

Abstract
Objective
To determine whether mutations in the genes for α-synuclein or β-synuclein are responsible for dementia with Lewy bodies (DLB), a disorder closely related to Parkinson disease (PD).
Methods
The authors ascertained 33 sporadic cases of DLB and 10 kindreds segregating DLB. DNA samples from the 43 index cases were screened for alterations in the genes for α-synuclein and β-synuclein, as α-synuclein alterations cause PD and β-synuclein may modulate α-synuclein aggregation and neurotoxicity.
Results
Two amino acid alterations were identified in unrelated DLB index cases: a valine to methionine substitution at codon 70 (V70M) and a proline to histidine substitution at codon 123 (P123H), both in the β-synuclein gene. These amino acid substitutions occur at conserved residues in highly conserved regions of the β-synuclein protein. Screening of at least 660 chromosomes from control subjects matched to the patients’ population groups failed to identify another V70M or P123H allele. Cosegregation analysis of an extended pedigree segregating the P123H β-synuclein alteration suggested that it is a dominant trait with reduced penetrance or a risk factor polymorphism. Histopathology and immunohistochemistry analysis of index case brain sections revealed widespread Lewy body pathology and α-synuclein aggregation without evidence of β-synuclein aggregation.
Conclusion
Mutations in the β-synuclein gene may predispose to DLB.
While Parkinson disease (PD) is a movement disorder with cognitive impairment as an occasional feature, distinct PD-related clinicopathologic entities with dementia as a presenting feature have been recognized.1,2 These patients have been lumped into the diagnostic category, dementia with Lewy bodies (DLB).3 Included in this diagnostic entity are patients with the “Lewy body variant of AD,” who display Alzheimer disease (AD) pathology with significant numbers of cortical and limbic system Lewy bodies, but do not show signs of parkinsonism.4 Another group of patients in the DLB diagnostic category have diffuse Lewy body disease, an atypical dementing illness with widespread PD-like Lewy body pathology, but no AD pathology.5,6
At present, the list of implicated genes for PD includes α-synuclein, parkin, ubiquitin C-terminal hydrolase L1 (UCH-L1), DJ-1, neurofilament medium chain, and synphilin-1.7–10 The discovery of missense mutations in the α-synuclein genes of patients with autosomal dominant PD was a particularly important observation,11 as studies of PD brains then demonstrated that α-synuclein (SNCA) is a major component of Lewy body inclusions.12 α-Synuclein belongs to a family of proteins, consisting of two other members, β-synuclein (SNCB) and γ-synuclein (SNCG).13 Given the wealth of data supporting a role for SNCB in modulating SNCA aggregation and toxicity,14–19 we reasoned that mutations in the α-synuclein gene (SNCA) or the β-synuclein gene (SNCB) might be responsible for the widespread Lewy body formation and extensive neurodegeneration seen in DLB patients. To test this hypothesis, we ascertained familial and sporadic cases of DLB and screened for mutations in SNCA and SNCB. We identified two nonconservative β-synuclein (SNCB) amino acid changes in unrelated DLB probands. Our findings raise the possibility that alterations in SNCB contribute to the production of Lewy body disorders.
Materials and methods
DLB patient ascertainment and evaluation
Twenty-two DLB sporadic patients and five DLB families were ascertained in Seattle, WA, under an approved University of Washington Medical Center (UWMC) human subjects protocol or in Niigata, Japan, after review board acceptance. Samples for the remaining 16 DLB cases were provided by Dr. Matthew Farrer of the Mayo Clinic (Jacksonville, FL). The probands underwent standard neurologic and psychiatric evaluation, and all probands met consensus clinical diagnostic criteria for DLB.3 Furthermore, 20 of the 33 sporadic cases and at least one patient from nine of the 10 families had brain pathology documenting Lewy bodies at autopsy. Clinical histories for deceased relatives were obtained from next-of-kin, and medical records were reviewed systematically in accordance with DLB consensus diagnostic criteria.3 Four hundred sixteen Caucasian subjects who presented to the Molecular Diagnosis unit for thrombophilia or cystic fibrosis carrier testing or as control subjects for an AD study were selected as normal control subjects at the UWMC, and 244 Japanese patients being evaluated for non-neurologic diseases were selected as control subjects in Niigata, Japan. Sporadic PD cases were similarly ascertained at both sites after clinical diagnosis by trained neurologists.
Sequencing of the α- and β-synuclein coding regions
Genomic DNA was extracted from lymphocytes, brain tissues, or cultured skin fibroblasts. Primer sequences (including three new primers: a5r, a7-2f, and a7-1r), expected product sizes, and annealing temperatures are given in table E-1 on the Neurology Web site (go to www.neurology.org). PCR products were purified using the QIAquick purification kit (Qiagen, Valencia, CA) prior to ABI prism BigDye Terminator cycle sequencing on an ABI-377 or ABI PRISM 3100 Genetic Analyzer.
PCR assays for the V70M and P123H alterations
As the P123H alteration creates a DraIII restriction site, we designed an assay to distinguish between normal and P123H samples based on DraIII restriction of PCR-amplified β-synuclein exon 5. To ensure that the PCR product is cut by DraIII, we engineered a single nucleotide alteration into the reverse primer to create a DraIII restriction site as a positive control for successful digestion, and used B5F as forward primer. V70M allele discrimination was performed on an ABI 7700 with different oligonucleotide probes and an exon 4–specific primer set using the TaqMan approach. Primers and probe sequences are given in table E-1 (go to www.neurology.org).
Immunohistochemistry
Antibodies specific for SNCA and SNCB were used to probe 6-μm sections that were deparaffinized in xylene followed by hydration in descending ethanol concentrations.20 The sections were then immunostained using anti-SNCA antibodies SNL-1 (1:5,000), Syn 43–57 (1:2,000), NAC (1:2,000), Syn 204 (1:2,000), Syn 202 (ascites, 1:20,000), or the anti-SNCB antibody 86–131 (1:2,000). Immunoreactivity was visualized with the avidin-biotin complex detection system (Vector Laboratories, Burlingame, CA) using 3,3-diaminobenzidine as the chromogen, and counterstained with hematoxylin.
Brain tissue fractionation
Cingulate, cerebellum, hippocampus, and occipital lobes (approximately 0.2 g) were homogenized in 10 mL/g high-salt (HS) buffer at pH 7.4 (50 mM Tris, 750 mM NaCl, 10 mM NaF, 5 mM EDTA, and protease inhibitor cocktail) and centrifuged at 40,000 rpm × 20 minutes at 4 °C. The pellets were re-extracted once with HS buffer, and myelin was floated off of the cerebellum sample. All pellets were then twice extracted in 10 mL/g HS containing 1% Triton X-100 (Triton X buffer), followed by twice extracting the pellets at room temperature in 10 mL/g Triton X buffer containing 2% sodium dodecyl sulfate (SDS) (SDS buffer). Finally, the resulting pellet was resuspended in 2 mL/g SDS buffer containing 8 M urea, with brief sonication.
Western blotting
Brain tissue fractionations were boiled for 10 minutes with 1X Laemmli buffer and resolved by 15% SDS-PAGE. The proteins were electrophoretically transferred onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH) and blocked with a 5% solution of powdered skimmed milk dissolved in Tris-buffered saline. The membranes were incubated with antibodies to SNCA (NAC) (1:500) or SNCB (86–131) (1:500).
Results
Sequence screen yields novel alterations in the β-synuclein gene
To determine whether a SNCA or SNCB alteration could be implicated in the pathogenesis of familial DLB, we sequenced the coding regions of these genes using primer sets that included adjacent untranslated and intronic regions. In the 43 index cases we analyzed, we found no significant alteration in the coding region of SNCA. However, we did detect a novel single nucleotide polymorphism (SNP) at nucleotide position 67 (A → G) in intron 5 of SNCA. Our sequence screening of SNCB similarly yielded a novel SNP in noncoding region DNA, in this case at nucleotide position 51 (C → T) in intron 6 of SNCB. We found no evidence for association of either noncoding SNP with DLB disease status (p = 0.55 and p = 0.84 by x2 analysis).
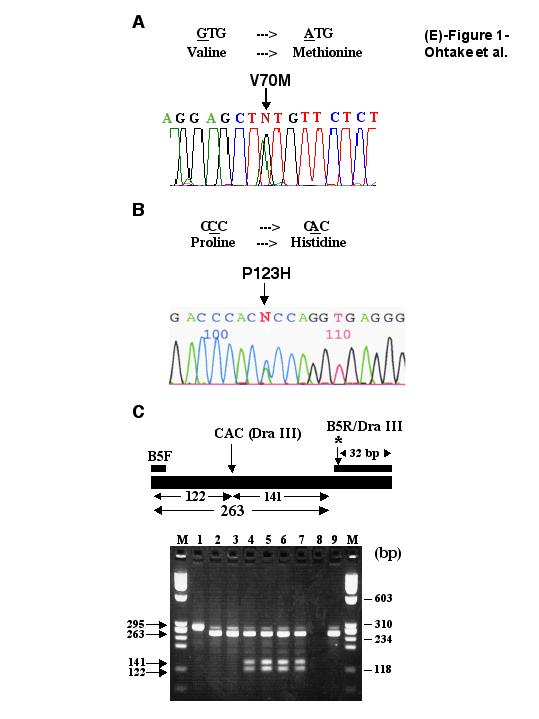
In addition to noncoding SNPs, our sequence screen identified two coding region alterations that stood out as particularly interesting: 1) an exon 4 SNCB codon 70 change (GTG → ATG; Val → Met), and 2) an exon 5 SNCB codon 123 change (CCC → CAC; Pro → His) (figure E-1, A and B; go to www.neurology.org). These nucleotide alterations yielded distinct amino acid substitutions in SNCB, leading us to designate them as the V70M and the P123H β-synuclein alleles. As numerous PD patients have been screened for SNCB coding region alterations,21,22 the absence of the V70M β-synuclein allele and the P123H β-synuclein allele from the Genbank SNP database suggested to us that these variants are not common SNPs. To determine whether the two SNCB alleles are instead uncommon nonsynonymous coding SNPs, we screened large numbers of unaffected control individuals from the two population groups (i.e., Caucasian and Japanese) from which the affected DLB cases were ascertained (table 1). As the P123H SNCB alteration creates a DraIII restriction site, we designed an assay for detection of this alteration in PCR-amplified SNCB exon 5 DNA (see figure E-1, C; go to www.neurology.org). Using this PCR-based assay and a V70M allele discrimination assay, we screened more than 330 control individuals (i.e., >660 chromosomes) for the V70M and the P123H SNCB alleles. We did not find these alterations in any control sample, nor in any sporadic PD patient (see table 1). These results suggest that the V70M and P123H SNCB alleles are unlikely to be coding SNPs.
Table 1.
Screening of patients and controls for β-synuclein alterations
| Nucleotide variant | G208A
|
C368A
|
||
|---|---|---|---|---|
| Amino acid alteration | V70M
|
P123H
|
||
| Alleles | G/G | G/A | C/C | C/A |
| Sporadic DLB | 33 | 1 | 33 | 0 |
| Familial DLB | 10 | 0 | 10 | 1 |
| Sporadic PD | 338 | 0 | 45 | 0 |
| Normal controls | 331 | 0 | 416 | 0 |
Values represent number of alleles.
DLB = dementia with Lewy bodies; PD = Parkinson disease.
Case summaries of the two DLB probands carrying SNCB coding region alterations
Patient A
The first case (V70M) was an 83-year-old man who noticed difficulties with moving his left arm at the age of 78 and gradually developed monotonous speech accompanied by a parkinsonian gait (table 2). Levodopa was effective for his motor symptoms, but his visual hallucinations worsened despite increasing amounts of carbidopa/levodopa. Motor function, especially of speech and swallowing, progressively deteriorated. He also displayed fluctuating dementia, visual hallucinations, and cognitive impairment. At age 83 years, upon hospital admission with a chief complaint of aspiration pneumonia, examination of the patient revealed evidence of dysarthria, dysphasia, bradykinesia, and rigidity and tremor of the left limbs. He was noted to walk with short steps and a stooped posture. The patient’s condition worsened until he became entirely bedridden and required tube feeding. He died of pneumonia the next year, but autopsy was not permitted.
Table 2.
Clinical features of DLB index cases
| Case | Age at onset, y | Age at death, y | Initial complaint | Timing of motor decline | Timing of dementia | Tremor | Delusion or hallucination |
|---|---|---|---|---|---|---|---|
| A | 78 | 84 | Bradykinesia | At onset | 5 years after onset | + | + |
| B | 61 | 79 | Memory loss | 12 y after onset | At onset | + | + |
DLB = dementia with Lewy bodies.
Patient B
The second case (P123H) was a 64-year-old man who presented with a 3-year history of mild dementia and deterioration in his handwriting (table 2). Early symptoms were atypical in this case, because of frontal lobe involvement manifesting as executive and language dysfunction. His cognitive difficulties progressed and were accompanied by significant depression with dysphoric mood. From age 71 to 77 years, periodic neurologic examinations documented his persistent motor apraxia and recurrent episodes of audio and visual hallucinations. At age 73 years, he displayed parkinsonism including prominent bradykinesia. The parkinsonian symptoms worsened over the next 5 years, and placement in a nursing home facility became necessary. The patient died of acute bronchopneumonia and multiple pulmonary emboli at age 79 years. Neuropathologic examination was consistent with DLB, neocortical subtype, with additional, significant changes of AD (Braak stage V) present. A clinical description of this case and of the neuropathologic findings has been published.23
Clinical and molecular evaluation of the familial DLB pedigree
While patient A was an apparently sporadic case of DLB, patient B is part of an extended pedigree (figure 1). The index case’s history, physical examination, and neuropathology studies were all consistent with a diagnosis of DLB, as was the clinical evaluation of his sister.23 The proband’s mother’s medical records provided evidence supporting a clinical diagnosis of possible DLB, because she met only two of the three necessary criteria. On the basis of all available clinical and autopsy records, only one of the patient’s maternal uncles fully satisfied the clinical diagnostic criteria for probable DLB (see figure 1). A number of first cousins and maternal aunts, however, could be designated as “possible affected” because they partially satisfied DLB clinical criteria. A full family history for the kindred of this proband has been described.23 As DNA was not available from the proband’s deceased mother and sister, we obtained samples from more distant surviving relatives of the proband. Screening of the DNA samples obtained from the proband’s three ascertained first cousins indicated that III-2 is homozygous normal, while III-5 and III-13 are heterozygous for the P123H SNCB allele. At the time of last examination, III-5 remains clinically unaffected but III-2 and III-13 have possible DLB. Clinical differentiation of DLB from AD or PD can be difficult, so the overlap in signs and symptoms between the two disorders means that some elderly individuals (such as III-2 or III-13) could be phenocopies.
Figure 1.
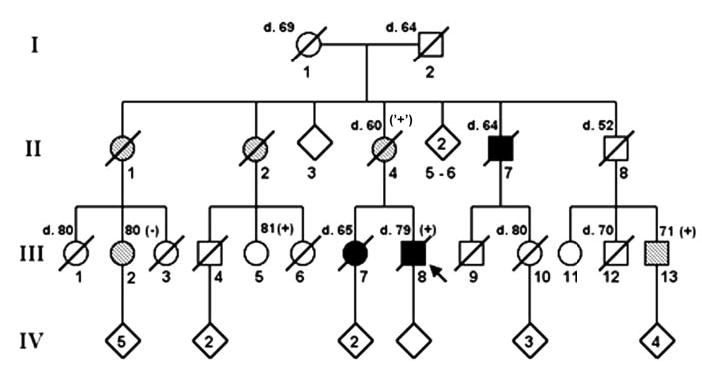
Extended pedigree of the dementia with Lewy bodies (DLB) family segregating the P123H SNCB alteration. Four generations of this family are shown with their DLB disease status indicated. For deceased members, the age at death is indicated above and to the left of their pedigree symbols. For living members of the pedigree for whom clinical and molecular information is available, their age at the time of ascertainment is indicated above and to the right of their symbol. PCR amplification of SNCB exon 5 followed by DraIII restriction digestion was performed for individuals III-2, III-5, III-8, and III-13. Individuals heterozygous for the P123H alteration are marked as (+), and individuals who are homozygous normal are marked as (−) above and to the right of their pedigree symbols (squares are males; circles are females). Unfilled symbols represent unaffected individuals, filled symbols correspond to affected individuals, and hatched symbols indicate possible DLB-affected individuals. The proband is marked with an arrow. The (‘+’) designation adjacent to the proband’s mother indicates her assumed P123H heterozygous status, the probability of which is 99.84% given the presence of the P123H alteration in her niece (III-2) and in her nephew (III-13), and assuming a P123H allele frequency of 1/500.
Functional implications of the V70M and P123H amino acid substitutions
As SNCA and SNCB share a high degree of amino acid sequence similarity, we compared the locations of the familial PD mutations in SNCA with the SNCB allele polymorphisms associated with DLB (figure 2A). While the V70M alteration resides in a synuclein protein family consensus repeat domain akin to the pathogenic SNCA alterations, P123H occurs at the carboxy-terminus. Despite being located at some distance from one another, the amino acids altered in our two DLB cases are extremely highly conserved residues based on a comparison of human SNCB protein sequence to the protein sequences of various mammals (see figure 2, B and C). Furthermore, the potential structural implications of such amino acid substitutions are significant. Valine is a branched chain amino acid, whereas methionine’s side chain is linear and contains a bulky sulfur group. Position 70 in SNCB occurs in a region that, by analogy with SNCA, could be important in determining the ternary structure of the protein, as residues 71–82 in SNCA are required for in vitro fibrillization.13 Proline is unique among all amino acids as its side chain is bonded to not only the α-carbon, but also to nitrogen, giving it a cyclic structure. Thus, proline typically plays an important role in influencing tertiary structure. Histidine, unlike proline, is highly hydrophilic because of its polar side chain. For these reasons, substitution of hydrophilic histidine for uncharged, sterically constrained proline at amino acid position 123 likely changes the ternary structure of SNCB. That this alteration involves a region of SNCB that is extremely highly conserved across vertebrate species further suggests that it may have functional consequences.
Figure 2.
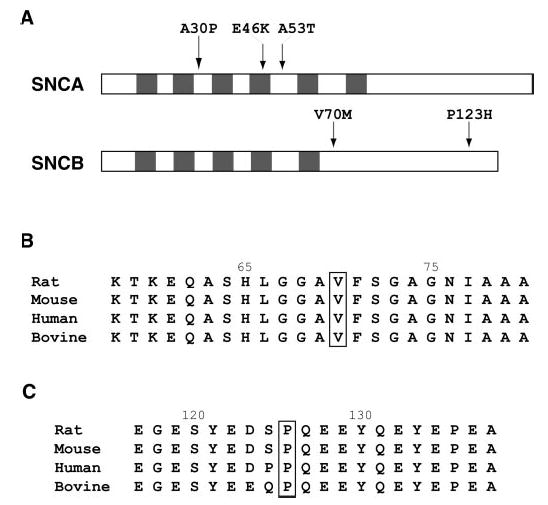
Locations and coding sequence conservation of the dementia with Lewy bodies–associated SNCB alterations. (A) Comparison of pathogenic SNCA mutation locations and putatively pathogenic SNCB alterations is shown. In the synuclein proteins, an 11-residue domain with a KTKEGV consensus repeat (shaded regions) has been proposed to exist and comprises the region to which all three known SNCA disease mutations localize. (B) Sequence comparison of the middle region of SNCB across rodent, human, and bovine species. The valine (V) at position 70 is boxed to indicate its location. As seen here, this valine residue is 100% conserved across a wide range of mammalian species, suggesting its potential functional importance. Genbank accession numbers are as follows: Q63754 (rat), AAK83238 (mouse), AAB30860 (human), and P33567 (bovine). (C) Sequence comparison of the carboxy-terminal region of SNCB across rodent, human, and bovine species. The proline (P) at position 123 is boxed to indicate its location. As shown here, the proline residue at position 123 is 100% conserved across a wide range of mammalian species, suggesting its potential functional importance. Accession numbers are as above.
P123H proband shows Lewy body pathology
Of the two DLB index cases found to possess SNCB alterations, only the DLB patient carrying the P123H SNCB alteration was available for further neuropathologic study. We began by immunostaining brain sections from this patient with an antibody specific for SNCA, and we observed extensive Lewy body pathology in regions of the hippocampus and amygdala (figure 3A), as well as in the substantia nigra (data not shown). We then surveyed hilar regions of the hippocampus and amygdala for SNCB pathology using an antibody specific for SNCB. As shown previously, Lewy bodies do not stain for SNCB (data not shown), and although we observed diffuse SNCB immunoreactivity in the hilus of the hippocampus and in the amygdala in the DLB proband, we did not detect any SNCB immunoreactive aggregates or inclusions (see figure 3, B and C). We then prepared HS and Triton fractions from hippocampal sections from the DLB proband. While insoluble SNCA could be detected in fractions run on polyacrylamide gels, all the detectable SNCB was present in the soluble fraction (see figure 3D; data not shown).
Figure 3.
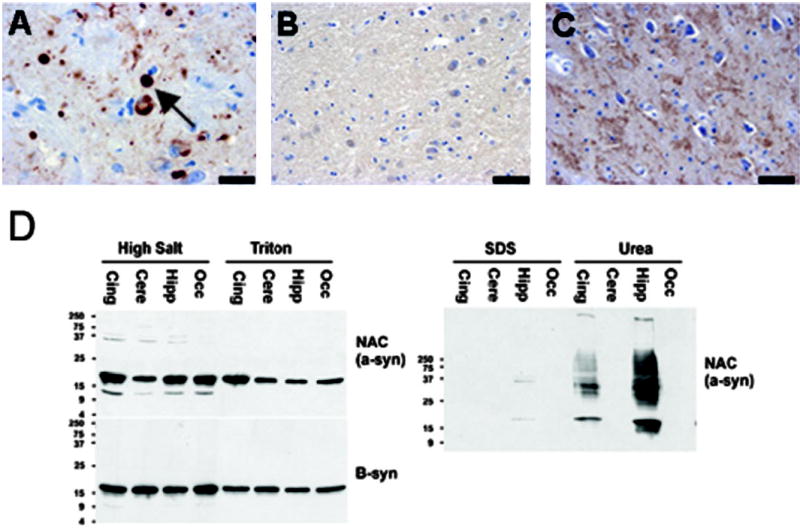
Neuropathology and fractionation studies of the P123H proband. (A) Immunostaining with an anti-SNCA antibody, Syn 202, reveals typical Lewy body pathology (marked by arrow) in the amygdala of the dementia with Lewy bodies (DLB) proband. Scale bar = 20 μm. (B) Immunostaining with a SNCB-specific antibody (β-syn 86–131) reveals diffuse reactivity throughout the neuropil of the DLB proband’s amygdala, but no β-synuclein aggregate staining is noted, as had been observed in SNCB aggregate-containing hippocampus sections, which served as a positive control. Scale bar = 40 μm. (C) Immunostaining with the same SNCB-specific antibody (β-syn 86–131) was then performed on sections from the hippocampus of the DLB proband. Diffuse immunoreactivity is again apparent throughout the hippocampal neuropil; however, punctate staining indicative of aggregates is not detected, as had been observed in SNCB aggregate-containing hippocampus sections, which served as a positive control. Scale bar = 40 μm. (D) Western blot analysis of synuclein soluble and insoluble fractions. Cingulate (Cing), cerebellum (Cere), hippocampus (Hipp), and occipital lobe (Occ) wedges from the brain of the DLB proband were fractionated to obtain insoluble and soluble protein lysates. Immunoblots of soluble high-salt and Triton extracts from these tissues were probed with an antibody against the NAC portion of SNCA or with an antibody specific for SNCB. Both SNCA and SNCB are abundant in these soluble fractions. Immunoblots of sodium dodecyl sulfate soluble (SDS) and SDS insoluble (Urea) extracts were similarly probed with these same two antibodies. Only insoluble SNCA was detected, and it was present in the cingulate and hippocampal extracts, not the cerebellar or occipital lobe extracts. No insoluble SNCB was detected (not shown).
Discussion
The pathologic hallmark of PD is the Lewy body, a spherical intracytoplasmic inclusion with a dense hyaline core and clear surrounding halo.1 DLB is pathologically related to PD because Lewy bodies are found throughout the cortex and limbic regions as well as in brainstem nuclei and the substantia nigra of brains from DLB patients. Although the molecular basis of PD remains elusive and the role of the Lewy body in PD pathogenesis is unclear, recent lines of evidence suggest that alteration of SNCA conformation is a key event in the pathogenic cascade. Hypothesized pathways for the production of putatively toxic SNCA protofibrils in PD include rare inheritance of mutant forms of SNCA, oxidative stress in the presence of cytoplasmic dopamine, and impaired proteosomal processing or metabolism of SNCA.24,25 In our DLB sporadic cases and kindreds, we theorized that an alteration of the SNCA or SNCB genes might cause or contribute to their phenotypes. Our sequence screening yielded nonconservative amino acid changes in the coding region of the SNCB gene in two DLB index cases. We have designated these alterations as the V70M and the P123H SNCB alleles.
One line of evidence for a polymorphism to be considered a pathogenic mutation would be its absence in the normal population. We tested more than 660 chromosomes from normal individuals of ethnic backgrounds comparable to each DLB proband, and we did not find a single instance of the V70M or P123H SNCB allele. These results suggest that the V70M and P123H alterations are unlikely to be coding SNPs. As stronger evidence for causality would be cosegregation of a disease allele with the disease phenotype within an affected kindred, we turned our attention to the remnants of an extended pedigree containing the DLB proband carrying the P123H allele. Our attempts to establish cosegregation, however, were inconclusive. Although possession of the P123H SNCB allele does not produce a disease phenotype in all individuals who carry it, three of four individuals who are heterozygous for the P123H allele in this DLB kindred are definitely or potentially DLB affected (II-2, III-8, and III-13). Thus, if the V70M or P123H SNCB alteration is pathogenic, our data suggest an autosomal dominant trait with reduced penetrance akin to BRCA1 mutations in breast–ovarian cancer or alternatively a risk factor polymorphism for disease similar to the Factor V Leiden R506Q alteration in venous thrombotic disease.26,27
Although the genetic evidence supporting a role for SNCB in causing DLB from this study is admittedly suggestive, the likelihood of these alterations being pathogenic in nature is reinforced by numerous independent studies that indicate a role for SNCB in the regulation of SNCA.14–19 Another exciting discovery relevant to DLB pathogenesis is the identification of a novel SNCA E46K alteration that cosegregates with the DLB phenotype in a multigeneration autosomal dominant kindred of Spanish ancestry.28 Recent studies of pedigrees segregating autosomal dominant DLB-like phenotypes also indicate that triplication of the SNCA gene can cause this disease,29,30 suggesting that SNCA overexpression can promote its pathogenic action. Our findings together with these studies lead us to propose a model for how alteration of SNCB results in DLB (figure 4). According to this model, an alteration in SNCB structure or function would accelerate the SNCA pathogenic cascade by preventing the normal inhibitory action of SNCB upon SNCA. The next step in assessing the potential pathogenicity of the V70M and P123H SNCB alterations will thus be to determine how SNCB modulates SNCA toxicity and the effect of mutated SNCB upon SNCA regulation. This work will be critical not only for confirming the role of these alterations in DLB, but also for understanding the molecular basis of all diseases that display Lewy body inclusions.
Figure 4.
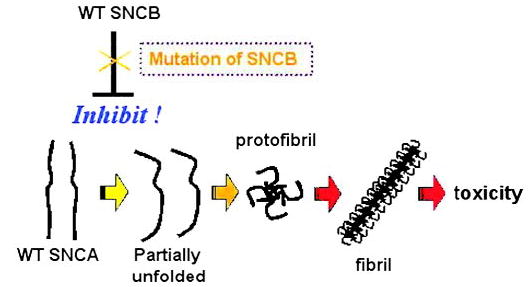
Model for the basis of SNCB pathogenicity in DLB. Conversion of wild-type SNCA to an altered conformer ultimately results in the formation of protofibrils and then fibrils yielding toxicity. Wild-type SNCB somehow modulates SNCA metabolism or function to inhibit this pathway, while SNCB mutation may alter or prevent this inhibitory effect, thereby predisposing to SNCA-mediated neurotoxicity. WT = wild-type.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank the patients and families who participated in this study; A. Smith and Y. Nakabayashi for technical assistance; E.J. Steinbart for patient sample ascertainment; Drs. S. Igarashi, M. Saito, M. Wakabayashi, M. Yamamoto, and N. Hattori for providing patient resources; and M. Hanson, A. Singleton, and M. Farrer, who provided DLB patient samples from the Udall Parkin-son’s Disease Center (Mayo Clinic, Jacksonville, FL).
Footnotes
Additional material related to this article can be found on the Neurology Web site. Go to www.neurology.org and scroll down the Table of Contents for the September 14 issue to find the title link for this article.
DLB patient samples were from the Udall Parkinson’s Disease Center (Mayo Clinic, Jacksonville, FL; NIH P01 grant NS-40256). A.R.L. was recipient of a Basic Biology of Aging pilot project grant from the Nathan Shock Center at the University of Washington and is a Paul Beeson Physician Faculty Scholar in Aging Research through the American Foundation for Aging Research. I.V.J.M. is funded by the Canadian Institutes of Health Research. V.M.Y.L. and J.Q.T. are supported by NIH grant AG-09215 and by a Pioneer Award from the Alzheimer’s Association.
Dr. Limprasert’s present address: Department of Pathology, Faculty of Medicine, Prince of Songkla University, Hat Yai, Songkhla 90112, Thailand.
References
- 1.Lang AE, Lozano AM. Parkinson’s disease: first of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 2.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 3.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 4.Hansen LA, Samuel W. Criteria for Alzheimer’s disease and the nosology of dementia with Lewy bodies. Neurology. 1997;48:126–132. doi: 10.1212/wnl.48.1.126. [DOI] [PubMed] [Google Scholar]
- 5.Okazaki H, Lipkin LE, Aronson SM. Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol. 1961;20:237–244. doi: 10.1097/00005072-196104000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73–S78. doi: 10.1046/j.1440-1789.2000.00301.x. [DOI] [PubMed] [Google Scholar]
- 7.Riess O, Jakes R, Kruger R. Genetic dissection of familial Parkinson’s disease. Mol Med Today. 1998;4:438–444. doi: 10.1016/s1357-4310(98)01343-4. [DOI] [PubMed] [Google Scholar]
- 8.Chung KK, Dawson VL, Dawson TM. New insights into Parkinson’s disease. J Neurol. 2003;250(suppl 3):III15–24. doi: 10.1007/s00415-003-1304-9. [DOI] [PubMed] [Google Scholar]
- 9.Lavedan C, Buchholtz S, Nussbaum RL, Albin RL, Polymeropoulos MH. A mutation in the human neurofilament M gene in Parkinson’s disease that suggests a role for the cytoskeleton in neuronal degeneration. Neurosci Lett. 2002;322:57–61. doi: 10.1016/s0304-3940(01)02513-7. [DOI] [PubMed] [Google Scholar]
- 10.Marx FP, Holzmann C, Strauss KM, et al. Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Hum Mol Genet. 2003;12:1223–1231. doi: 10.1093/hmg/ddg134. [DOI] [PubMed] [Google Scholar]
- 11.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 12.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavedan C. The synuclein family. Genome Res. 1998;8:871–880. doi: 10.1101/gr.8.9.871. [DOI] [PubMed] [Google Scholar]
- 14.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186 –190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 15.Kahle PJ, Neumann M, Ozmen L, et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mori F, Nishie M, Yoshimoto M, Takahashi H, Wakabayashi K. Reciprocal accumulation of beta-synuclein in alpha-synuclein lesions in multiple system atrophy. Neuroreport. 2003;14:1783–1786. doi: 10.1097/00001756-200310060-00005. [DOI] [PubMed] [Google Scholar]
- 17.Rockenstein E, Hansen LA, Mallory M, Trojanowski JQ, Galasko D, Masliah E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res. 2001;914:48–56. doi: 10.1016/s0006-8993(01)02772-x. [DOI] [PubMed] [Google Scholar]
- 18.Uversky VN, Li J, Souillac P, et al. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. Beta-synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 20.Giasson BI, Jakes R, Goedert M, et al. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson’s disease. J Neurosci Res. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 21.Lincoln S, Crook R, Chartier-Harlin MC, et al. No pathogenic mutations in the beta-synuclein gene in Parkinson’s disease. Neurosci Lett. 1999;269:107–109. doi: 10.1016/s0304-3940(99)00420-6. [DOI] [PubMed] [Google Scholar]
- 22.Lavedan C, Buchholtz S, Auburger G, et al. Absence of mutation in the beta- and gamma-synuclein genes in familial autosomal dominant Parkinson’s disease. DNA Res. 1998;5:401–402. doi: 10.1093/dnares/5.6.401. [DOI] [PubMed] [Google Scholar]
- 23.Bonner LT, Tsuang DW, Cherrier MM, et al. Familial dementia with Lewy bodies with an atypical clinical presentation. J Geriatr Psychiatry Neurol. 2003;16:59–64. doi: 10.1177/0891988702250585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 25.Chung KKK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 2001;24(11 suppl):S7–S14. doi: 10.1016/s0166-2236(00)01998-6. [DOI] [PubMed] [Google Scholar]
- 26.Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med. 2001;3:139–148. doi: 10.1097/00125817-200103000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratton MR. Recent advances in understanding of genetic susceptibility to breast cancer. Hum Mol Genet. 1996;5:1515–1519. doi: 10.1093/hmg/5.supplement_1.1515. [DOI] [PubMed] [Google Scholar]
- 28.Zarranz J, Alegre J, Gomez-Esteban J, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 29.Farrer M, Kachergus J, Forno L, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 30.Singleton AB, Farrer M, Johnson J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.