

Summary
N-acetylglucosaminyltransferase V (GnT-V) is an enzyme involved in the biosynthesis of asparagine-linked oligosaccharides. It is responsible for the transfer of N-acetylglucosamine (GlcNAc) from the nucleotide sugar donor, UDP-GlcNAc, to the 6 position of the α-1–6 linked Man residue in N-linked oligosaccharide core structures. GnT-V up-regulation has been linked to increased cancer invasiveness and metastasis and, appropriately, targeted for drug development. However, drug design is impeded by the lack of structural information on the protein and the way in which substrates are bound. Even though the catalytic domain of this type II membrane protein can be expressed in mammalian cell culture, obtaining structural information has proved challenging due to the size of the catalytic domain (95 kDa) and its required glycosylation. In this manuscript, we present an experimental approach to obtaining information on structural characteristics of the active site of GnT-V through the investigation of the bound conformation and relative placement of its ligands, UDP-GlcNAc and β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)-β-D-GlcpOOctyl. Nuclear magnetic resonance (NMR) spectroscopy experiments, inducing transferred nuclear Overhauser effect (trNOE) and saturation transfer difference (STD) experiments, were used to characterize the ligand conformation and ligand-protein contact surfaces. In addition, a novel paramagnetic relaxation enhancement experiment using a spin-labeled ligand analogue, UDP-TEMPO, was used to characterize the relative orientation of the two bound ligands. The structural information obtained for the substrates in the active site of GnT-V can be useful in the design of inhibitors for GnT-V.
Keywords: UDP-N-acetylglucosamine, glycosyltransferase, NOE, STD, spin-label
Introduction
Proteins are often characterized as to function, connected to disease, and validated as potential drug targets prior to complete physical characterization. This is the case for the N-acetylglucosaminyltransferase V (GnT-V, also called GnT-VA),1,2,3,4 which is clearly linked to the progression of carcinomas by a number of studies, including those with GnT-V null mice that show inhibition of breast carcinoma progression and metastasis.5 GnT-V has been sequenced, expressed, and kinetically evaluated using various substrates, but no structural information exists. While drugs can be developed in the absence of structural information, this lack of knowledge certainly impedes progress. Ideally, three dimensional structures of proteins are obtained through investigations of the protein itself using X-ray crystallography or NMR spectroscopy, but these approaches often prove challenging. In this manuscript we explore the possibility of obtaining structural information about the active site of GnT-V indirectly through investigation of the conformation and relative placement of its natural ligands and analogues of those ligands. The approach is based on widely used NMR techniques for the characterization of ligand conformation (transfer nuclear Overhauser effect, trNOE, spectroscopy) and ligand-protein contact surfaces (saturation transfer difference, STD, spectroscopy). It also incorporates a novel approach to the relative placement of ligands using spin-labeled analogues of one ligand to restrict placement of a second ligand.
GnT-V and its family members represent challenging targets for structural characterization for a number of reasons. GnT-V is classified as a family 18 glycosyltransferase in the CAZy database (Carbohydrate Active Enzymes database, http://www.cazy.org/).6,7 Currently, this family has no structural representatives in the Protein Data Bank.8 GnT-V and other members of family 18 are type II membrane proteins. Membrane proteins are challenging for structural characterization; fortunately, the catalytic domain of GnT-V without the membrane domain has been successfully expressed and purified.9 The catalytic domain has three, occupied N-glycosylation consensus sites, at least some of which are necessary for maintaining activity.10 To attain proper glycosylation it is necessary to express the protein in mammalian cells. The resulting heterogeneous glycosylation and the limited yields in mammalian cell expression are an obstacle to the production of crystals suitable for X-ray crystallography. Heterogeneity of glycosylation is less of an obstacle to NMR structure determination. However, even a minimal construct for the catalytic domain of 66 kDa (produced by proteolysis) is large for structure determination by NMR. Also, the need to express GnT-V in mammalian cells makes uniform 13C, 15N isotope labeling used in NMR structure determination prohibitively expensive, and 2H labeling used for work on large proteins nearly impossible.
With respect to substrate interaction as a potential probe of active site structure, GnT-V offers a number of opportunities. GnT-V catalyzes the transfer of an N-acetylglucosamine (GlcNAc) from the nucleotide sugar donor, UDP-GlcNAc, to the 6 position of the α-1–6 linked Man residue in an oligosaccharide containing the native β-D-GlcpNAc-(1→2)-α-D-Manp-(1→3)-[β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)]-D-Manp motif, or a close synthetic analogue. The addition occurs with inversion of configuration at the anomeric site, consistent with a mechanism in which both substrates occupy the active site simultaneously. In our case we will use a synthetic acceptor analogue discovered by Srivastava, et al., β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)-β-D-GlcpOOctyl, in place of native acceptors that are more difficult to obtain.11 The synthetic acceptor contains the required β-D-GlcpNAc-(1→2)-α-D-Manp but has a (1→6) linked Glc rather than Man due to increased ease of synthesis. The Glc is linked to an octyl chain that allows for separation of product in the GnT-V activity assay by reverse-phase solid-phase extraction,12 but is also required for enzyme transfer, since acceptors lacking the octyl group show very little or no transfer of substrate. The donor substrates used were the native UDP-GlcNAc and an analogue that has a TEMPO derivative in place of the GlcNAc moiety. The presence of both, rather large, donor and acceptor substrates presents the opportunity of probing a substantial portion of the protein’s active site pocket.
The dominant methods for determining bound conformations and binding motifs of substrates include trNOE spectroscopy and STD spectroscopy. Use of trNOEs has been extensively reviewed and there are a number of applications to carbohydrates bound to proteins.13 trNOEs are acquired using standard 2D NOE sequences to monitor correlations among ligand resonances and provide distance constraints between proximate pairs of protons. Use in cases where ligands are rapidly exchanging between bound and free forms is particularly advantageous because excess ligand (10–30 X) can be used to improve sensitivity while NOEs from the bound state still dominate the observed average. This weighted average occurs because of the linear dependence of magnetization transfer rates on correlation times for large systems. STD spectroscopy is of more recent vintage and is used to identify binding epitopes on the surfaces of ligands. Spectra are collected with and without saturation of regions of the spectrum containing only protein resonances. In the presence of excess ligand, differences between the spectra primarily show resonances belonging to ligand protons in close proximity to protein protons. STD depends on the efficient spin diffusion of magnetization among protons within large proteins and the transfer of this magnetization from protein to ligand protons in a 1/r6 dependent fashion.
The third method used for investigation of ligand geometry is intended to retrieve information on the relative placement of the two ligands in the binding pocket. In principle, this could be determined using inter-ligand NOEs from the trNOE experiments, but these data are limited by the requirement of very close approach and reduction of signal due to partial occupations of each site. An alternate approach uses perturbation of spin relaxation by the presence of an unpaired electron on one of the ligands. Relaxation of spins on the other ligand is enhanced with the same 1/r6 distance dependence as an NOE, but because the magnetic moment of an electron is on the order of 1000 times that of a proton, the distance range is larger. The unpaired electron in our case was introduced by replacing the GlcNAc in UDP-GlcNAc with a nitroxide containing TEMPO moiety. This compound is a novel addition to a set of tools that might be used to investigate properties of a number of glycosyltransferases. Use of TEMPO analogues to retrieve structural constraints is well established in systems where the nitroxides are covalently attached to a protein and amide protons of the protein are observed; here distances between protons and nitroxide as large as 20 Å have been characterized.14 In our case we detect relaxation effects by introducing spin relaxation delays in the preparation period of 1H-13C HSQC experiment of rapidly exchanging ligands. The 2D HSQC experiment provides the resolution needed to individually monitor effects on a large number of sites on the acceptor in the presence of the TEMPO analogue of the sugar donor.
Modeling of all data collected provides a picture of bound ligand geometry that constrains the active site of a structurally uncharacterized protein. In principle, the geometry could be used to begin to rationally design inhibitors that could modulate activity of an enzyme whose activity can regulate the invasiveness and metastatic potential of malignant cells.
Results
Binding Epitopes from Saturation Transfer Difference
Saturation transfer difference spectroscopy was used to determine the binding epitopes of the acceptor, the donor (UDP-GlcNAc), and the non-paramagnetic form of the donor analogue (UDP-TEMPOH). The results of these experiments are illustrated in Figures 1, 2, and 3. Each figure includes a reference spectrum of the compound, the STD spectrum of the compound in the presence of GnT-V, and the normalized STD percentages. The indicated assignments of resonances were completed using COSY and TOCSY as well as reference to literature assignments.15,16 The background signal from GnT-V was subtracted from each STD spectrum to improve the baseline and analysis of the spectra. The highest STD percentages for UDP-GlcNAc and UDP-TEMPOH are localized on the uridine portion of the molecules with values greater than 60% for H6, H5, H1’, H2’, and H3’. A saturation frequency of 7.1 ppm was used instead of 0.0 ppm for the UDP-TEMPOH experiment to avoid saturating impurities in the UDP-TEMPOH sample that have resonances near 0.0 ppm. To rule out the possibility that this change alters response, a STD experiment with UDP-GlcNAc was performed with 7.1 ppm irradiation. The STD values were very similar to the values obtained at 0.0 ppm (data not shown).
Figure 1.
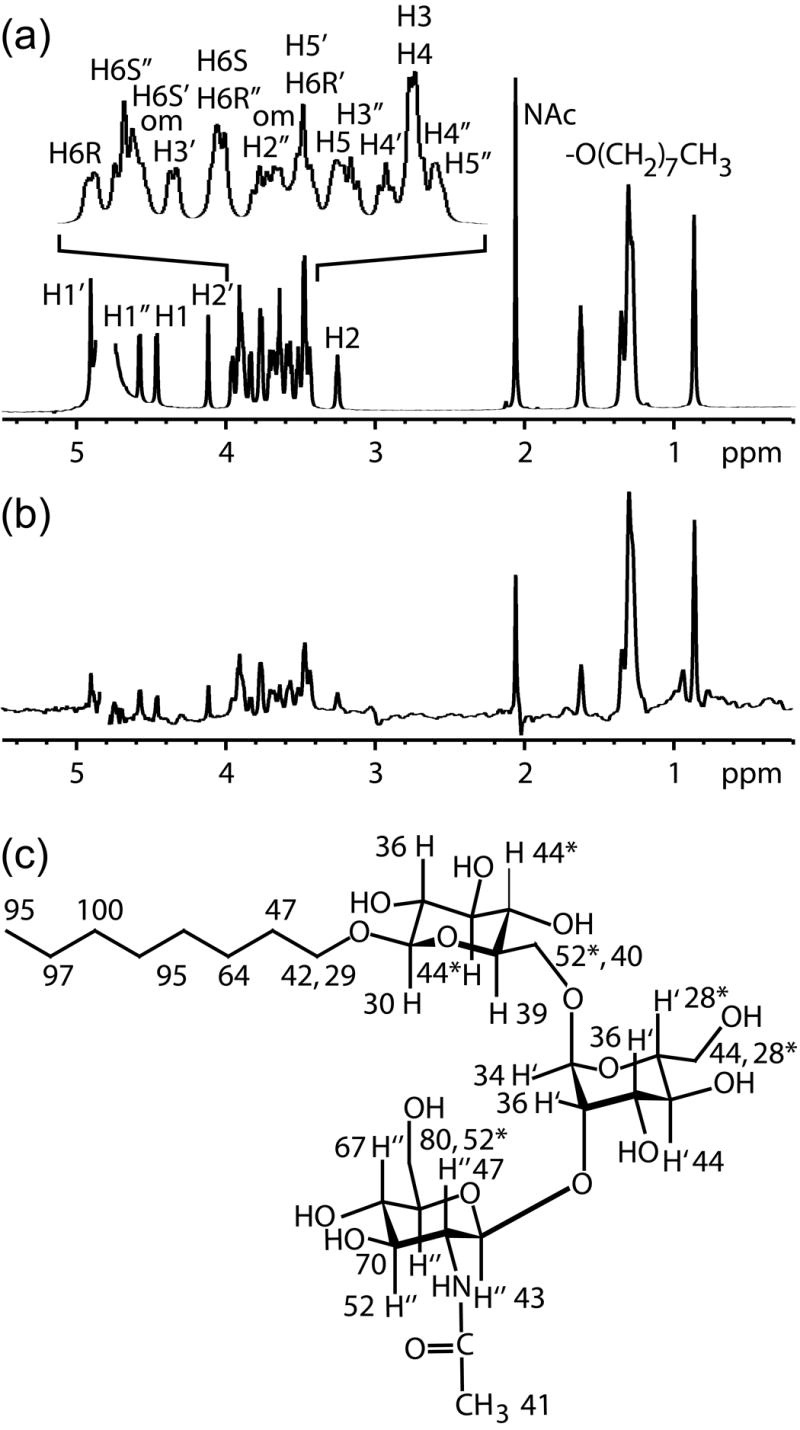
(a) The reference 1D 1H NMR spectrum of the acceptor in deuterated buffer solution including assignments. An exponential weighting function of 5 Hz was applied to mimic the linewidth of the acceptor in the STD spectrum. Chemical shift overlap of the octyl chain methylenes, H3/H4, H5’/H6R’, and H6S/H6R” pairs could not be resolved at 900 MHz. (b) The STD spectrum of the acceptor with GnT-V using a 100 Hz Gaussian saturation pulse train of 500 ms centered at 0.0 ppm. (c) The normalized percentages calculated from the STD and reference spectra of the acceptor are superimposed on the chemical structure of the acceptor.
Figure 2.
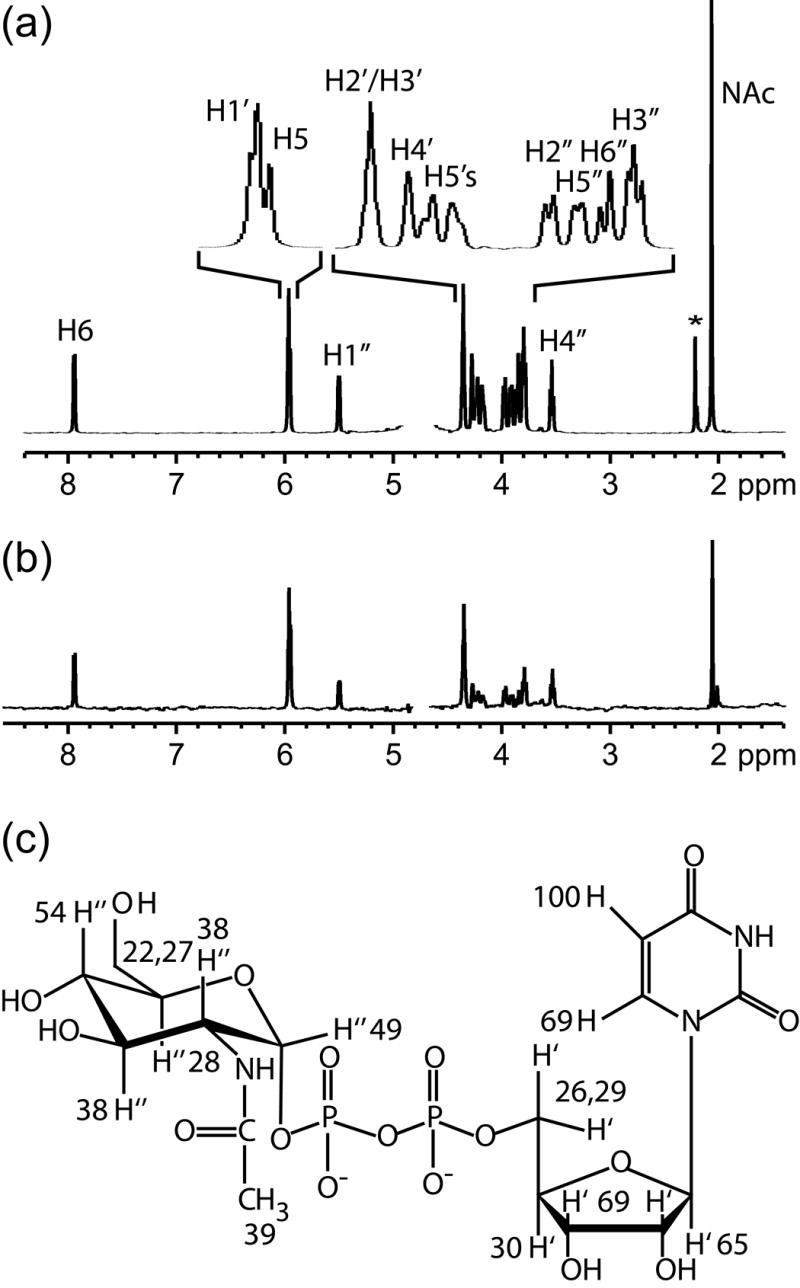
(a) The reference 1D 1H NMR spectrum of UDP-GlcNAc in deuterated buffer solution. The asterisk indicates acetone, which was used as a chemical shift reference. An exponential weighting function of 4 Hz was applied to mimic the linewidth of UDP-GlcNAc in the STD spectrum. (b) STD spectra of UDP-GlcNAc with GnT-V using a 100 Hz Gaussian saturation pulse train of 2.0 s centered at 0.0 ppm. (c) The normalized percentages calculated from the STD and reference spectra of UDP-GlcNAc are superimposed on the chemical structure of UDP-GlcNAc.
Figure 3.
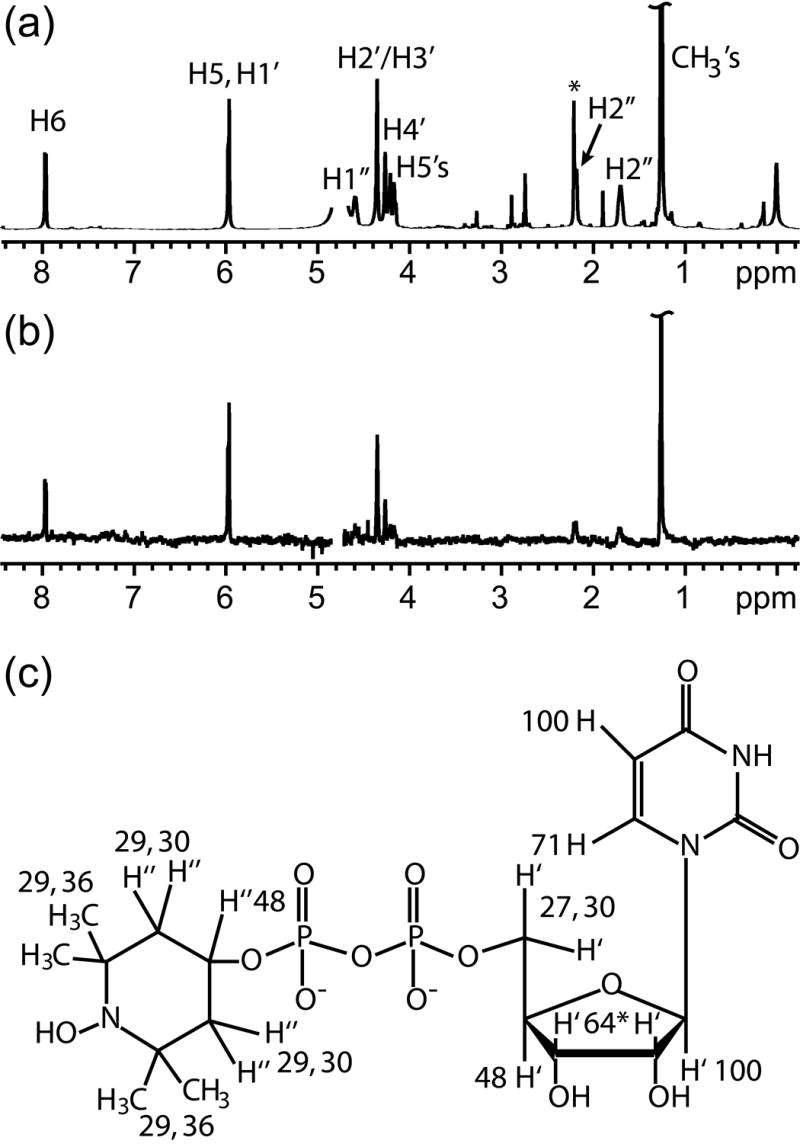
(a) The reference 1D 1H NMR spectrum of UDP-TEMPOH in deuterated buffer solution. The asterisk in the spectrum indicates acetone, which was used as a chemical shift reference. An exponential weighting function of 2 Hz was applied to mimic the linewidth of UDP-TEMPOH in the STD spectrum. (b) The STD spectrum of UDP- TEMPOH with GnT-V. (c) The normalized percentages calculated from the STD and reference spectra of UDP- TEMPOH are superimposed on the chemical structure of UDP- TEMPO. Peaks from H2’ and H3’ are overlapped giving an average STD value.
The results for the acceptor indicate that the highest STD percentages are localized on the GlcNAc ring and octyl chain with contacts greater than 50% at H3”, H4”, H5”, H6S”, H6R”, and at the protons on the terminal 6 carbons of the octyl chain [–(CH2)5CH3]. An additional STD experiment was performed with the acceptor in which UDP was added. A high concentration (10 mM UDP) was used to maximize the occupancy since the Kd is 3.8 mM.17 Otherwise, identical conditions were used as in the acceptor STD experiment. The results of this experiment are compared to the acceptor results in Figure 4. The STD contacts of the acceptor to GnT-V do not change significantly when UDP is present.
Figure 4.
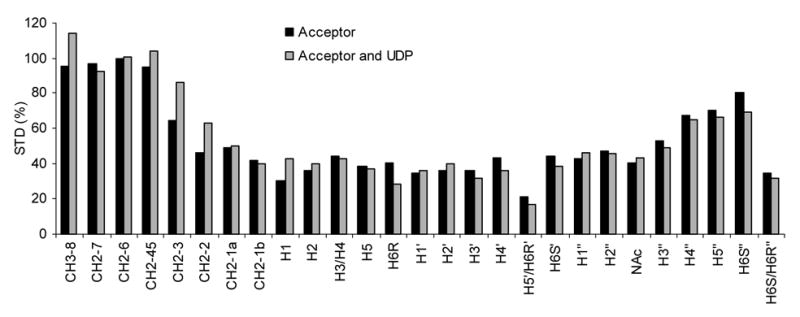
STD data for the acceptor plus GnT-V without (black) and with UDP (gray). Each set was normalized to 100% using the peak height of the acceptor octyl chain methylene signal (CH2-6). The STD patterns are similar for both conditions suggesting little conformational change around the acceptor binding pocket due to UDP binding.
Bound Conformations from Transferred NOE
Intramolecular 1H-1H distances for the donor and acceptor substrate separately bound to GnT-V were determined using 1D trNOE and 2D trNOESY experiments. 2D NOESY spectra taken of UDP-GlcNAc with and without GnT-V are included as supplementary data. NOEs in the presence of GnT-V are negative, as expected in a case where contributions from the bound form dominate. A minimum of structural data is available in this case with only a single conformationally variant NOE constraint observed for pairs of protons on different rings of UDP-GlcNAc. This constraint is between uracil H6 and ribose H2’. Qualitatively this supports an anti conformation for the nucleoside glycosidic bond. Intensities can be more quantitatively interpreted in terms of distances using 1D NOE spectra in which NOE mixing times are varied. Such spectra were collected for both free and bound UDP-GlcNAc and data are shown in Figure 5. Assuming a rigid model and using the H5-H6 distance and NOE data as a reference, the distances between H6 and H2’ for free UDP-GlcNAc and UDP-GlcNAc in the presence of GnT-V were both determined to be 2.5 +/− 0.1 Å.
Figure 5.
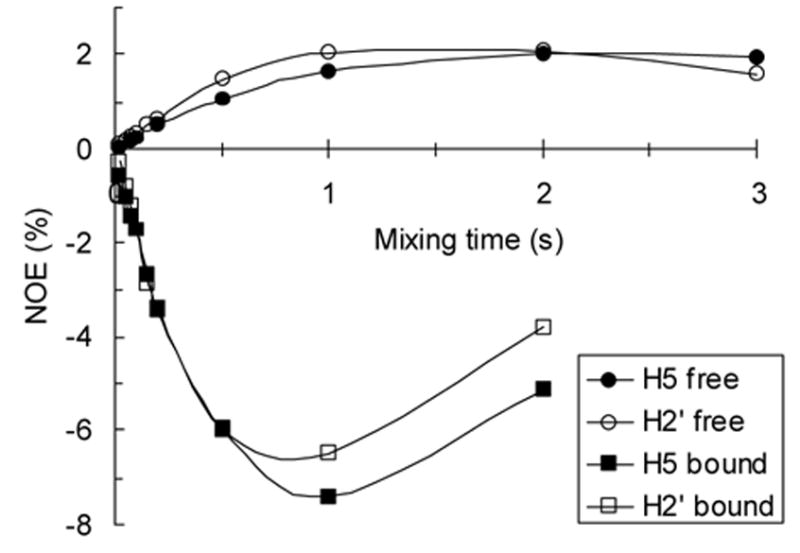
NOE buildup curves for contacts to uracil-H6 in the absence (free, circles) and presence (bound, squares) of GnT-V. The initial slopes of the curves were used to calculate the distance between uracil-H6 and ribose-H2’ in the free and bound states using the distance between uracil-H6 and -H5 as a reference. Smooth curves were added to data points for better visualization.
trNOESY spectra of UDP-TEMPOH with GnT-V were collected to verify binding and identify distance constraints. Like UDP-GlcNAc, negative NOEs were observed for UDP-TEMPOH in the presence of GnT-V, indicating that UDP-TEMPOH binds GnT-V. The only distance constraint observed was again between H6 and H2’. The distance was calculated from trNOESY spectra similarly to the UDP-GlcNAc calculation and was determined to be 2.5 +/− 0.1 Å. As with UDP-GlcNAc, no NOEs were observed between the TEMPOH ring and the uridine portion of the UDP-TEMPOH molecule.
NOESY spectra were collected on the acceptor with and without GnT-V. Figure 6 shows an expansion of the glycosyl region; the full spectra are available as supplementary materials. Due to the overlap of several resonances of the acceptor and interference from protein resonances, the spectra were collected at 900 MHz. Based on their positions and relatively narrow lines, the interfering protein signals must arise from glycosyl residues on the protein which exhibit significant flexibility relative to the protein. These background signals were identified by observing the protein alone at a mixing time of 200 ms as shown in Figure 6c. The chemical shifts of the background signals are typical of oligosaccharides containing GlcNAc, Man, Gal, and NeuAc, which are constituents of GnT-V’s glycosylation.10 The GnT-V background NOESY spectrum was carefully compared to the trNOESY acceptor spectrum. Crosspeaks with significant overlap with protein signals were not used in the analysis of intermolecular 1H-1H distances of the acceptor.
Figure 6.
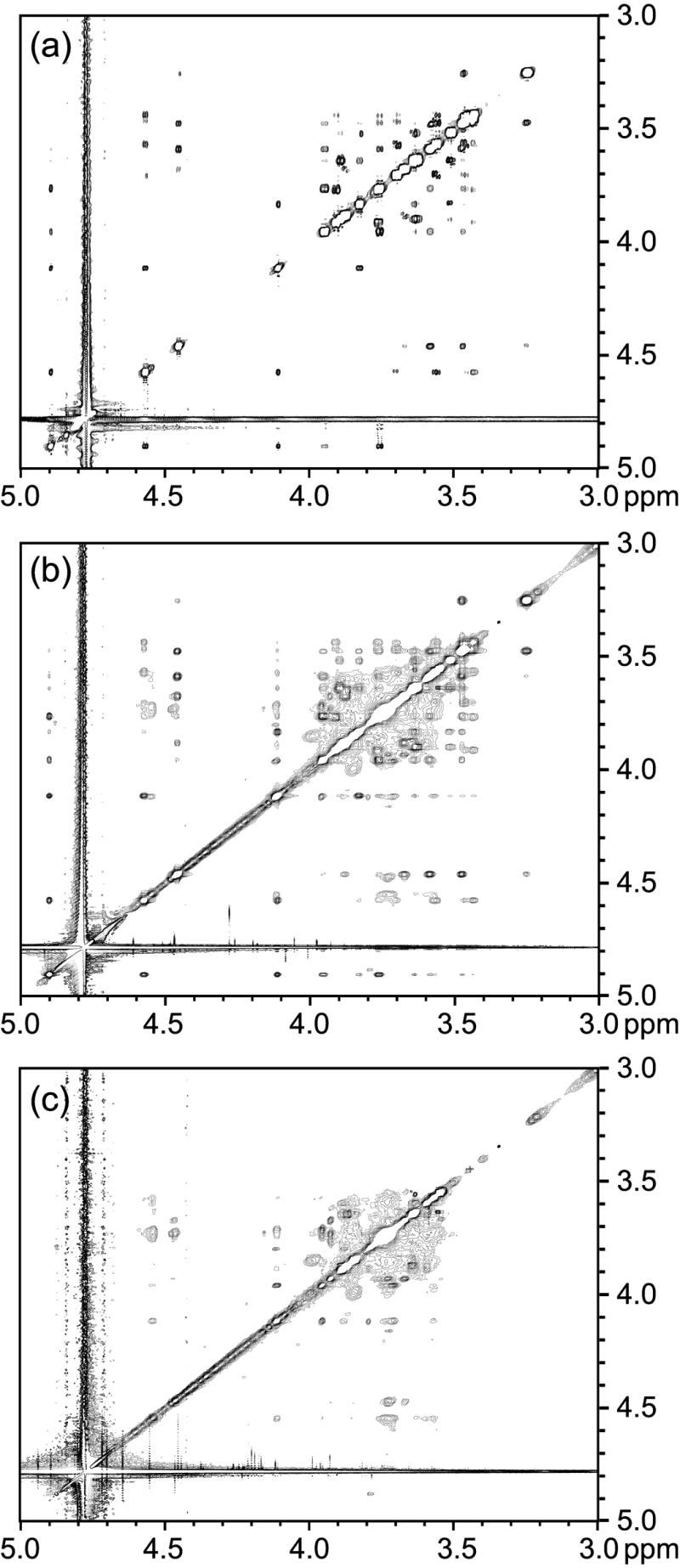
Expanded (carbohydrate region) NOESY spectra of (a) the acceptor, (b) the acceptor in the presence of GnT-V, and (c) GnT-V background. These allow assessment of conformational changes on binding to GnT-V.
Unlike the donor spectra, the NOESY spectrum of the acceptor free in solution (Figure 6a) gives some negative (sugar residues) NOEs in addition to positive (octyl chain) NOEs. The intensities are also small, being 1–2 % at a mixing time of 800 ms. It can be inferred that the correlation time of the acceptor free in solution is approximately 1.2 ns, which is a little long for a molecular weight of 658 g/mole. It has been suggested that the acceptor forms micelles due to the hydrophobic octyl chain, and some tendency to formation of micelles could lengthen the effective correlation time. Nevertheless, due to their small size, the negative NOEs of the free acceptor do not contribute significantly to the NOEs of the GnT-V bound acceptor. The negative NOEs for the H1’-H2’ pair in the presence of GnT-V (Figure 6b) become approximately 40 times larger for intra-ring interactions showing the expected enhancement from bound ligand contributions (the time evolution data for the H1’-H2’ pair is provided as supplemental material). Even after discounting crosspeaks from the protein, Figure 6b shows a number of crosspeaks not observed in the free acceptor. They offer the possibility of extracting additional distance constraints that can be used in modeling a bound conformation. Because of the possibility of step-wise diffusion of magnetization while the acceptor is bound to GnT-V we used data from short mixing times (50 – 150 ms) that gave no indication of delayed build-up in later points. In addition, only the shorter constraints of the two methylene protons, Glc H6S and H6R, were used; this compensates for efficient spin diffusion between these closely spaced protons. Because of missing information on the possible location of protein protons that could act as part of the spin system, no attempts were made to model spin diffusion more precisely.18 The distance constraints that could be determined from the trNOESY spectra of the acceptor with GnT-V are listed in Table 1.
Table 1.
1H-1H distances determined from trNOEs for the acceptor in the presence of GnT-V.a
| O-Octyl | Glc | Man' | GlcNAc” | Distance(Å) |
|---|---|---|---|---|
| CH2-1b | H1 | --- | --- | 3.0 |
| CH2-1a | H1 | --- | --- | 3.0 |
| CH2-2 | H1 | --- | --- | 3.5 |
| --- | H5 - H6(S) | --- | --- | 2.2 |
| --- | H1 - H6(S) | --- | --- | 3.3 |
| --- | H6(S) | H1' | --- | 2.5 |
| --- | H6(S) | H2' | --- | 2.9 |
| --- | H4 | H3' | --- | 3.0 |
| --- | H5 | H1' | --- | 3.5 |
| --- | H4 | H1' | --- | 3.9 |
| --- | H6(S) | --- | H1” | 3.0 |
| --- | H6(S) | --- | NAc | 3.6 |
| --- | --- | H4' - H6'(S) | --- | 2.9 |
| --- | --- | H2' | H1” | 2.6 |
| --- | --- | H1' | H1” | 2.7 |
| --- | --- | H2' | H5” | 3.5 |
| --- | --- | H1' | NAc | 3.5 |
| --- | --- | H3' | H1” | 3.6 |
| --- | --- | H6'(S) | NAc | 3.6 |
| --- | --- | H2' | H4” | 3.7 |
| --- | --- | H1' | H5” | 4.1 |
| --- | --- | H2' | NAc | 4.8 |
| --- | --- | H4' | NAc | 4.9 |
| --- | --- | --- | H5” - H6”(R) | 2.5 |
| --- | --- | --- | H5” - H6”(S) | 2.5 |
| --- | --- | --- | H4” - H6”(S) | 3.1 |
| --- | --- | --- | H2” - NAc | 4.0 |
| --- | --- | --- | H1” - NAc | 4.5 |
| --- | --- | --- | H3” - NAc | 4.8 |
Only intermolecular and sugar methylene proton NOEs were included to simplify the table. Intramolecular NOEs were used as reference values to calculate the tabulated NOE values.
Structural Modeling of the Acceptor
Simulated annealing was used to refine the structure of the bound-state acceptor using the NOE constraints listed in Table 1. From the 100 structures generated, the 10 lowest overall energy conformations were selected. The average torsion angles and standard deviations are listed in Table 2 for the 10 structures (see Materials and Methods sections for torsion angle definitions). An ensemble of structures is illustrated in Figure 7. The structures were aligned based on the sugar ring atoms. The RMSD for the sugar ring atoms after alignment of the rings is 0.17 Å. The RMSD for all sugar atoms is 0.56 Å and for all atoms including the octyl chain is 2.04 Å.
Table 2.
The average torsion angles and standard deviation of the 10 acceptor structures produced by simulated annealing with NOE refinement.
| Residue or Linkage | Torsion angle | Average (degrees) | Standard deviation (degrees) |
|---|---|---|---|
| GlcNAc | χ5 | −160 | 83 |
| GlcNAc | χ2a | 78 | 11 |
| GlcNAc | χ2b | 10 | 5 |
| GlcNAc(1–2)Man | φ | −107 | 7 |
| GlcNAc(1–2)Man | ψ | −84 | 2 |
| Man | χ5 | −39 | 59 |
| Man(1–6)Glc | φ | 116 | 3 |
| Man(1–6)Glc | ψ | 119 | 6 |
| Man(1–6)Glc | ω | 60 | 3 |
| Glc(1-O)Octyl | φ | −111 | 43 |
| Glc(1-O)Octyl | ψ | −126 | 56 |
Figure 7.
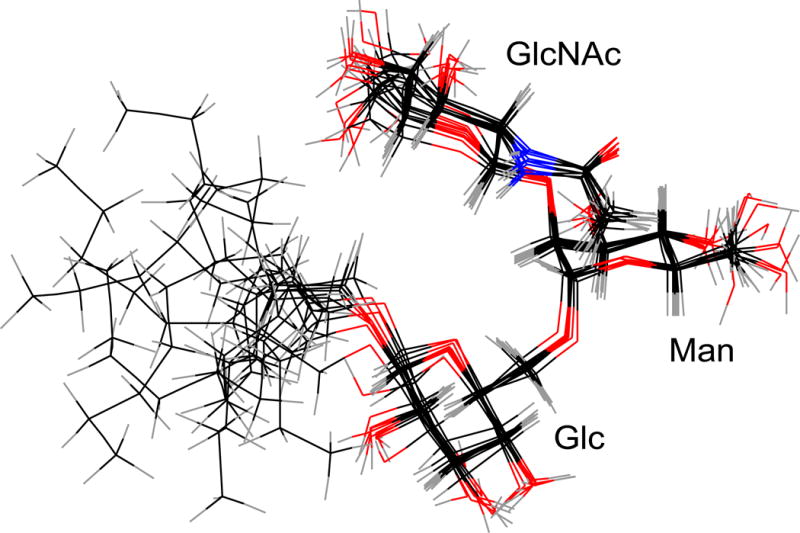
The 10 lowest energy structures from NOE restrained simulated annealing were aligned using the sugar ring atoms.
Placement of Donor and Acceptor by Paramagnetic Relaxation Enhancement
The paramagnetic relaxation enhancement of the bound acceptor due to bound UDP-TEMPO was measured by comparing the longitudinal relaxation rates (1/T1) of the acceptor protons in a control and relaxation-enhanced sample. The relaxation-enhanced sample contained GnT-V and a near saturating amount of UDP-TEMPO, while the control sample contained the acceptor, GnT-V, and an appropriate amount of a soluble paramagnetic species presumed not to bind to the protein (4-hydroxy-TEMPO). While applications of paramagnetic relaxation enhancement on protein protons by nitroxides covalently attached to protein are now routine, several difficulties arise when measuring perturbations of rates for protons on an exchanging ligand in the presence of a second exchanging ligand carrying a nitroxide group. These difficulties necessitated a rather complex experimental protocol. First, we chose to measure longitudinal relaxation rates rather than the transverse relaxation rates that are measured in many experiments on simple protein systems. Exchanging ligands often suffer chemical exchange broadening of resonances that can be difficult to separate from paramagnetic transverse relaxation effects. The longitudinal relaxation effects are small at the high fields needed to resolve various resonances, but they are not contaminated by these additional effects. Second, the control experiment must have the protein, GnT-V, present. Quite aside from the perturbations by nitroxide, the protein affects relaxation of protons on the acceptor by changing the correlation time. And third, the control experiment must have an equivalent amount of nitroxide in solution since chance encounters of unbound acceptor and unbound donor analogue contribute to the relaxation of protons on the acceptor. A relatively high concentration of UDP-TEMPO was used to maximize the percentage of GnT-V occupied (estimated at 70% at 10 mM UDP-TEMPO). This high concentration, in turn, required careful consideration of an appropriate control for nitroxide induced relaxation in solution. The solution effects of UDP-TEMPO were experimentally found to result in a 3-fold decrease in the T1 values of the acceptor protons. The concentration of hydroxy-TEMPO required to match the T1 reduction was found to be 6.85 mM, somewhat lower than expected, possibly because of incomplete oxidation of the UDP-TEMPO to nitroxide.
The change in relaxation rates on comparing UDP-TEMPO to 4-hydroxy-TEMPO samples are plotted in Figure 8 for the GlcNAc, Man, and Glc residues of the acceptor. A positive change indicates paramagnetic perturbation from bound UDP-TEMPO to the bound acceptor. Data for the octyl chain was not included since the trNOESY and refined structures suggest that this residue is flexible in the binding pocket of GnT-V; and, paramagnetic relaxation enhancement of these flexible residues would be heavily biased towards close, intermittent contacts with UDP-TEMPO. The paramagnetic perturbation from UDP-TEMPO is most apparent around the Man and Glc protons and apparent to a lesser extent around the GlcNAc protons. The N-acetyl methyl protons and H5’ could not be measured in the 1H-13C HSQC due to overlap from GnT-V oligosaccharide signals.
Figure 8.
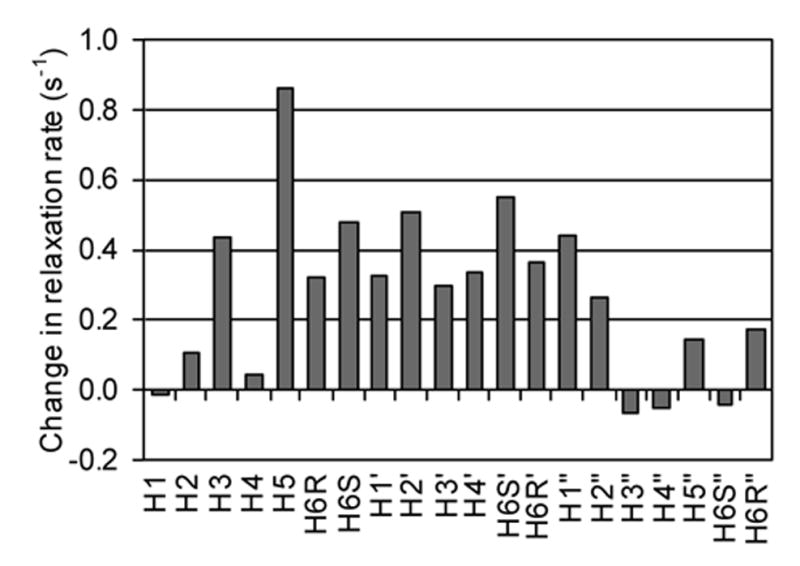
The difference in the longitudinal relaxation rates (1/T1) of the acceptor in the control and experimental samples. A positive change indicates an increase in the longitudinal relaxation rate of the acceptor from paramagnetic relaxation enhancement by bound UDP-TEMPO.
The change in the longitudinal relaxation rate for the sugar residues is actually small, ranging from −0.06 to 0.86 s−1. This is partly due to the fact that paramagnetic effects on longitudinal relaxation rates by nitroxides are inherently small at high magnetic fields, and partly due to the dependence on the fraction of the acceptor bound to GnT-V at the same time as UDP-TEMPO. Assuming a Kd of 12.8 μM19 for the acceptor and 4.4 mM for UDP-TEMPO (UDP-GlcNAc Kd = 4.4 mM),17 the experimental sample of 155 μM GnT-V, 2 mM acceptor, 10 mM UDP-TEMPO would give only 5.3% of the acceptor bound to UDP-TEMPO-occupied GnT-V. If a fraction of the UDP-TEMPO was in the reduced form, then this percentage would be even less. It would clearly be advantageous to have nitroxide labeled analogues with higher affinities for these experiments. Nevertheless, some useful data can be extracted. The propagated standard error is 0.23 s−1 based on the standard error from the exponential fit of the peak volume data from the incremented time delay 1H-13C HSQC spectra. The enhanced relaxation of H3, H5, and H6S on the Glc residue and H6S and H2 on the Man residue are clearly significant, as are the absence of significant effects on the GlcNAc residue. These data are useful in placing the donor relative to the acceptor in the active site.
Discussion
Binding Epitopes of Donor and Acceptor
Important parts of the binding epitope of UDP-GlcNAc are clearly indicated in saturation transfer difference data for this ligand. More efficient magnetization transfers indicate short ligand proton - protein proton distances for the uracil H6 and H5 and ribose H1’, H2’, and H3’ protons, and less efficient magnetization transfers indicate somewhat longer distances for protons in the GlcNAc ring. The close contacts for the uracil ring and its attached ribose are consistent with evidence that UDP serves as an efficient competitive inhibitor of GnT-V with a Kd of 3.8 mM.17 The lesser contacts with the GlcNAc residue are consistent with the necessity of leaving at least the anomeric region of this ring exposed for addition to the acceptor. The binding epitope determined for UDP-GlcNAc bound to GnT-V supports increasing evidence that the uridine portion acts as a recognition motif for glycosyltransferases utilizing UDP-sugar donor substrates while recognition of the sugar portion provides specificity.20
The binding epitope of UDP-TEMPOH was determined in order to verify binding to GnT-V and preservation of a binding epitope similar to that of UDP-GlcNAc. The reduced form of UDP-TEMPO was used because the nitroxyl radical form is paramagnetic and the 1H signals from UDP-TEMPO are greatly attenuated due to paramagnetic relaxation. Since the reduction results only in the addition of a single hydrogen to the nitroxide oxygen, the results on UDP-TEMPOH are assumed to yield a good representation of the UDP-TEMPO epitope. The results for the STD experiment with UDP-TEMPOH shown in Figure 3 indicate strong contacts to the uracil H6 and H5 and ribose H1’, H2’, and H3’ protons. The values across the molecule are similar to those of UDP-GlcNAc with the exception of R1’ which shows a higher STD contact in UDP-TEMPOH. The STD enhancement of R1’ may be due to differences in the off-rates of UDP-TEMPOH compared to UDP-GlcNAc, and with this single exception, the overall similarity of the binding epitope supports the assumption that UDP-TEMPOH binds to the catalytic donor site of GnT-V.
The binding epitope of the acceptor is also well defined by STD data. The closest protein proton – ligand proton distances for the acceptor are localized on the β-(1→2)-GlcNAc ring and the octyl chain. The strong STD contacts of the octyl chain may be a result of interaction with a hydrophobic pocket in the acceptor catalytic site of GnT-V or even non-specific interactions with other sites on the protein. In either case, the octyl chain is not a structurally important element of the native acceptor. The β-(1→2)-GlcNAc ring contacts are more interesting. The closest contacts to GnT-V protons on the β-(1→2)-GlcNAc ring are H3”, H4”, H5”, and H6S” (see Figure 1) with relative STD percentages of 52, 67, 70, and 80%. Compared to the STD values for the Man and Glc rings which range from 22 to 44%, the contacts to the GlcNAc ring are significant. The presence of β-(1→2)-GlcNAc at the non-reducing terminus of the acceptor substrate is known to be essential for activity with GnT-V.21 Additionally, trNOESY data comparing acceptor analogues with and without a terminal GlcNAc, namely β-D-GlcpNAc-(1→2)-α-D-Manp-(1→3)-[β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)]-D-Manp and α-D-Manp-(1→3)-[α-D-Manp-(1→6)]-α-D-ManpOMe, indicate that the β-(1→2)-GlcNAc is important for binding to GnT-V at least in the fast exchange regime (data not shown). Furthermore, structure activity relationships determined by Kanie, et al. show that OH-3”, OH-4”, and OH-6” of this acceptor are key recognition elements by GnT-V.22,23 Modification of these hydroxyl groups to NH2 and NHAc groups resulted in 23 to 230-fold higher Km values for the 3”-modified and 4”-modified acceptor analogues and no activity up to 10 mM for the 6”-modified acceptor analogues. The STD data indicating very close GnT-V proton distances to H6S” (80% STD) would be consistent with an inability to modify this site; lesser distances for H3” and H4” (52% and 67% STD, respectively) suggest proximities where small modifications might improve protein interaction. Taken together, the data indicate a sterically restricted interaction between the catalytic site of GnT-V and the 3”, 4”, 5”, and 6” end of the GlcNAc ring.
Weaker STD contacts are seen for the Man and Glc rings. The weak Man contacts, particularly around the H5’ and H6’ sites are again consistent with the need to keep this region open to addition of a GlcNAc from the acceptor. However, these weaker STD signals, particularly for the acceptor, require careful interpretation. STD contacts can be strongly influenced by spin diffusion during the time spent in the protein binding site. We do not have independent information on the lifetime of the acceptor in the binding site, but the low dissociation constant (Kd = 12.8 μM) would support a longer residence time for the acceptor than the donor. Spin diffusion can distribute saturation occurring at a strongly interacting site to adjacent sites in the ligand, tending to reduce STD variations across the molecule. We also have to be careful in interpreting that lack of STD intensity as a lack of protein contact. Longer contacts such as those mediated by hydrogen bonding interactions are not easily observed. In our case, deuteriums from the solvent replace the protons actually in the hydrogen bonds. Hence, any inter-proton distance interpretations need to stay at a qualitative level.
Nevertheless, the qualitative results we do have can be supported with data from other sources. For example, there are structure-activity relationships to support modest interactions at the Man and Glc rings. While, removal of the Glc ring from the acceptor has been shown to produce a very poor substrate for GnT-V, modifications to the Glc ring are well-tolerated by GnT-V. For example, O-benzylating the hydroxyl groups of Glc is allowed (Km increases by a factor of 2), indicating little steric restriction around the Glc residue by GnT-V.24 Likewise, modification of the OH-3’ to O-methyl in the Man residue has only a small effect on the activity of GnT-V.25 O-methylation of OH-4’ results in no activity, but does not affect its binding.26 Given the lower saturation transfers to the Glc and Man rings and the possibility that residual intensities are partly from spin diffusion effects, the STD results support these activity relationships. The absence of close contacts with GnT-V around OH-3’ or OH-4’would allow their modification and create an ‘open’ space that is the catalytic pocket for the donor substrate, UDP-GlcNAc.
The STD results on the acceptor in the presence of UDP are of some additional interest. The mechanism for glycosylation by GnT-V was suggested by Zhang, et al. to be an ordered reaction where UDP-GlcNAc binds first, resulting in a conformational change of the protein followed by sequential binding of the acceptor substrate.17 Both UDP and UDP-GlcNAc were suggested to induce a similar conformation change. Therefore, we might expect somewhat different STD patterns for the acceptor in the absence and presence of UDP. However, no significant differences in STD data in the presence and absence of UDP were found (see Figure 4). This could simply mean that UDP does not induce the same change that UDP-GlcNAc does, or that the STD of the acceptor is not sensitive to the structural change. The result does not, of course, rule out conformational changes in parts of the binding site that are not in close contact with the acceptor.
Conformation of Donor and Acceptor
Intramolecular 1H-1H distance constraints determined using trNOE and trNOESY experiments on UDP-GlcNAc bound to GnT-V provide a reasonable picture of the bound conformations of the donor. Only one restraining trNOE was observed for both UDP-GlcNAc, and that was between H6 and H2’. It corresponds to a distance of 2.5 Å. This is consistent with the anti glycosidic torsion angle known to be adopted by most pyrimidine nucleosides in solution;27 thus there is little conformational change of the uridine portion of UDP-GlcNAc compared to free UDP-GlcNAc. The fact that no NOE contacts are observed between the uridine portion of the molecule and the GlcNAc prevents a complete conformational determination, but it does eliminate a folded conformation that would have short distances between protons on the respective moieties as was recently observed for UDP-Gal bound to a galactosyltransferase in the presence of Mg2+.28 These results suggest that GnT-V binds in a low energy conformation such as those found in solution or sugar nucleotide containing crystal structures.
For the acceptor, the conformational data from transferred NOE experiments is quite rich. Most of the constraints (see Table 1) involve the Man and GlcNAc rings, and in particular the N-acetyl group. These restrain the bound conformation of the Man and GlcNAc rings to a well defined conformation. Constraints to the Glc ring exist, but most of the constraints involve the exocyclic methylene protons which are subject to efficient spin-diffusion due to the short distance between the methylene protons. Only constraints to the methylene showing the strongest NOE were used (H6S) assuming the NOE to H6R was heavily contaminated by spin diffusion. Nevertheless, even with a reduced set of constraints, the Glc ring is restrained with respect to the Man and GlcNAc rings. There are no contacts between the octyl chain and the Man or the GlcNAc rings, suggesting isolation of these two sugar rings from the octyl chain in the bound conformation. To get a picture of the conformation of the acceptor in the active site of GnT-V, distance constraints derived from the NOEs were used to refine a structure using simulated annealing.
The results from the simulated annealing of the acceptor using the trNOE distance constraints resulted in a distinct conformation with little variation between the 10 lowest energy structures. The standard deviations of the torsion angles for the β-D-GlcpNAc-(1→2)-α-D-Manp linkage, the α-D-Manp-(1→6)-β-D-Glcp linkage, and the N-acetyl group are very low ranging from 2 – 11 degrees. Less well-defined are the exocyclic OH-6 groups of GlcNAc and Man and the Glc-octyl chain linkage. The OH-6’ of Man is important because it is the site of catalytic addition of a GlcNAc residue by GnT-V. The torsion angle, χ5, confines the position of OH-6’ to -39 degrees with a standard deviation of 59 degrees. This deviation is small enough to restrict the rotation to the tg rotamer conformation (-60 degrees) (see Materials and Methods sections for a description of the naming conventions).
To assess whether the above structure is reasonable, comparisons to other carbohydrate structures and structure activity relationships were made. The GlyTorsion database was used to compare known carbohydrate structures with the refined bound-state acceptor structure.29 The database reports on torsion angles from structures in the Protein Data Bank (PDB).8 No data was available for α-D-Manp-(1→6)-β-D-Glcp linkages since this is not a typical linkage. However, data on α-D-Manp-(1→6)-β-D-Manp is available for comparison. The difference between Man and Glc is a change in the stereocenter of the C2 ring atom; thus, a change from Glc to Man is not expected to significantly affect the torsion angles of the (1→6) linkage. Plots of the φ, ψ, andω torsion angles for the α-D-Manp-(1→6)-β-D-Glcp linkage of the refined acceptor are shown in Figures 9a and 9b along with the GlyTorsion data for α-D-Manp-(1→6)-β-D-Manp linkage. Even though the φ and ψ torsion angles for the refined acceptor structure do not fall within the most concentrated areas of GlyTorsion data, they do fall within the range of torsion angles for known structures. The ω torsion angles lie in one of the two highly concentrated areas at 60 degrees, the gg conformation. This conformation is also supported by solvated molecular dynamics simulations30 and 1H NMR spectroscopy31 of methyl α-D-glucopyranoside, which predict that the gg conformation is the most highly populated. A plot of the φ and ψ torsion angles for the β-D-GlcpNAc-(1→2)-α-D-Manp linkage and the χ2a and χ2b exocyclic angles for the N-acetyl group of β-D-GlcpNAc are included in the supplementary materials. The values for the refined structures fall within the range of values found in the PDB for both the β-D-GlcpNAc-(1→2)-α-D-Manp linkage and the χ2a and χ2b exocyclic angles, indicating that the GlcNAc and Man rings are in a reasonable geometry with respect to each other. These results indicate that the refined conformation of the bound-state acceptor is a reasonable structure.
Figure 9.
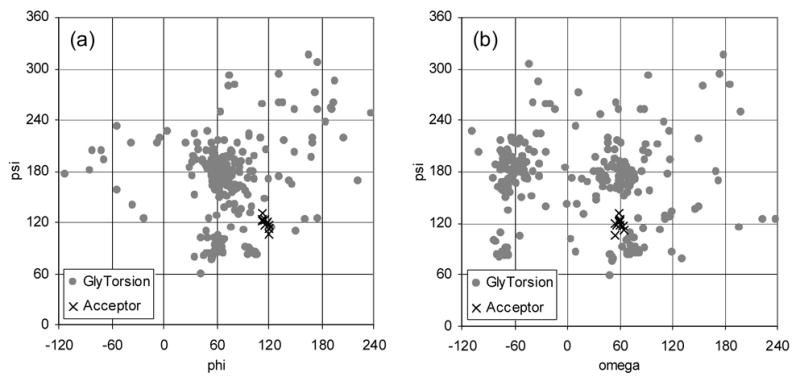
Plot of the φ, ψ, andω torsion angles for the α-D-Manp-(1→6)-β-D-Manp linkages from the GlyTorsion database (gray circles) and the α-D-Manp-(1→6)-β-D-Glcp linkage of the NOE refined structures of the acceptor (black x).
In addition to agreeing with known structural data, the conformation of the bound-state acceptor structure agrees with known structure activity relationships. The conformation of the α-D-Manp-(1→6)-β-D-Glcp linkage is supported by data from Bock, et al., who showed a strong preference for the gg conformation of the acceptor analogue by GnT-V over the gt conformation using restricted ω torsion angle acceptor analogues.16 The acceptor structure is also consistent with the acceptor analogue modifications described earlier in the binding epitope discussion. Even the large modifications such as O-benzylating the hydroxyl groups of Glc would be spatially allowed based on the proposed acceptor structure.24 One feature of the refined bound-state acceptor structure is seemingly inconsistent with structure activity relationship data. This feature involves the position of OH-6’ of the Man residue, which, based on our data, is predicted to be in the tg rotamer conformation. This conformation has been shown to be a weakly populated conformation based on solvated molecular dynamics calculations using the model compound, methyl α-D-glucopyranoside.30 However, in the absence of modeled water, the tg rotamer conformation is of low energy due to intramolecular hydrogen bonding between OH-6 and O-4. Thus, our predicted acceptor structure indicates that OH-6’ and O-4’ of the Man residue may be forming a hydrogen bond in the bound-state. Structure activity relationship of OH-4’ derivatives from Khan, et al. showed increased GnT-V activity with a 4’-deoxy acceptor analogue, and no activity, but retention of binding capacity with the 4’-methyl acceptor analogue.25 These results were interpreted as no catalytically important role for OH-4’, since the 4’-deoxy derivative was active. In our structure it may be that the tg conformation persists in the bound-state acceptor structure because the binding pocket of GnT-V is incomplete without the sugar donor.
Paramagnetic relaxation enhancement
The difference in the longitudinal relaxation rate between experimental and control samples were plotted in Figure 8 for the Man, GlcNAc, and Glc residues. As discussed in the results section, the errors in these experiments are relatively high, but the enhanced relaxation of H3, H5, and H6S on the Glc residue and H6S’ and H2’ on the Man residue in particular are significant. This enhancement is consistent with approach of the TEMPO moiety from a direction away from the GlcNAc and on the face of the Man ring between H6S’ and Glc H5. The result is also consistent with STD data that suggests that the GlcNAc residue is in close contact with the GnT-V surface, and thus, removed from easy contact with UDP-TEMPO. In addition, this approach does not interfere with space around OH-4’, which we propose to be in close proximity with the nucleophilic residue of GnT-V responsible for the activation of O-6’.
In Figure 10, we have assembled a model that is consistent with data on bound conformations, STD contacts, and relative placement of ligands. This can be compared with the crystal structure of β1,6-N-acetylglucosaminyltransferase (C2GnT-L) with its acceptor, β-D-Galp-(1→3)-D-GalpNAc.32 Like GnT-V, it is an inverting glycosyltransferase, metal ion independent, uses UDP-GlcNAc as a donor, and creates a β-D-GlcpNAc-(1→6) glycosidic bond. In the case of C2GnT-L, the glycosidic bond is made to the GalNAc residue of the core 1 O-glycan, β-D-Galp-(1→3)-α-D-GalpNAc-O-Ser/Thr. As predicted for glycosyltransferases of the inverting type, a nucleophilic residue, glutamate 320, forms hydrogen bonds with GalNAc OH-4 and OH-6 in the C2GnT-L/acceptor structure. Similarly, a glutamate residue was added to our model in Figure 10 to coordinate with Man OH-4’ and OH-6’. This scenario is consistent with weak STD contacts to the Man ring since deuterated hydrogen bonds would result in long protein proton – acceptor proton distances. It is also consistent with structure activity relationship data, which shows an ability to sterically disable the catalytic reaction at OH-4’ with methylation. The structure of UDP-GlcNAc used in the model was taken from the crystal structure of MurG with UDP-GlcNAc.33 MurG is a metal-independent N-acteylglucosaminyltransferase, as is GnT-V, and the only enzyme of its type with a structure including bound UDP-GlcNAc. The MurG UDP-GlcNAc structure is consistent with the structural data determined for GnT-V UDP-GlcNAc including the trNOE distance constraint between H6 and H2’ and the lack of distance constraints between the GlcNAc ring and uridine portion. UDP-GlcNAc was modeled below and in front of the Man plane based on results from the paramagnetic relaxation data from the spin-labeled donor analogue, UDP-TEMPO. While this model is consistent with the data presented, the relative placement of UDP-GlcNAc is speculative due to the large error in the paramagnetic relaxation enhancement experiment. In addition, it should be noted that while the placement of the GlcNAc ring in the model is dependent on the experimental data presented, the relative position of the uridine diphosphate in the model is unknown and may differ from the MurG UDP-GlcNAc structure due to the high degree of flexibility about the phosphate residues. The native acceptor structure was used in the model to illustrate the additional volume of the native acceptor compared to the acceptor analogue. The three sugars corresponding to the acceptor analogue were modeled with the torsion angles determined from the NOE refinement and are indicated in Figure 10 with thick bonds. The model is consistent with the STD data for the acceptor. Space around the GlcNAc residue allows for protein contacts to be modeled as indicated by the dashed lines. In addition, the lack of STD contacts to the Glc residue (Man(Glc) residue in Figure 10) can be justified because space around this residue would be necessary to accommodate the native acceptor with its two additional glycosidic chains. The model is a culmination of data presented here and data acquired by others relating to GnT-V activity and glycosyltransferase structure and mechanisms. It is intended to be used to form hypotheses to test for more structural information on GnT-V and to design effective inhibitors for the treatment of carcinoma progression.
Figure 10.
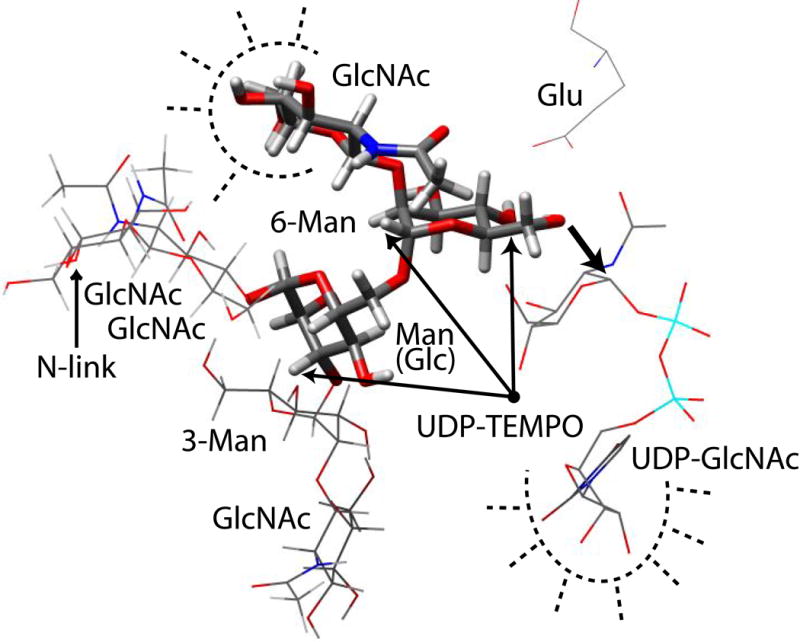
The native N-linked oligosaccharide acceptor, β-D-GlcpNAc-(1→2)-α-D-Manp-(1→3)-[β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)]-α-D-Manp-(1→4)-β-D-GlcpNAc-(1→4)-β-D-GlcpNAc, is used to illustrate the structural information gained from this study. The three sugars highlighted with thick bonds represent the portion of the native acceptor retained in the analogue used in this study. The placement of the other sugars in the native acceptor structure illustrates the additional volume needed for the native acceptor compared to the analogue. Saturation transfer difference results are illustrated with dashed lines to indicate proximate residues from GnT-V. A single point is drawn to indicate the approximate location of the nitroxide radical of UDP-TEMPO based on the paramagnetic relaxation data. The structure of UDP-GlcNAc was taken from the crystal structure of MurG-UDP-GlcNAc33 and was placed such that the anomeric carbon is positioned for attack by Man O-6’ and the OH-4 of UDP-GlcNAc is near the UDP-TEMPO nitroxide point. A glutamate residue was added on the opposite plane of the Man ring from the UDP-GlcNAc approach positioned to extract the proton from Man OH-6’.
In conclusion, we have provided structural characterization of the bound substrates of GnT-V and their structural interaction with the active site of GnT-V. Data from trNOEs were successfully used to determine the structures of the bound-state of UDP-GlcNAc and the acceptor analogue, β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)-β-D-GlcpOOctyl. The donor analogues show the typical anti glycosidic torsion angle for the uridine residues and indications of a linear configuration for the molecules with no interaction between the GlcNAc and the uridine. The trNOEs for the acceptor were successfully used with simulated annealing to refine a structure where the GlcNAc, Man, and Glc rings are well-defined in the active site of GnT-V. Binding epitopes for the donor analogues were consistent with binding of glycosyltransferases to nucleotide sugars with close contacts from the binding pocket of the GnT-V to the uridine residues. The binding epitope of the acceptor indicated close contacts between the GlcNAc ring and GnT-V, consistent with known substrate activity of GnT-V and structure-activity relationships of the acceptor analogue with GnT-V. A novel paramagnetic relaxation enhancement experiment was performed to determine the distance and relative orientation of the donor and acceptor substrate pair in the active site of GnT-V. Even though spin diffusion effects prevent a quantitative analysis of the latter data, a qualitative interpretation, combined with the conformations and protein surface contacts determined for the two ligands, leads to a model that is consistent with mechanistic expectations. The model also provides a basis for the design of inhibitors that may span parts of both ligands, and may make contacts with more extended regions of the protein binding site.
Materials and Methods
Preparation of protein samples
Recombinant soluble human GnT-V was expressed in Chinese hamster ovary (CHO) cells and purified from concentrated media as described previously.9,10 The activity of GnT-V was determined by radiochemical assay as described previously.12 Gel electrophoresis indicated that 80% of the purified GnT-V was the full length 95 kDa construct while the remaining 20% was a known, active proteolytic fragment of 66 kDa.12 To prepare samples for NMR spectroscopy, purified GnT-V was dialyzed and concentrated in NMR sample buffer (50 mM potassium phosphate (pH 6.5), 100 mM NaCl, D2O). Coomassie Plus reagent and an albumin standard were purchased from Pierce Biotechnology, Inc. and used to determine the concentration of GnT-V using a spectrophotometric (595 nm) protein assay. Concentrations of GnT-V in the range of 50 – 250 μM were used for the experiments.
Small molecule components including donor and acceptor analogues were prepared as stock solutions at 30 – 50 mM concentrations for addition to GnT-V samples. Uridine 5’-diphosphate sodium salt (UDP), uridine 5′-diphospho-N-acetylglucosamine sodium salt (UDP-GlcNAc), and 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (4-hydroxy-TEMPO) were purchased from Sigma-Aldrich. β-D-GlcpNAc-(1→2)-α-D-Manp-(1→3)-[β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)]-D-Manp and α-D-Manp-(1→3)-[α-D-Manp-(1→6)]-α-D-ManpOMe were purchased from Dextra Laboratories Ltd. The acceptor substrate of GnT-V, β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)-β-D-GlcpOOctyl,11 was purchased from Rose Scientific Ltd. 6-Acetamido-6-deoxycastanospermine was purchased from Industrial Research Ltd and used as a β-N-acetylglucosaminidase inhibitor to protect the acceptor substrate from degradation due to a contaminating hexosaminidase.34 Inhibitor concentrations of 10 mole percent relative to GnT-V were used in all samples containing GnT-V and acceptor substrate. The activity of GnT-V was not affected by the inhibitor.
IUPAC conventions were used throughout the text to define the dihedral angles of the acceptor substrate, β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)-β-D-GlcpOOctyl: φ = O5(i)-C1(i)-On(i − 1)-Cn(i −1), ψ = C1(i)-On(i −1)-Cn(i −1)-C(n −1)(i −1), ω = O6(i)-C6(i)-C5(i)-C4(i), where (i) indicates a given residue and (n) a ring position.35 Exocyclic torsion angles are named χn, where (n) is the ring position of the side group; for example, χ5 refers to the torsion angle for the exocyclic 6-hydroxymethyl groups on GlcNAc and Man (C4-C5-C6-O6). The exocyclic torsion angles for the N-acetyl group on GlcNAc are χ2a (C1-C2-NNAc-CNAc) and χ2b (C2-NNAc-CNAc-ONAc). The gg, gt, tg naming convention was used to describe the α-D-Manp-(1→6)-β-D-Glcp ω rotamer conformation and the χ5 rotamer conformations. Each character refers to a torsion angle being either gauche (g) or trans (t), where the first character describes the O6(i)-C6(i)-C5(i)-O5(i) dihedral angle and the second describes the O6(i)-C6(i)-C5(i)-C4(i) dihedral angle.
Synthesis of UDP-TEMPO
Uridine 5′-diphospho-4-O-2,2,6,6-tetramethylpiperidine 1-oxyl (UDP-TEMPO) was synthesized following the procedure reported by Berliner and Wong with some experimental modifications.36 In particular, it was observed that the 300 MHz NMR of two commercial samples of 4-phosphonooxy-TEMPO in D6 DMSO show that the product had degraded. Therefore this intermediate was synthesized following published procedures.37
Uridine 5’-monophosphomorpholidate-4-morpholine-N,N’-dicyclohexylcarboxamidine salt was purchased from Sigma. Other chemicals used in the synthesis were purchased from Aldrich. Pyridine was dried by distillation over calcium hydride and kept on activated molecular sieves. Reactions were run under argon. Solvents, water and pyridine were evaporated under reduced pressure with a bath temperature under 32°C. Iatrobeads 6RS-8060 used in the purification were purchased from Bioscan. Thin layer chromatograms were run on Merck Kieselgel 60 F254 aluminium coated plates.
2,2,6,6-Tetramethyl-4-phosphopiperidine-1-oxyl pyridinium salt (4-phosphonooxy-TEMPO)
Barium cyanoethylphosphate hydrate (4 g, 12.4 mmol) was run through a column of Dowex 50 x 4 (H+ form, about 65 g) and eluted with water. The appropriate fractions were freeze dried and the residue co-evaporated three times with dry pyridine providing the pyridinium salt (2.89 g). 4-hydroxy TEMPO (0.516 g, 3.0 mmol) was added to a solution of the above material in dry pyridine (15 mL). The pyridine was evaporated and the operation repeated two times. Dicyclohexyl carbodiimide (3.0 g, 15 mmol) was added to a solution of the above recovered mixture in dry pyridine (15 mL). After 2 days at room temperature, TLC (80:20 CHCl3, CH3OH) indicated the disappearance of the starting 4-hydroxy TEMPO. Water (5 mL) was added and the mixture was vigorously stirred and then filtered. The flask and the precipitate were further washed with water (20 x 2 mL). Lithium hydroxide (0.6 g) was then added and the mixture refluxed for 1 h. The insoluble material was removed by filtration, half of the water was evaporated, the mixture further filtered, and the solution finally concentrated by evaporation.
The above material was run through a column of Dowex 50 x 4 (pyridinium form, 30 g) with water as eluant and the recovered orange fractions evaporated to provide a residue which was dried in vacuo. The recovered material dissolved in the strict minimum amount of methanol was loaded onto a column of Iatrobeads (15.5 g) and the product eluted with a 70:30:4 mixture of chloroform, methanol and water to provide the expected material as an orange sticky solid (0.260 g, 21 %). 1H-NMR resonance assignments are included in the supplementary materials. Integrals from the 1H-NMR spectrum indicated that the material was likely to be a di-pyridinium salt.
Uridine 5’-diphosphate-4(2,2,6,6-tetramethylpiperidine-1-oxyl) ammonium salt (UDP-TEMPO)
The above nitroxide intermediate (0.260 g, 0.685 mmol) was evaporated 3 times with 5 mL of dry pyridine. UMP-morpholidate (0.646 g, 0.942 mmol) and tetrazole (3.2 mL of a 3% solution in acetonitrile, 1.1 mmol) were also evaporated 3 times with dry pyridine and added to the UMP-morpholidate with 5 mL of dry pyridine. The mixture was stirred at 25°C for 5 days. TLC (chloroform, methanol water 80:20:1) indicated the disappearance of the TEMPO-phosphate. The pyridine was evaporated in vacuo and the crude material divided in two portions separately purified by ion exchange chromatography on DOWEX 1 x 8 (200 mesh, HCO2−, 12 x 1 cm) and eluted with a gradient of NH4+ HCO2− (0.1–0.6 M). The recovered fractions were checked by TLC (3:2:1 isopropanol, ammonium hydroxide, water) and the results indicated a mixture of the expected product and hydrolyzed UDP. Fractions containing the upper spot were pooled, co-evapotated with an excess of distilled water and finally freeze dried providing 0.324 g dry yellowish solid. UDP-TEMPO was finally obtained after chromatography of the previous solid material (0.241 g) directly on a small column of Iatrobeads (3.5 g) packed in 90:10 isopropanol and ammonium hydroxide. Elution with a 80:20:1 mixture of isopropanol, ammonium hydroxide and water separated some impurities (TLC 3:2:1 of the same mixture) from the UDP-TEMPO (0.118 g, ammonium salt, yellowish solid). The recovered material was divided in two portions and further chromatographed on Biogel P2 (75 x 1.5 cm) using water as eluant. Appropriate yellowish fractions were collected and freeze dried providing the UDP-TEMPO (96 mg, ammonium salt, yellowish dry foam). 1H-NMR resonance assignments are included in the supplementary materials.
A reduced form of UDP-TEMPO (UDP-TEMPOH), where the oxyl radical is reduced to a hydroxyl group, was prepared by the addition of phenylhydrazine to 2.4 mg of UDP-TEMPO in deuterated methanol. Reduction was monitored with 1H NMR by the appearance of signals from UDP-TEMPOH. Water was added to the sample and the phenylhydrazine was extracted under argon with ethyl ether and dicholormethane. After the sample was dried, 2.0 mg of UDP-TEMPOH was recovered and analyzed with 1H NMR to verify complete removal of phenylhydrazine and reduction.
NMR Spectroscopy
Literature-based NMR resonance assignments of UDP-GlcNAc and β-D-GlcpNAc-(1→2)-α-D-Manp-(1→6)-β-D-GlcpOOctyl were confirmed using 1H-1H correlation spectroscopy (COSY), 1H-1H total correlation spectroscopy (TOCSY), and 1H-13C HSQC experiments.15,16
The binding epitope of the acceptor, UDP-GlcNAc, and UDP-TEMPOH were determined using the NMR saturation transfer difference (STD) experiment. Individual samples were prepared in 50 mM potassium phosphate, pH 6.5 (not corrected for deuterium), 100 mM NaCl, D2O buffer. To produce spectra for protein background subtraction, additional samples of GnT-V were prepared and run at identical experimental conditions as the ligand experiments. Reference spectra of each ligand in buffer were also collected using the standard single pulse-acquire 1H experiment. STD experiments generally follow those described by Mayer and Meyer except protein background was subtracted rather than using a T1ρ filter.38 Other experimental conditions and parameters for the STD experiments for the acceptor, UDP-GlcNAc, and UDP-TEMPOH are listed in Table 3. A Gaussian π pulse was used in the pulse train to saturate the protein in each experiment.
Table 3.
NMR and sample conditions for STD experiments.
| Substrate | Acceptor | Acceptor with 10 mM UDP | UDP-GlcNAc | UDP-TEMPOH |
|---|---|---|---|---|
| Concentration (mM) | 2.0 | 2.0 | 2.5 | 1.5 |
| GnT-V concentration (μM) | 100 | 100 | 100 | 60 |
| Magnet strength (MHz) | 900 | 900 | 600 | 600 |
| Saturation bandwidth (Hz) | 100 | 100 | 100 | 50 |
| Saturation frequency (ppm) | 0.0 | 0.0 | 0.0 | 7.1 |
| Saturation timea (s) | 0.5 | 0.5 | 2.0 | 1.9 |
The total saturation time includes the delay (1 ms) between the Gaussian pulses in the cycled pulse train.
Transferred nuclear Overhauser effect (trNOE) experiments were used to determine intramolecular distances for the acceptor, UDP-GlcNAc, and UDP-TEMPOH in the presence of GnT-V. Samples were prepared with free substrates and the substrates with GnT-V. Sample and NMR experimental conditions for the trNOE and trNOESY experiments are listed in Table 4. Both experiments included a z-filter for zero quantum coherence suppression which reduces line-shape distortions at short mixing times.39 The analysis of the 1D trNOE spectra of UDP-GlcNAc and 2D trNOESY spectra of UDP-TEMPOH involved volume integration of H6, H5, and R2’. The percent NOE was calculated for the H6-H5 and H6-H2’ pairs and plotted against the mixing time. The initial slopes of the NOE buildup curves and the H6-H5 reference distance of 2.43 Å were used to calculate the H6-H2’ distance. The analysis of the trNOESY spectra of the acceptor with GnT-V involved assignment of the crosspeaks and volume integration using NMRPipe40 and NMRView,41 respectively. 1H-1H distances within each sugar ring were taken from an energy minimized structure of the acceptor to use as reference distances. The inter-ring and exocyclic group distance constraints were calculated using the reference distances and the slopes from NOE buildup curves (peak volume versus mixing time).
Table 4.
NMR and sample conditions for trNOE and trNOESY experiments.
| Sample | UDP-GlcNAc | UDP-GlcNAc/GnT-V | UDP-GlcNAc | UDP-GlcNAc/GnT-V | Acceptor | Acceptor/GnTV | GnT-V | UDP-TEMPOH/GnT-V |
|---|---|---|---|---|---|---|---|---|
| Substrate concentration (mM) | 5 | 1.15 | 5 | 1.5 | 2 | 2 | 1.5 | |
| GnT-V concentration (μM) | 46 | 60 | 100 | 100 | 60 | |||
| Experiment | 2D NOESY | 2D NOESY | 1D NOEa | 1D NOEa | 2D ZQ filtered NOESY | 2D ZQ filtered NOESY | 2D ZQ filtered NOESY | 2D ZQ filtered NOESY |
| Magnet strength (MHz) | 500 | 500 | 600 | 600 | 900 | 900 | 900 | 600 |
| Temperature (ºC) | 25 | 25 | 10 | 10 | 25 | 25 | 25 | 25 |
| Mixing times (ms) | 100, 300, 500, 1000 | 100, 300, 500, 1000 | 25, 50, 75, 100, 150, 200 | 25, 50, 75, 100, 150, 200 | 100, 200, 300, 400, 500 | 50, 75, 100, 125, 150 | 200 | 50, 100, 300, 500 |
1D trNOE experiment was performed with selective saturation of uracil H6 with a 50 Hz bandwidth selective pulse.
Simulated Annealing
Xplor-NIH was used to refine the structure of the bound-state acceptor substrate using the NOE constraints listed in Table 1 and simulated annealing.42 Parameter and topology files were generated using standard files from Xplor-NIH for the carbohydrate portion of the acceptor and PRODRG for the octyl chain.43 A standard simulated annealing protocol from the Xplor-NIH on-line manual was used to refine the structure. A starting temperature of 1000 K was used with 2 fs time steps for the 12 ps high-temperature stage and 6 ps cooling stage. 100 structures were generated and the 10 lowest energy structures were selected. No NOE violations (outside of ± 0.5 Å) occurred for the 10 lowest energy structures.
Paramagnetic Relaxation Enhancement
Paramagnetic relaxation enhancement of the bound acceptor protons induced by bound UDP-TEMPO was measured by comparing the proton T1 relaxation times of an experimental and control sample. The experimental sample was 155 μM GnT-V, 2 mM acceptor, and 10 mM UDP-TEMPO. The control sample was 155 μM GnT-V, 2 mM acceptor, and 6.9 mM 4-hydroxy-TEMPO. The effective T1 relaxation times of the experimental and control samples were determined using a 1H-13C heteronuclear single quantum correlation (HSQC) experiment with recycle delay times of 0.25, 0.5, 1, and 2 seconds (including acquisition time). An effective T1 value for each sample and each cross-peak was determined by fitting the peak volume versus overall delay time (t) with the exponential equation: c*[1−exp(−t/T1)]. The HSQC experiment used was a modified version of Varian’s ProteinPack HSQC with gradients for 13C/1H chemical shift correlation. The experiment was optimized for natural abundance carbohydrates by replacing the selective carbon pulses with hard pulses and using the sensitivity enhancement option. WURST decoupling was used with a bandwidth of 32 kHz during the 50 ms acquisition time. A 13C sweep width of 6 kHz was used resulting in folding of the anomeric and octyl chain peaks with 64 time increments. The 1H spectral window (6.8 kHz) was centered at the water peak and the width adjusted to contain all acceptor peaks. The HSQC spectra were processed with NMRPipe40 with a 20 Hz Gaussian weighting function along the direct dimension to smooth the highly J-coupled peaks to one line. Linear prediction and a cosine squared weighting function were used along the indirect dimension to improve the signal-to-noise ratio. The peak volumes were measured using NMRView.41
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health’s National Center for Research Resources, RR005351, and National Institute for General Medical Sciences, GM033225.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yamashita K, Tachibana Y, Ohkura T, Kobata A. Enzymatic basis for the structural changes of asparagine-linked sugar chains of membrane glycoproteins of baby hamster kidney cells induced by polyoma transformation. J Biol Chem. 1985;260:3963–3969. [PubMed] [Google Scholar]
- 2.Pierce M, Arango J. Rous sarcoma virus-transformed baby hamster kidney cells express higher levels of asparagine-linked tri- and tetraantennary glycopeptides containing [GlcNAc-beta (1,6)Man-alpha (1,6)Man] and poly-N-acetyllactosamine sequences than baby hamster kidney cells. J Biol Chem. 1986;261:10772–10777. [PubMed] [Google Scholar]
- 3.Pierce M, Arango J, Hindsgaul O. Activity of UDP-GlcNAc:alpha-mannoside beta(1,6)N-acetylglucosaminyltransferase (GnT V) in cultured cells using a synthetic trisaccharide acceptor. Biochem Biophys Res Commun. 1987;146:679–684. doi: 10.1016/0006-291x(87)90582-1. [DOI] [PubMed] [Google Scholar]
- 4.Kaneko M, Alvarez-Manilla G, Kamar M, Lee I, Lee JK, Troupe K, Zhang WJ, Osawa M, Pierce M. A novel beta(1,6)-N-acetylglucosaminyltransferase V (GnT-VB) Febs Letters. 2003;554:515–519. doi: 10.1016/s0014-5793(03)01234-1. [DOI] [PubMed] [Google Scholar]
- 5.Guo HB, Lee I, Kamar M, Pierce M. N-acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J Biol Chem. 2003;278:52412–52424. doi: 10.1074/jbc.M308837200. [DOI] [PubMed] [Google Scholar]
- 6.Coutinho PM, Henrissat B. Carbohydrate-active enzymes: an integrated database approach. In: Gilbert HJ, Davies G, Henrissat B, Svensson B, editors. Recent Advances in Carbohydrate Bioengineering. The Royal Society of Chemistry; Cambridge: 1999. pp. 3–12. [Google Scholar]
- 7.Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. J Mol Biol. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- 8.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Zhang N, Adler B, Browne J, Freigen N, Pierce M. Preparation of antisera to recombinant, soluble N-acetylglucosaminyltransferase V and its visualization in situ. Glycoconjugate J. 1995;12:813–823. doi: 10.1007/BF00731243. [DOI] [PubMed] [Google Scholar]
- 10.Kamar M, Alvarez-Manilla G, Abney T, Azadi P, Kumar Kolli VS, Orlando R, Pierce M. Analysis of the site-specific N-glycosylation of β1,6-N-acetylglucosaminyltransferase V. Glycobiology. 2004;14:583–592. doi: 10.1093/glycob/cwh062. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava OP, Hindsgaul O, Shoreibah M, Pierce M. Recognition of oligosaccharide substrates by N-acetyl-glucosaminyltransferase-V. Carbohyd Res. 1988;179:137–161. doi: 10.1016/0008-6215(88)84115-6. [DOI] [PubMed] [Google Scholar]
- 12.Shoreibah MG, Hindsgaul O, Pierce M. Purification and characterization of rat kidney UDP-N-acetylglucosamine:α-6-D-mannoside β-1,6-N-acetylglucosaminyltransferase. J Biol Chem. 1992;267:2920–2927. [PubMed] [Google Scholar]
- 13.Siebert HC, Jimenez-Barbero J, Andre S, Kaltner H, Gabius HJ. Describing topology of bound ligand by transferred nuclear Overhauser effect spectroscopy and molecular modeling. In: Lee YC, Lee RT, editors. Recognition of Carbohydrates in Biological Systems, Methods in Enzymololgy. Vol. 362. Academic Press; London: 2003. pp. 417–434. [DOI] [PubMed] [Google Scholar]
- 14.Liang BY, Bushweller JH, Tamm LK. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J Am Chem Soc. 2006;128:4389–4397. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramakrishnan V, Teng Q, Adams MWW. Characterization of UDP amino sugars as major phosphocompounds in the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 1997;179:1505–1512. doi: 10.1128/jb.179.5.1505-1512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bock K, Duus JØ, Hindsgaul O, Lindh I. Analysis of conformationally restricted models for the (1→6)-branch of asparagine-linked oligosaccharides by NMR-spectroscopy and HSEA calculation. Carbohyd Res. 1992;228:1–20. doi: 10.1016/s0008-6215(00)90544-5. [DOI] [PubMed] [Google Scholar]
- 17.Zhang N, Peng KC, Chen L, Puett D, Pierce M. Circular Dichroic Spectroscopy of N-Acetylglucosaminyltransferase V and its substrate interactions. J Biol Chem. 1997;272:4225–4229. doi: 10.1074/jbc.272.7.4225. [DOI] [PubMed] [Google Scholar]
- 18.Moseley HNB, Curto EV, Krishna NR. Complete relaxation and conformational exchange matrix (CORCEMA) analysis of NOESY spectra of interacting systems- 2-dimensional transferred NOESY. J Mag Res B. 1995;108:243–261. doi: 10.1006/jmrb.1995.1129. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Palcic MM, Schriemer DC, Alvarez-Manilla G, Pierce M, Hindsgaul O. Frontal affinity chromatography coupled to mass spectrometry for screening mixtures of enzyme inhibitors. Anal Biochem. 2001;299:173–182. doi: 10.1006/abio.2001.5417. [DOI] [PubMed] [Google Scholar]
- 20.Blume A, Angulo J, Biet T, Peters H, Benie AJ, Palcic M, Peters T. Fragment-based screening of the donor substrate specificity of human blood group B galactosyltransferase using saturation transfer difference NMR. J Biol Chem. 2006;281:32728–32740. doi: 10.1074/jbc.M600424200. [DOI] [PubMed] [Google Scholar]
- 21.Cummings RD, Trowbridge IS, Kornfeld S. A mouse lymphoma cell line resistant to the leukoagglutinating lectin from Phaseolus vulgaris is deficient in UDP-GlcNAc:α-D-Mannoside β1,6 N-acetylglucosaminyltransferase. J Biol Chem. 1982;257:13421–13427. [PubMed] [Google Scholar]
- 22.Kanie O, Crawley SC, Palcic MM, Hindsgaul O. Acceptor-substrate recognition by N-acetylglucosaminyltransferase-V: Critical role of the 4”-hydroxyl group in β-D-GlcpNAc-(1–2)-α-D-Manp(1–6)-β-D-Glcp-OR. Carbohyd Res. 1993;243:139–164. doi: 10.1016/0008-6215(93)84087-m. [DOI] [PubMed] [Google Scholar]
- 23.Kanie O, Crawley SC, Palcic MM, Hindsgaul O. Key involvement of all three GlcNAc hydroxyl groups in the recognition of β-D-GlcpNAc-(1–2)-α-D-Manp(1–6)-β-D-Glcp-OR by N-acetylglucosaminyltransferase-V. Bioorg Med Chem. 1994;2:1231–1241. doi: 10.1016/s0968-0896(00)82074-x. [DOI] [PubMed] [Google Scholar]
- 24.Linkder T, Crawley SC, Hindsgaul O. Recognition of the acceptor β-D-GlcpNAc-(1–2)-α-D-Manp(1–6)-β-D-Glcp-OR by N-acetylglucosaminyltransferase-V: None of the hydroxyl groups on the Glc-residue are important. Carbohyd Res. 1993;245:323–331. doi: 10.1016/0008-6215(93)80081-o. [DOI] [PubMed] [Google Scholar]
- 25.Khan SH, Duus JØ, Crawley SC, Palcic MM, Hindsgaul O. Acceptor-substrate recognition by N-acetyl-glucosaminyltransferase-V: role of the mannose residue in β DGlcNAc(1–2)αDMan(1–6)βDGlcOR. Tetrahedron: Asymmetry. 1994;5:2415–2435. [Google Scholar]
- 26.Khan SH, Crawley SC, Kanie O, Hindsgaul O. A trisaccharide acceptor analog for N-acetylglucosaminyltransferase V which binds to the enzyme but sterically precludes the transfer reaction. J Biol Chem. 1993;268:2468–2473. [PubMed] [Google Scholar]
- 27.Patel DJ, Kozlowski SA, Nordheim A, Rich A. Right-handed and left-handed DNA – studies of B-DNA and Z-DNA by proteon nuclear Overhauser effect and P NMR. P Natl Acad Sci USA. 1982;79:1413–1417. doi: 10.1073/pnas.79.5.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angulo J, Langpap B, Blume A, Biet T, Meyer B, Krishna NR, Peters H, Palcic MM, Peters T. Blood group B galactosyltransferase: Insights into substrate binding from NMR experiments. J Am Chem Soc. 2006;128:13529–13538. doi: 10.1021/ja063550r. [DOI] [PubMed] [Google Scholar]
- 29.Lütteke T, Frank M, von der Lieth CW. Carbohydrate Structure Suite (CSS): analysis of carbohydrate 3D structures derived from the PDB. Nuleic Acids Res. 2005;33:D242–246. doi: 10.1093/nar/gki013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirschner KN, Woods RJ. Solvent interactions determine carbohydrate conformation. PNAS. 2001;98:10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bock K, Duus JØ. A conformational study of hydroxymethyl groups in carbohydrates investigated by 1H NMR spectroscopy. J Carb Chem. 1994;13:513–543. [Google Scholar]
- 32.Pak JE, Arnoux P, Zhou S, Sivarajah P, Satkunarajah M, Xing X, Rini JM. X-ray crystal structure of leukocyte type core 2 β1,6-N-acetylglucosaminlytransferase. J Biol Chem. 2006;281:26693–26701. doi: 10.1074/jbc.M603534200. [DOI] [PubMed] [Google Scholar]
- 33.Hu Y, Chen L, Ha S, Gross B, Falcone B, Walker D, Mokhtarzadeh M, Walker S. Crystal structure of the MurG:UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc Natl Acad Sci USA. 2003;100:845–849. doi: 10.1073/pnas.0235749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu PS, Kang MS, Sunkara PS. A potent inhibitor of β-acetylglucosaminidases: 6-acetamido-6-deoxycastanospermine. Tetrahedron Lett. 1991;32:719–720. [Google Scholar]
- 35.IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN) Symbols for Specifying the Conformation of Polysaccharide Chains. Eur J Biochem. 1983;131:5–7. doi: 10.1111/j.1432-1033.1983.tb07224.x. [DOI] [PubMed] [Google Scholar]
- 36.Berliner LJ, Wong SS. Conformational investigations of bovine lactose synthetase. I Manganese(II) and spin-labeled uridine 5'-diphosphate binding to bovine galactosyltransferase. Biochemistry-US. 1975;14:4977–4982. doi: 10.1021/bi00693a029. [DOI] [PubMed] [Google Scholar]
- 37.Weiner H. Interaction of a spin-labeled analog of nicotinamide adenine dinucleotide with alcohol dehydrogenase. I Synthesis, kinetics, and electron paramagnetic resonance studies. Biochemistry-US. 1969;8:526–533. doi: 10.1021/bi00830a011. [DOI] [PubMed] [Google Scholar]
- 38.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 39.Thrippleton MJ, Keeler J. Elimination of zero-quantum interference in two-dimensional NMR spectra. Angew Chem Int Ed. 2003;42:3938–3941. doi: 10.1002/anie.200351947. [DOI] [PubMed] [Google Scholar]
- 40.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 41.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 42.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR Molecular Structure Determination Package. J Magn Res. 2003;160:66–74. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 43.Schuettelkopf AW, van Aalten DMF. PRODRG - a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.