hsiao K, Pazhanisamy S, Galullo V, Bemis GW, Fitzgibbon MJ, Caron PR, Murcko MA, Su MS. The structural basis for the specificity of pyridinylimidazole inhibitors of p38 MAP kinase. Chem. Biol. 1997;4:423–431. doi: 10.1016/s1074-5521(97)90194-0. [DOI] [PubMed] [Google Scholar]
- 36.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 38.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]