

Abstract
The three-dimensional structures of the viral capsid of three AAV serotypes have previously been determined by X-ray crystallography or cryo-electron microscopy (cryo-EM). These studies of AAV and similar studies of autonomous parvoviruses have yielded important structural information of the virions in a low-energy conformation. However, there is little information on the structural properties of AAV virions in solution under physiological conditions. We demonstrate that proteolytic digestion of AAV2 virions with trypsin results in cleavage at a specific site on the capsid surface while the capsid remains intact. The products of digestion were mapped using unique antibodies, protein sequencing, mass spectroscopy, and 3D structure modeling to a region on a surface loop that is common to all three AAV2 structural proteins. Empty AAV2 capsids could be distinguished from full (DNA-containing) capsids, having an increased susceptibility of VP2 to trypsin, and more rapid digestion by chymotrypsin. Proteolytic analysis utilizing trypsin or chymotrypsin was also capable of distinguishing AAV2 from AAV1 and AAV5, as seen by differential susceptibility and unique fragment patterns. These data demonstrate a novel approach for studying the structure of AAV capsids in solution, and should be valuable in the testing and engineering of AAV vectors for gene transfer.
Keywords: adeno-associated virus, structure, capsid, proteolysis, serotype
INTRODUCTION
Adeno-associated viral (rAAV) vectors can mediate safe gene transfer for long-term correction of genetic diseases in animal models, and have been shown to persist and are safe in humans following delivery to lung, sinus, skeletal muscle, brain, and liver [1–5]. Transient correction of hemophilia B following hepatic artery administration of a rAAV2-hFIX vector has been reported [5] and for cystic fibrosis (CF), a phase II aerosol trial showed evidence of a decrease in pulmonary inflammation and an increase in pulmonary function [6]. AAV serotype 2 (AAV2) is the prototypical AAV that has been studied most comprehensively and more than 100 serotypes of AAV have been identified [7–10]. The primate and human AAVs fall into nine antigenically different genetic clades where a >20% sequence difference exists among the viruses grouped into the clades, suggesting that they were distantly separated during evolution. Representative members of each clade have distinct host range and tissue tropism properties [7], with comparative gene transfer studies in vitro and in animal models showing dramatic differences in the transduction efficiency and cell specificity dictated by the AAV capsid [10–13].
The non-enveloped T=1 icosahedral AAV2 capsid consists of 60 subunits comprised of three viral structural proteins, VP1 (87kD), VP2 (72kD) and VP3 (63kD), in an approximate ratio of 1:1:10. The less abundant capsid proteins, VP1 and VP2, share the same C-terminal amino acid sequence with VP3 but have additional N-terminal sequences. The unique N-terminal region of VP1 has been shown to possess phospholipase A2 activity and is required for infectivity [14]. The role of VP2 has been suggested to be either in nuclear transport of VP3 or in particle assembly [15, 16]. However, Warrington et al. [17], demonstrated that VP2 is non-essential for virus assembly and infectivity, and that VP3 is sufficient to assemble a DNAse resistant particle that is non-infectious. Capsid assembly has been shown to occur in the nucleus [18] and pulse-chase experiments show that preformed empty capsids are the precursors for DNA packaging where the genome is inserted by a replication-dependent mechanism [19] that results in genome-full capsids (i.e. virions).
The three-dimensional (3D) structures of several autonomous parvoviruses [20–22], plus those of AAV2 [23, 24], AAV4 [25], and AAV5 [26] have been determined by X-ray crystallography or cryo-electron microscopy (cryo-EM). Recent studies of the AAV2 capsid by cryo-EM have suggested the location of the unique N-terminus of VP1 and the overlapping N-terminus of VP2 [27]. All of the parvoviral capsid proteins have a core eight-stranded (designatedβB-βI) antiparallel β-barrel structure that forms the contiguous shell, with loop insertions between the strands forming the capsid surface [28]. The major surface features of empty or full (DNA-containing) parvovirus capsids include depressions at the icosahedral 2-fold and surrounding the 5-fold symmetry axes, and protrusions at or surrounding the 3-fold axis. Structural and mutational analyses clearly show that parvoviral host tropism and antigenic differences arise from variations in surface amino acids [29] with the majority of surface variable loop regions are near the 2- and 3-fold axes [25].
Basic amino acid residues located on the floor and wall of the valley between the protrusions at the 3-fold axis of the capsid, including R585 and R588 [30, 31] have been shown to be required for AAV2 to bind heparan sulfate proteoglycan (HSPG), one of its cellular receptors. These arginine residues are unique to AAV2 and are not found in serotypes 1 or 3–11. Serotypes other than AAV2 interact with different cell surface molecules [32, 33]. AAV1, which is ~83% identical to AAV2, does not bind heparin sulfate [34], but utilizes sialic acid for infection [35]. Like AAV1, AAV4 and AAV5 also utilize sialic acid for infection[33]. Recently, Grieger, et al., [36] identified four basic regions of the AAV2 capsid that are conserved in AAV serotypes 1–11 and play a role in infectivity and virion assembly.
The 3D structures available for autonomous parvoviruses and AAV serotypes provide a “snapshot” of the capsid topology in a low energy conformation. However, given the intense study of AAV serotypes and their development as viral vectors for gene transfer, our knowledge about their viral capsid structure in solution is limited. Here we begin to characterize the AAVs in solution using proteolysis, specific antibodies, and mass spectrometry to map differences and similarities in their capsid structure. We observe differences in protease susceptibility between full and empty capsids, and between AAV1, AAV2, and AAV5 - serotypes that are actively under development for gene therapy applications.
RESULTS AND DISCUSSION
Trypsin digestion of AAV2 virions generates a unique pattern
Historically, AAV has been considered to be resistant to proteolysis by trypsin [37]. However, rAAV2-GFP vectors purified using the classic Trypsin/DOC/CsCl technique (see Materials and Methods for details) showed additional bands to VP1, VP2, and VP3 when probed with anti-AAV2 polyclonal sera on Western blots (Fig. 1A). These proteolytic fragments co-purified with the intact virus, a result comparable to that reported for wild-type (wt) AAV2 virions purified using a similar protocol for crystallographic studies [38]. To determine if the inclusion of trypsin in the purification protocol was responsible for the additional bands, AAV2-GFP vector virions were purified by the F-T/Iodixanol/Heparin method in the absence of trypsin [39] and subjected to proteolytic digestion with trypsin as described in Materials and Methods. After digesting purifiedfull rAAV2-GFP capsids with trypsin for 24 hours, we observed a limited number of fragments that were similar to those of capsids purified by the classic Trypsin/DOC/CsCl technique (Fig. 1B). Trypsin is predicted to cleave denatured AAV2 VP1 69 times based on primary sequence analysis (data not shown), with VP2 and VP3 sharing many of the same potential tryptic sites due to the overlap of these protein sequences.
Fig. 1. Trypsinized AAV2 capsids generate novel fragments. A. Trypsin used during purification.
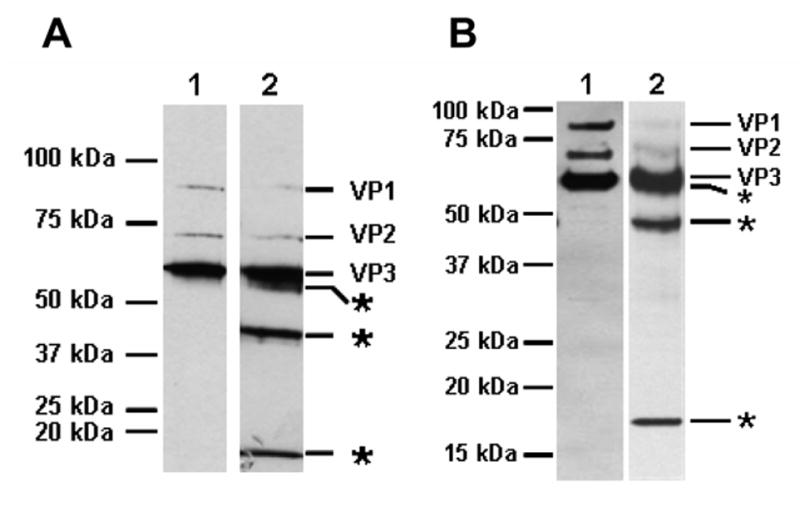
Lane 1, AAV2-GFP cell lysates made in the presence of deoxycholate (DOC) and centrifuged on CsCl density gradients (DOC/CsCl). Lane 2, AAV2-GFP cell lysates made in the presence of 0.02% trypsin and DOC (30 minutes at 37°C), and purified on CsCl density gradients (Trypsin/DOC/CsCl). The peak fraction of full capsids was denatured and separated on a 10% SDS-PAGE gel, blotted, and probed with anti-AAV polyclonal antisera. B. Trypsinization of purified AAV2. Purified rAAV2-GFP virions (F-T/iodixanol/Heparin, Lane 1) were treated with 0.02% trypsin for 24 hours (Lane 2), and the capsid proteins were separated on a 12.5% SDS-PAGE gel, blotted and probed with anti-AAV2 polyclonal antisera. The positions of molecular weight standards are indicated on the left side and the tryptic fragments are indicated on the right side with asterisks.
The tryptic fragments were mapped using a variety of antisera to AAV2 (polyclonal, A1, B1, A69, [18, 40]) (Fig. 2). The removal of the ~15–20 kDa C-terminal VP1,2,3T fragment from VP1, VP2, and VP3 generates the VP1T, VP2T, and VP3T truncated capsid proteins, respectively, as detected by anti-AAV2 polyclonal antisera (Fig. 2, lane 2). VP1T, VP2T, and VP3T no longer harbor the B1 epitope which is found in the VP1,2,3,T fragment (Fig. 2, lane 4), however, the N-terminal specific antibody A1 recognizes VP1T (Fig. 2, lane 8), and A69 recognizes VP1T and VP2T (Fig. 2, lane 6) confirming cleavage at the C-terminus. VP1T migrates slightly slower than VP2 (Fig. 2, lanes 2, 6, and 8). Residual undigested VP3 protein remains following treatment (Fig. 2, lane 2 and 4) as does residual VP1 (Fig. 2, lanes 2, 4, 6, and 8), but VP2 is almost completely converted to VP2T (Fig. 2, lanes 4 and 6). In the undigested starting material (Fig. 2, lane 1) six fragments are detected that migrate between 25 and 50kDa; these breakdown products are likely the result of cellular proteolytic activity present in the freeze-thaw lysate made during this purification of AAV, and are seen to varying degrees when preparing other parvovirus capsids (data not shown). Full rAAV2-GFP capsids purified by a variety of methods (including Trypsin/DOC/CsCl/Heparin, DOC/CsCl/Heparin, Trypsin/DOC/CsCl, F-T/Iodixanol/Heparin, F-T/CsCl/Heparin, and DOC/CsCl) produce the same banding pattern following trypsin digestion (see below). The VP1,2,3T C-terminal fragment migrates to a position corresponding to a molecular weight between 15 and 20kDa when resolved by SDS-PAGE (Fig. 1B). The protein fragments identified with the different antisera combined with their molecular weight (Fig. 2) narrows the cleavage site to the common C-terminal end of VP1, VP2, and VP3 near the heparin binding domain.
Fig. 2. Tryptic mapping of full AAV2 capsids.
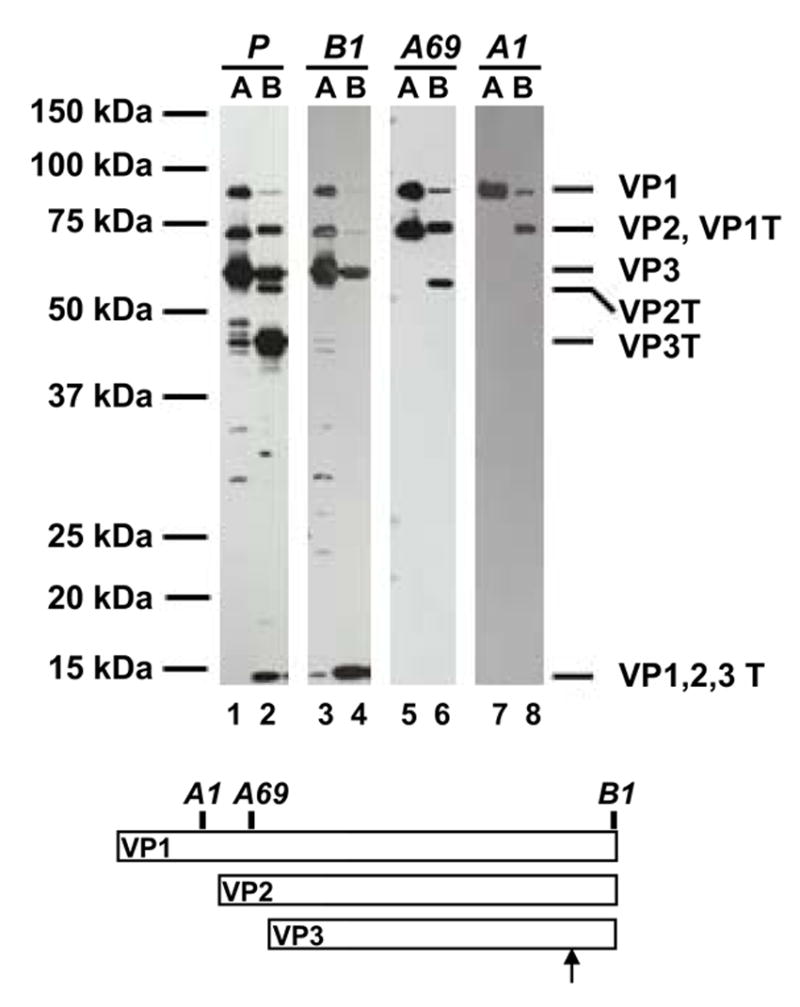
Purified full AAV2 virions (F-T/iodixanol/Heparin) were treated with trypsin, and the capsid proteins were probed with the indicated anti-AAV antibodies (AAV2 polyclonal, A1, B1, A69). The epitope locations of the monoclonal antibodies and the likely trypsin digestion site are indicated on the schematic diagram. A, AAV2 full capsids; B, AAV2 full capsid digested with trypsin for 24hours. The assignment of the tryptic fragments are given on the right. VP1T is the digestion product of VP1, VP2T is the digestion product of VP2, VP3T is the digestion product of VP3, and VP1,2,3T is the common C-terminal trypsin digestion product of VP1, VP2, and VP3. Trypsin did not cross react with these antibodies (data not shown).
Fine mapping of the trypsin cleavage site
Theoretical prediction of tryptic sites in the capsid G-H loop [25], comprised of amino acid (aa) residues 416 to 645, and fragment masses for AAV2 VP1 (Table 1) indicated that trypsin digestion at amino acids R566, R585 or R588 would produces fragments of 18.8, 16.8, and 16.4kDa, respectively, which are within the range expected for the VP1,2,3T C-terminal fragment that is recognized by the B1 antibody (Fig. 1B and Fig. 2, lane 4). The mass of this fragment was observed to be 16,461Da from MALDI-TOF analysis and is close to the theoretical mass of the C-terminal peptide that results from cleavage at R588 (Table 1). The R588 tryptic site was confirmed by N-terminal sequencing which mapped residues 589QAATADVNTQGV600 as the terminal peptide for the VP1,2,3T fragment.
Table 1.
Predicted AAV VP1 tryptic fragment mass from cleavage in the G-H loop.
| Fragment | Average Mass (Da) | Peptide |
|---|---|---|
| VP1 | 81944.65 | All Cys in reduced form |
| 1 – 549 | 61112.40 | VP1 start to 549 |
| 137 – 549 | 45757.24 | VP2 start to 549 |
| 203 – 549 | 39231.12 | VP3 start to 549 |
| 550 – 735 | 20850.26 | C terminal end |
| 1 – 556 | 61912.28 | VP1 start to 556 |
| 137 – 556 | 46557.12 | VP2 start to 556 |
| 203 – 556 | 40031.00 | VP3 start to 556 |
| 557 – 735 | 20050.38 | C terminal end |
| 1 – 566 | 63128.65 | VP1 start to 566 |
| 137 – 566 | 47773.49 | VP2 start to 566 |
| 203 – 566 | 41247.37 | VP3 start to 566 |
| 567 – 735 | 18834.01 | C terminal end |
| 1 – 585 | 65176.85 | VP1 start to 585 |
| 137 – 585 | 49821.69 | VP2 start to 585 |
| 203 – 585 | 43295.57 | VP3 start to 585 |
| 586 – 735 | 16785.81 | C terminal end |
| 1 – 588 | 65504.19 | VP1 start to 588 (VP1T) |
| 137 – 588 | 50149.03 | VP2 start to 588 (VP2T) |
| 203 – 588 | 43622.91 | VP3 start to 588 (VP3T) |
| 589 – 735 | 16458.47 | C terminal end (VP1,2,3T) |
| 1 – 609 | 67743.68 | VP1 start to 609 |
| 137 – 609 | 52388.52 | VP2 start to 609 |
| 203 – 609 | 45862.40 | VP3 start to 609 |
| 610 – 735 | 14218.98 | C terminal end |
Trypsin cleavage sites of AAV2 VP1 were determined using the program Peptide Cutter found on the ExPASy server (www.expasy.org). Based on the sites where trypsin cleaves, these sequences were entered into the program Peptide Mass on the ExPASy server and the mass of each trypsin fragment was determined.
Trypsin-treated virions remain intact
The presence of tryptic fragments found with Trypsin/DOC/CsCl purified rAAV2-GFP virions (Fig. 1A, lane 2) indicated that the cleaved products remained tightly associated with the capsids. The buoyant densities of these and DOC/CsCl purified capsids (Fig. 1A, lane 1) are similar at ~1.40g/cm3, indicating similar protein and DNA compositions. The value is slightly less than the density of wtAAV2 (1.41–1.45g/cm3) because the rAAV2-GFP vector is smaller (4331 nucleotides) than the wtAAV genome. Negative stain EM analysis of the trypsin-treated and untreated purified rAAV2-GFP samples confirmed the intact nature of the treated capsids (Fig. 3A). However, the negative staining pattern suggested a difference between the samples in their permeability to uranyl acetate (UA), indicating a possible structural rearrangement or flexibility due to the cleavage event.
Fig. 3. Trypsinized AAV2 virions remain intact. A. Electron microscopy.
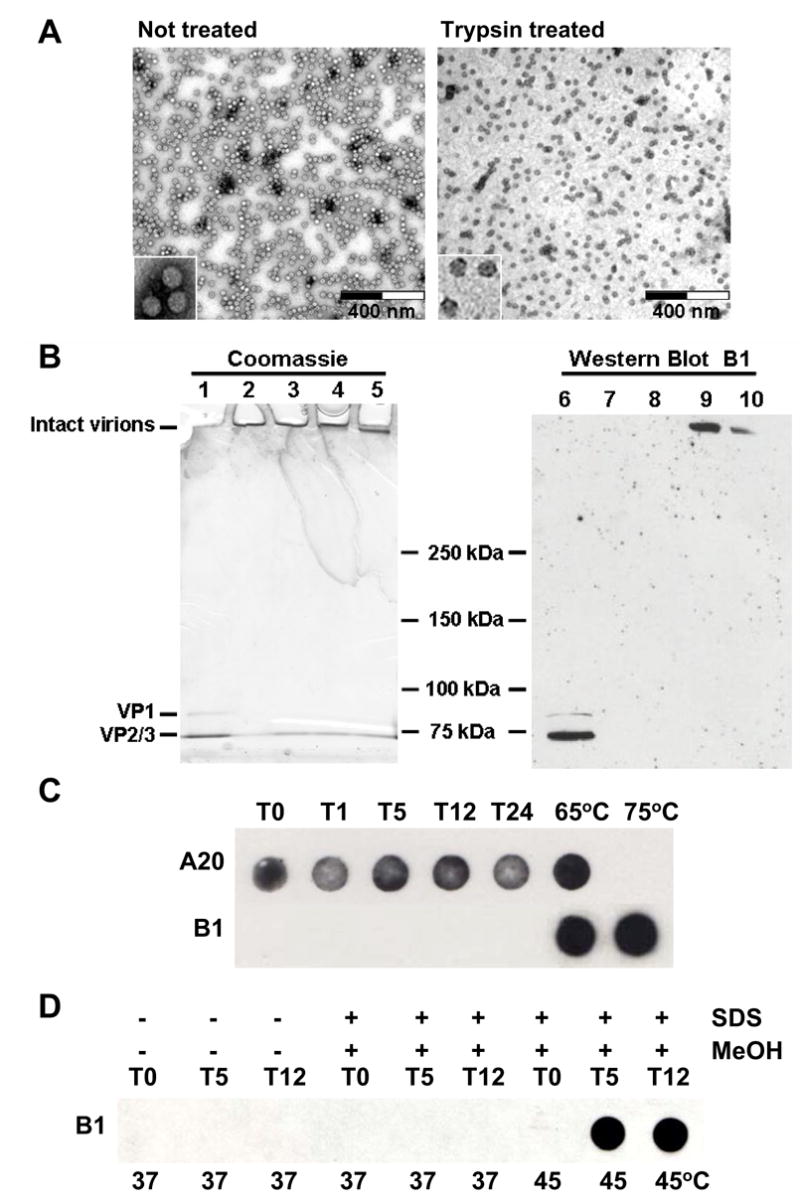
Negative stain electron micrographs of untreated rAAV2-GFP capsids (F-T/iodixanol/Heparin) or trypsin-treated rAAV2-GFP virions. The samples were viewed on a Hitachi H-7000 transmission electron microscope at 30,000X magnification. Each high-powered insert is 70,000X. B.Semi-native polyacrylamide gel analysis. Semi-native gel of untreated (DOC/CsCl/Heparin) and trypsin-treated rAAV2-GFP virions stained with Coomassie Blue (left panel) or Western blotted and probed with B1 antibody (right panel). Lanes 1 and 6, rAAV2-GFP (not treated with trypsin) was boiled in SDS and DTT (denatured); Lanes 2 and 7, rAAV2-GFP incubated at 37°C for 24 hours without trypsin (non-denatured); Lanes 3–5 and 8–10, rAAV2-GFP treated with trypsin at 37°C for 1, 5 and 24 hours, respectively (non-denatured). C. Immuno dot-blot. rAAV2-GFP virions were digested with trypsin and samples were taken at the indicated time points (hours). An immuno dot-blot was performed on undigested sample (T0), as well as sample digested for 1, 5, 12, or 24 hours with trypsin (T1, T5, T12, T24, respectively). Duplicate samples were probed with either A20 or B1 antibodies. Also included are native virions heated at 65°C or 75°C for 30 minutes, treatments that are known to expose the B1 epitope [27]. D. Immuno dot-blot with SDS and MeOH. To mimic the Western transfer conditions of Fig. 3C, AAV2 virions (T0) were digested with trypsin for 5 and 12 hours (T5 and T12, respectively), and treated with 0.1% SDS and 20% MeOH for 2 hours at 37°C or 45°C, transferred to nitrocellulose, and probed with the B1 antibody.
To further confirm that the virions were still intact following trypsinization, untreated and trypsin-treated rAAV2-GFP virions were analyzed by semi-native gel electrophoresis (see Materials and Methods) followed by Coomassie staining or Western blotting. Untreated rAAV2-GFP virions or those treated for 1 hour are unable to migrate in polyacrylamide gels under semi-native conditions (Fig. 3B, lanes 2, 3, 7, and 8), however, samples treated with trypsin for 5 or 24 hours were able to migrate slightly into the gel (Fig. 3B, lanes 4, 5, 9, and 10). Capsid protein bands from virions denatured by heating in the presence of SDS and β-mercaptoethanol were observed migrating near the dye front in the gel (Fig. 3B, lanes 1 and 6). When analyzed by Western blotting, the rAAV2-GFP virions treated for 5 and 24 hours with trypsin were recognized by B1 (Fig. 3B, lanes 9 and 10) confirming that the cleaved 147aa VP1,2,3T C-terminal peptide remains associated with the capsid and that the penetration of the virions into the gel was not due to a significant loss in molecular weight (~5 x 106Da), but may be due to increased flexibility.
Finally, native immuno dot-blot analysis with the A20 anti-capsid antibody that recognizes intact capsids [40], showed that the virions remain intact following 24 hours of trypsin digestion (Fig. 3C). However, unlike the Western analysis where the B1 epitope was detected following 5 hours of trypsin digestion (Fig 3B), the B1 epitope was not detected in the immuno dot-blot analysis of trypsinized virions. B1 does not recognize intact capsids [18] because it binds a linear epitope (residues 726–733) that is only partially exposed on the capsid surface near the 2-fold axis of symmetry ([24] and Figure 7D). As a control, the B1 epitope was detected in native undigested virions heated to 65°C as previously reported by Kronenberg, et al. [27] (Fig 3C). To explore this difference, the immuno dot-blot was repeated, but following trypsin digestion, the virions were treated with SDS, MeOH and mild heating to mimic the transfer of the proteins to nitrocellulose for Western blotting (Fig. 3D). The B1 epitope is not recognized until the virions are trypsinized and treated with SDS and MeOH at 45°C. Undigested control virions treated with SDS and MeOH at 45°C, and trypsinized virions treated with SDS and MeOH at 37°C were not detectable by the B1 antibody.
Fig. 7. Capsid structures. A. Capsid monomer.
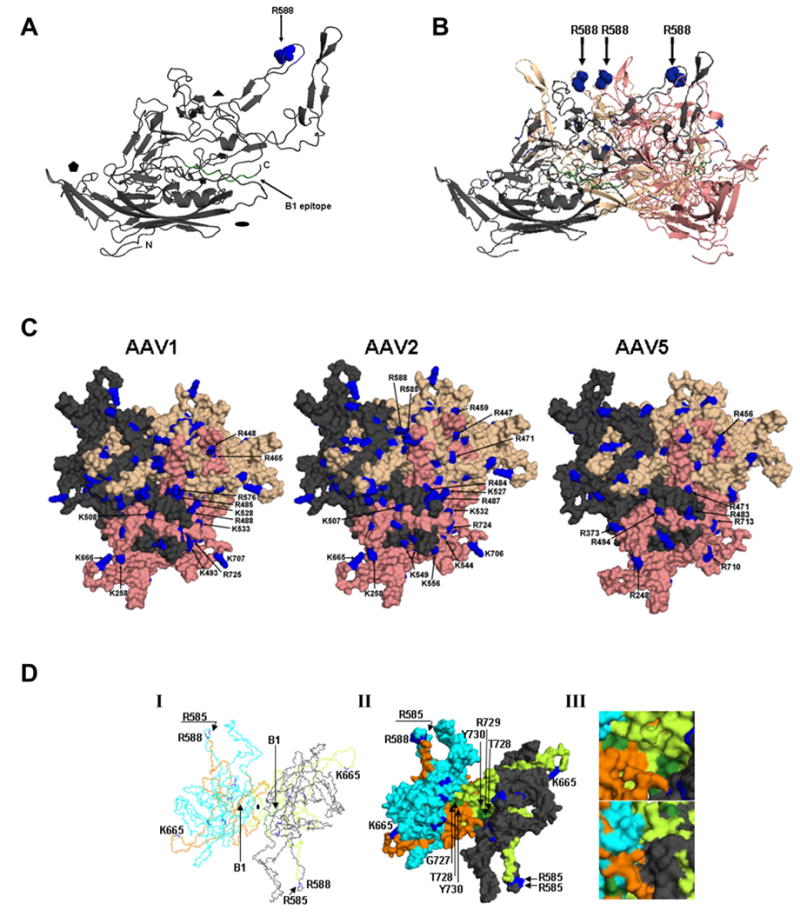
Ribbon representation of an AAV2 VP3 monomer (amino acids 217–735) rotated 90° from standard icosahedral orientation. β-strands and α-helices are represented as arrows and coils, respectively. Small arrows indicate the location of R588 (blue spheres) and the B1 epitope at the extreme C-terminus (green). The 2-fold (oval), 3-fold (triangle) and 5-fold (pentamer) axes are indicated. B. Side view of an AAV2 VP3 trimer. Amino acids R585 and R588 in the three G-H loops from three VP3 monomers (gray, salmon, wheat) are represented as blue spheres and indicated by arrows. C. Capsid surface basic amino acids. AAV1, AAV2, and AAV5 capsid surface structures at the 3-fold axis of symmetry with the indicated basic amino acids of the salmon reference monomer highlighted in blue. D. AAV2 VP3 dimer viewed down the 2-fold axis. (I) Polyglycine trace, (II) Surface, and (III) Close-up view of the surface exterior (top) and interior (bottom) of an AAV2 VP3 dimer (cyan and gray) showing the locations of R588 and the B1 epitope (color scheme as in A, B, and C). The residues in the G-H loop are colored limon and orange in the reference monomer and 2-fold related monomer, respectively. The coordinate files used in A-D were based on the X-ray crystallographic structure of AAV2 [24] (PDB accession No. 1LP3) and homologous models were generated for AAV1 and AAV5 using a structure-based alignment with AAV2. Coordinate files for the homologous models were generated using VIPER [50]. Figures were generated using PyMOL.
Taken together, the data indicate that the digested AAV2 capsids appear intact by EM, semi-native gel electrophoresis where significant loss in molecular weight was not observed, buoyant density on cesium chloride gradients, and recognition by the anti-capsid A20 antibody. The internal disposition of the majority of the C-terminal proteolytic fragment (~70% of residues 589–735, Fig. 7) ensures that the fragment remains associated with the capsid in 3.3M CsCl during purification (Fig. 1), in the 0.1% SDS used in semi-native gel electrophoresis (Fig. 3B), in 0.1% SDS and 20% MeOH used in the immuno dot-blot (Fig. 3D), and during heparin chromatography (Fig 4, lane 1 below). However, heating the trypsinized virions in the presence of 0.6M DTT for mass spectroscopy and N-terminal sequencing, or heating in the presence of SDS and reducing agent for denaturing gel electrophoresis was able to dissociate the VP1,2,3T fragment from the core capsid. Since no cysteine residues are found in the VP1,2,3T C-terminal fragment, the interaction of this fragment with the capsid is due to non-covalent interactions.
Fig. 4. Mildly trypsinized but not fully trypsinized virions bind heparin.
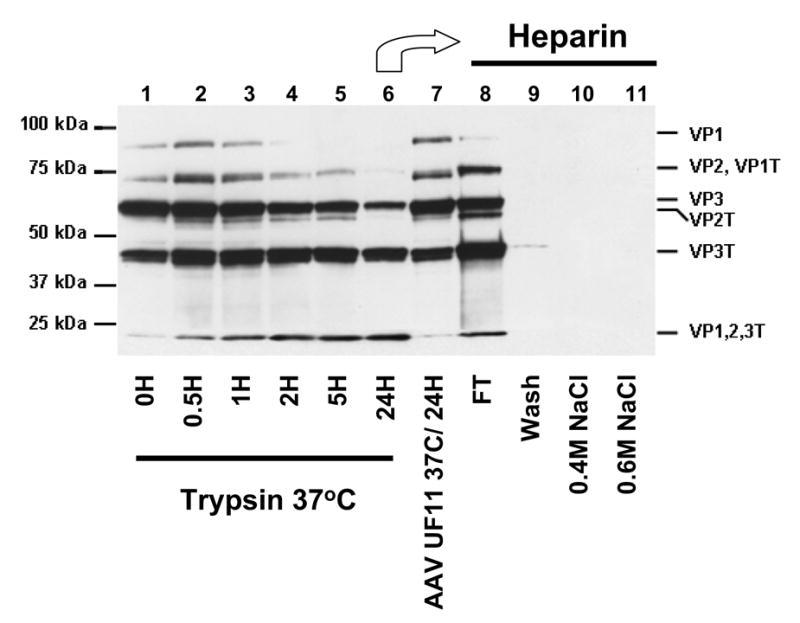
AAV2-GFP purified by trypsin/DOC/CsCl/heparin (lane 1) was further treated with 0.02% trypsin for 30 minutes (lane 2), 1 hour (lane 3), 2 hours (lane 4), 5 hours (lane 5), or 24 hours (lane 6). As a control, the purified virus from lane 1 was incubated for 24 hours at 37°C without trypsin (lane 7). Virus treated for 24 hours with trypsin (lane 6) was loaded onto a heparin affinity column: Lane 8, flow-through; Lane 9, wash; Lane 10, elution with 0.4M NaCl; Lane 11, elution with 0.6M NaCl. Capsid proteins were probed with anti-AAV2 polyclonal antisera. The position of the tryptic fragments are given on the right as indicated in Figure 2.
The crystal structure of AAV2 [24] was determined for capsids that contained VP cleavage fragments that co-purified and migrated similarly to the bands designated here as VP2T, VP3T and VP1,2,3T (Figs. 1 and 2), although they were not mapped [38]. However, the crystal structure of AAV2 did not show a break in the polypeptide chain at position R588. This suggests that the majority of the 60 R588 sites remain uncleaved by the mild trypsin treatment used during purifications reported here, and for samples used for crystallization of AAV2 [38], which generated the additional bands observed in Figures 1A and 4. The cleaved capsids thus maintain the properties and structure of the native virus in the crystalline state but appear to have increased flexibility as shown here by differential staining in EM, penetration of semi-native gels, and exposure of the B1 epitope in the presence of SDS and MeOH at 45°C (Fig. 3A, 3B and 3D). For future studies, it may be prudent to avoid incorporating proteases into AAV purification schemes for structural studies or gene therapy applications where proteolysis may not only alter the capsid structure, but also chromatographic properties and vector potency. In retrospect, the use of trypsin during purification of rAAV2 vectors and the heat inactivation of the helper adenovirus at 56°C (which can expose the N-terminus of AAV VP1 [27]) may have been responsible for the low vector titers and low transduction efficiencies obtained during the initial evaluations of this vector system.
Receptor binding and infectivity are reduced following treatment with trypsin
To further confirm the tryptic cleavage of the heparin binding site at R588, rAAV2-GFP virions purified in the presence of trypsin using heparin column chromatography (Trypsin/DOC/CsCl/Heparin) (Fig. 4, lane 1) were treated with trypsin for up to 24 hours (Fig. 4, Lanes 2 to 6). The virions treated for 24 hours (Fig. 4, lane 6) were found in the flow through when reloaded onto a heparin sulfate affinity column (Fig. 4, lane 8). This lack of heparin binding occurs despite the fact that the cleaved protein fragments remained associated with the capsid (Fig. 3B and 3D). Elution of the column with 0.4M and then 0.6M NaCl (undigested AAV2 elutes from heparin-sepharose at ~300mM NaCl) did not elute any detectable virus (Fig. 4, lanes 10 and 11). The loss of capsid interactions with heparin was further confirmed by infectivity assays which showed a 3–4 log reduction of infectivity over the course of the trypsin digestion (Fig. 6D). This observation was similar to the reduction in infectivity reported for a R588A mutation [31]. Thus, while the mild trypsinization of cell extracts during purification does not appear to be detrimental to the heparin interaction (Fig. 4, lane 1), prolonged digestion of the purified virus capsid must cleave most, if not all, of the 60 R588 sites found on the capsid surface, preventing heparin binding and infection, and increasing flexibility. Loss of infectivity could be due to direct disruption of the heparin binding sites and/or global structural alterations to the virion. Since residual AAV2 infectivity remained following trypsin treatment, the cleaved virus may still be able to inefficiently bind cellular HSPG or interact directly with the cellular co-receptors.
Fig. 6. AAV2, AAV1 and AAV5 capsids can be distinguished proteolytically. A. Trypsin time course.
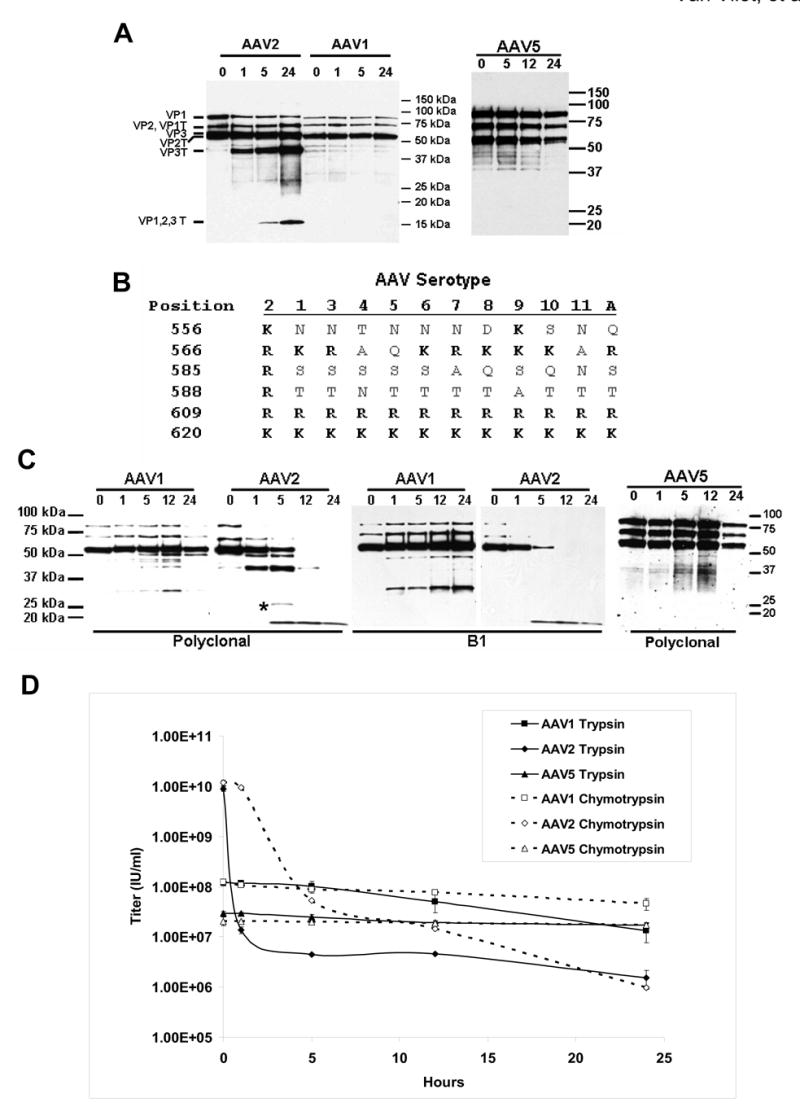
Purified AAV1 (F-T/iodixanol/Q), AAV2 (F-T/iodixanol/heparin), and AAV5 (F-T/iodixanol/Q) virions were incubated at 37°C without trypsin (0), or treated with 0.02% trypsin for the time (hours) indicated above each lane. The capsid proteins of AAV1 and AAV2 were probed with anti-AAV2 polyclonal and AAV5 was probed with anti-AAV5 polyclonal sera. B.Alignment. Amino acid alignment of the G-H loop [25] region 556–620 of AAV serotypes 1 to 11 and Avian AAV. Arginine or Lysine (R or K) residues, the target residues for trypsin, are shown in bold. Amino acid position numbering is based on VP1 of AAV2. C. Chymotrypsin time course. Purified AAV1, AAV2, and AAV5 were incubated at 37°C without chymotrypsin (0) or digested with chymotrypsin at 37°C for the time (hours) indicated above each lane. The capsid proteins of AAV1 and AAV2 were probed with anti-AAV2 polyclonal or B1 antisera, and AAV5 was probed with anti-AAV5 polyclonal sera. The asterisk indicates an additional AAV2 band observed with chymotrypsin that was not seen following trypsin digestion in Fig. 6A. D. Infectivity. Infectious titering of rAAV1-GFP, rAAV2-GFP, and rAAV5-GFP virions was performed following digestion with trypsin or chymotrypsin by infecting C12 cells in the presence of Adenovirus (reported as the average ± SDEV from three separate infections). AAV1 and AAV5 infect C12 cells less efficiently than AAV2 [49].
Proteolytic digestion distinguishes genome-full and empty capsids
To determine if genome-full capsids (i.e. virions) and empty capsids can be distinguished by proteolysis, purified full rAAV2-GFP capsids and empty AAV2 capsids were digested with trypsin. Similar to the differential proteolytic sensitivity reported for members of the autonomous parvoviruses [41], the full and empty AAV2 capsids differed in their sensitivity to trypsin cleavage (Fig. 5). The unique proteolytic fragment (VP2T) derived from VP2 of the full capsids was seen migrating slightly ahead of VP3 when probed with polyclonal sera (Fig. 5A, lane 2) or the A69 antibody (Fig. 5A, lane 10), but was not observed in the digested empty capsids (Fig. 5A, lanes 3 and 11). In addition to the absence of the VP2T fragment in empty capsids, full length VP2 was not detected, indicating complete digestion of this protein (Fig. 5A, lanes 3 and 11). A trypsin digestion timecourse showed that VP2 and the VP2T fragment are more sensitive to digestion in empty capsids compared to full capsids (Fig. 5B). During the timecourse, less VP2T is seen in empty capsids than full capsids, and by 12 hours VP2T is not detected in empty capsids, whereas the VP2T fragment is still detected in full capsids even after 24 hours of digestion. Chymotrypsin was also able to differentiate between the full rAAV2-GFP capsids and the AAV2 empty capsids (Fig. 5C). The empty capsids were digested more rapidly than full capsids, as observed with trypsin treatment, but with chymotrypsin all of the VPs were digested within 2 hours.
Fig. 5. Proteolysis distinguishes full and empty AAV2 particles. A. Trypsin.
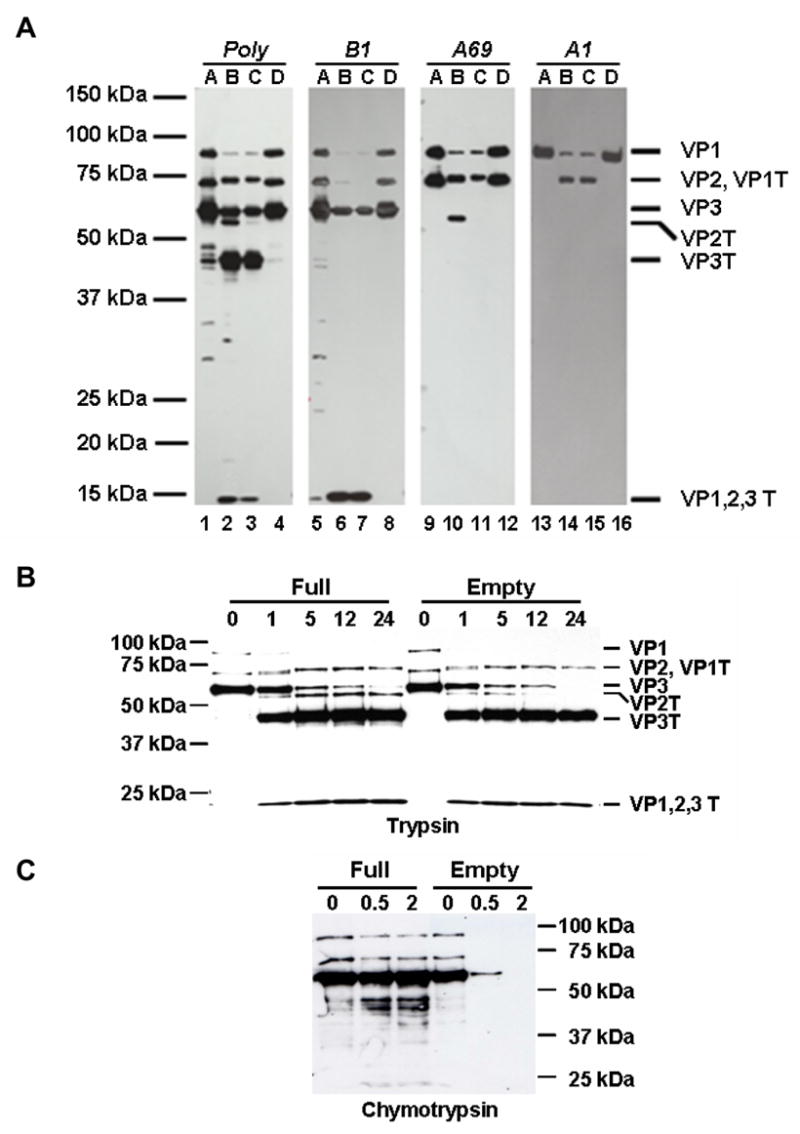
Purified full (F-T/iodixanol/heparin) and empty AAV2 capsids were treated with 0.02% trypsin, and the capsid proteins were probed with the indicated antibodies (AAV2 polyclonal, A1, B1, A69). Lanes A, AAV2 full capsids; lanes B, AAV2 full capsids digested with trypsin for 24 hours; lanes C, AAV2 empty capsids digested with trypsin for 24 hours; lanes D, AAV2 empty capsids. B. Trypsin time course. Purified full (F-T/iodixanol/heparin) and empty AAV2 virions were treated with trypsin for the time (hours) indicated above each lane and probed with anti-AAV2 polyclonal antisera. C. Chymotrypsin time course. Purified full (F-T/iodixanol/heparin) and empty AAV2 capsids were treated with chymotrypsin for the time (hours) indicated above each lane and the capsid proteins were probed with anti-AAV2 polyclonal antisera.
Comparison of the proteolytic susceptibility of full and empty AAV2 capsids indicates that there are recognizable structural differences between these capsids in solution that involve VP2. This may be due to stabilization of the capsid by packaged DNA, in addition to different conformations of the capsid surface loops. In addition, the AAV2 VP1 and VP2 N-termini become accessible in full capsids at 65°C, as A69 and A1 antibody reactivity is seen in the 110S (full) virus species, but not in empty capsids [27, 18]. Furthermore Bleker et al. [42] were able to cleave the N-termini of AAV2 VP1 with trypsin following heating of full capsids resulting in a fragment that maintained the A69 and B1 antibody epitopes. Since we do not detect the VP2 or VP2T bands following digestion of empty capsids, trypsin digestion for 24 hours may degrade the VP2 protein to the point where it is not recognized by either the polyclonal, B1, or A69 antibodies. Alternatively, the cleavage of both the VP2 C- and N-termini of empty capsids may result in a fragment that is indistinguishable from VP3T. Since differences in the parvovirus capsid surface, consisting of the overlapping capsid protein region, are indistinguishable between full and empty capsid structures determined using X-ray crystallographic or cryo-EM techniques [28, 29], the use of proteolysis and specific antigen mapping of cleavage products provides a means to distinguish these species in solution. In addition, proteolytic distinction between full and empty capsids can be used in conjunction with quantitative methods to determine full/empty capsid ratios [43].
Proteolytic digestion distinguishes AAV serotypes
To further confirm that R588 is the specific site on the AAV2 capsid surface susceptible to tryptic cleavage, we compared the suceptibility of rAAV1-GFP, rAAV2-GFP, and rAAV5-GFP virions to proteolytic cleavage (Fig. 6A). AAV1 provided an example of a highly homologous serotype to AAV2 (83%) that does not contain R588 and AAV5 provides an example of a less homogolous (59%) serotype to AAV2 (Fig. 6B). As seen in Figure 6A, AAV1 and AAV5 are resistant to trypsin digestion after 24 hours of incubation, even though AAV1 VP1 has 65 potential cleavage sites and AAV5 VP1 has 60 potential cleavage sites; further supporting R588 as the specific trypsin cleavage site in AAV2. The lack of trypsin digestion of AAV1 at other tryptic sites within the C-terminal stretch, for example, K567, R610, and K621 was not unexpected because of the lack of AAV2 digestion at the equivalent locations, R566, R609, and K620 (Fig. 6B).
A difference in the susceptibility of the AAV1-GFP, AAV2-GFP, and AAV5-GFP virions to proteolytic cleavage was also evident from digestion with chymotrypsin. Chymotrypsin digestion of AAV2 (Fig. 6C) generated similar fragments to those seen with trypsin treatment (Fig. 6A) but a novel 27kDa fragment was detected by polyclonal antisera at 5 hours that was not B1 antibody reactive. AAV1 was more resistant to chymotrypsin, with all fragments of AAV2 being undetectable, except for the C-terminal 18kDa fragment, at 24 hours. Five C-terminal AAV1 fragments were generated at 12 hours, ranging in size from 30kDa to 50kDa, that are recognized by polyclonal sera; two of these fragments harbor the B1 antibody epitope, with the 30kDa fragment as the most C-terminal fragment (Fig. 6C). Based on primary sequence, chymotrypsin is expected to cleave denatured AAV1 VP1 148 times and AAV2 VP1 145 times, but as shown, only a limited number of cleavage sites are accessible. Additionally, AAV5 with 139 potential cleavage sites was resistant to chymotrypsin (Fig. 6C). The proteolytic data was confirmed by correlating the effect of digestion with infectivity (Fig. 6D) where the infectious titers of the vectors following 24 hours of digestion with trypsin or chymotrypsin resulted in AAV2 losing 3–4 logs of infectivity, but AAV1 and AAV5 lost less than 1 log, indicating that chymotrypsin cleavage of AAV1 is at a site(s) less important for infectivity.
The predictive power of the primary sequence and 3D structure data is limited with respect to identifying protease cleavage sites on intact AAV capsids. Analysis of the primary sequence of AAV1, AAV2, and AAV5 predicted numerous tryptic and chymotryptic sites, but the actual proteolysis resulted in cleavage at a very limited number of sites. Of note are the surface accessible R and K residues in the G-H loop that are common in AAV1, AAV2, and AAV5 (Fig. 7C), but are not cleaved by trypsin, which may be due to inaccessibility or steric hindrance in solution. Access to these sites could be dictated by their stable or transient presence on the virion surface, and, in some cases, surface loops in solution may not be exactly positioned as in the 3D structures.
Proteolytic analysis complements 3D-structure analysis
The available crystal structure of AAV2 [24] and homologous VP3 models generated for AAV1 and AAV5, based on a structural alignment with AAV2 [25], allows visualization of cleavage sites onto the capsid surface (Fig. 7). The R588 trypsin cleavage site is located on the capsid surface in the “finger-like” projections surrounding the 3-fold axis, and forms part of the basic patch that is responsible for AAV2’s heparin binding [30, 31]. Comparison of AAV1 and AAV5 with AAV2 capsids in this region clearly shows that the cleavage is specific, since adjacent basic residues are not susceptible, and AAV1 and AAV5, which do not contain R588, are resistant. Capsid protein loops, including the G-H loop, are able to tolerate differences in conformation when the AAV capsid structures are compared [25]. These loops are intertwined to form the characteristic AAV surface protrusions that decorate the core capsid and provide the immunogenic, tropic, and proteolytic determinants. The cleaved AAV2 C-terminal fragment, residues 589 to 735, is able to remain associated with the capsid because the cleavage site is on the outer surface of a 3-fold protrusion, not part of the eight-stranded β-barrel core that remains intact following trypsin cleavage. The B1 epitope (residues 726–733) is located on the “wall/floor” of the dimple at the 2-fold axis, close to a “buried” stretch of G-H loop amino acids leading up to the tryptic cleavage site (Fig. 7D I-III). Interestingly, the B1 epitope, found on the thinnest region of the AAV2 capsid, contains residues that are exposed on both the interior and exterior surfaces (Fig. 7D III).
Conclusions and practical applications
The isolation of numerous AAV serotypes [7] and the observation that each virus has unique cellular recognition and transduction phenotypes dictated by the capsid protein sequence has generated a need to fully characterize the capsids of these viruses, including their structural features. Here we use proteolytic digestion combined with available information on antigenic epitopes to analyze the AAV capsid in solution. We show that proteolysis is able to generate a characteristic cleavage pattern that can be used to distinguish full from empty AAV2 capsids and differentiate between two highly homologous serotypes, AAV1 and AAV2, as well as between less homologous serotypes (AAV5). The size and sequence of the proteolytic products in addition to the origin of the products (i.e. VP1, VP2, or VP3) were determined. Furthermore, available structural information enabled the 3D visualization of the location of the AAV2 cleavage and antigenic sites on the surface of the virion.
AAV2 is currently being evaluated in a variety of human clinical trials (summarized in [44]) and clinical trials are being planned that utilize other serotypes. Prior to use in the clinic, an important product release test is the confirmation of product identity to comply with current Good Manufacturing Practices (cGMP) regulations. Identity release testing involves not only confirmation of the vector genome that is packaged, but also the serotype of the capsid. Since immunologic reagents generated to one serotype have the potential to cross react with other serotypes, screening different proteases on different AAV serotypes may generate a capsid fingerprint database that could distinguish the more than 100 AAV serotypes isolated [7]. An assay capable of distinguishing different serotypes that utilizes the commercially available reagents applied in these studies will be useful to the gene therapy community.
In addition, vector targeting has gained a great deal of interest, and is intended to reduce non-specific uptake by non-target organs and increase the efficiency of uptake into target tissue. Cell-specific epitopes and receptors have been engineered into the AAV capsids for efficient targeting [45–47]. The sites in the capsid sequence that can accommodate insertions have been identified mainly through mutagenesis studies. In the absence of 3D structure, proteolytic mapping, as is demonstrated in these studies, can identify exposed, flexible loops of the AAV capsid. These regions can be tested for their ability to accommodate epitope/peptide insertions. Having an additional approach to identifying insertion sites in the capsid of any AAV serotype can aid in the generation of new targeting vectors.
MATERIALS AND METHODS
Viruses and cell lines
Full (DNA-containing) rAAV2-GFP vector virions were produced by transient transfection of HEK293 cells, lysed with 0.5% deoxycholate (DOC), treated with 0.02% trypsin (Gibco) for 30 minutes at 37°C, and purified on sequential CsCl density gradients (designated as Trypsin/DOC/CsCl) [48] or using DOC and CsCl without trypsin (designated as DOC/CsCl). All purification methods incorporated the use of Benzonase at 50U/ml for 30 minutes at 37°C and in some cases additionally utilized a final heparin chromatography step for concentration [39] (designated as Trypsin/DOC/CsCl/Heparin and DOC/CsCl/Heparin). rAAV2-GFP, rAAV1-GFP, rAAV5-GFP vector virions were also produced by transient transfection of HEK293 cells, lysed by freeze/thaw, and purified on iodixanol gradients followed by heparin affinity or Q-sepharose chromatography (designated as F-T/Iodixanol/Heparin or F-T/Iodixanol/Q [39]). Alternatively, rAAV2-GFP and rAAV1-GFP vector virions were purified by freeze/thaw and CsCl gradients followed by heparin affinity or Q-sepharose chromatography (designated as F-T/CsCl/heparin or F-T/CsCl/Q). Infectivity of the AAV vectors were assayed on C12 cells in the presence of adenovirus as previously described [39]. Empty AAV2 capsids were made by infecting HEK293 cells with an Adenoviral vector expressing the AAV2 cap ORF (MOI=2). The cells were lysed by 3 freezing/thawing cycles, the cell debris was removed by centrifugation (3000g for 10 minutes), and DOC was added to the supernatant to a final concentration of 0.5%. The supernatant was digested with 50U/ml benzonase for 30 minutes at 37°C, filtered (Acrodisc 25mm PF, Pall), and purified by heparin affinity chromatography. Virions and empty capsid preparations were dialyzed into 50mM Tris-Cl pH 8 containing 100mM NaCl using a 10,000 MWCO membrane (Pierce), aliquoted, and stored frozen at −20°C or −80°C.
Proteolytic digestion
0.8μg of virions (equivalent to ~1.2X1011 capsids) were digested with 5μg (0.02% final concentration) of trypsin (Gibco) or 80ug of α-chymotrypsin (Sigma) in a 25μl reaction at 37°C for up to 24 hours. The products of proteolysis were denatured at 100°C using Laemmli sample buffer at final concentrations of 1% SDS and 655mM β-mercaptoethanol, and separated by SDS-PAGE. After transferring the proteins to nitrocellulose (25mM Tris/192mM Glycine/0.1% (w/v) SDS/20%MeOH for 2 hours at 0.5 Amp in a Criterion-transblot apparatus (Bio Rad) that reached 45°C), they were probed with antisera to AAV2 (polyclonal, A1, B1, A69 [18, 40]) and AAV5 from Progen at dilutions of 1:250 (polyclonal) to 1:2000 (monoclonal). The AAV2 polyclonal antiserum cross-reacts with other AAV serotypes, but cross-reacts weakly with AAV5 under our conditions. Based on the peptides used to generate the monoclonal antisera, B1 (antibody epitope IGTRYLTR) recognizes AAV1–3, and AAV5–10; A69 (antibody epitope LNFGQTGDADSV) is specific for AAV2; and A1 (antibody epitope KRVLEPLGL) recognizes AAV1, AAV2, AAV4, AAV7, AAV8, AAV10, and AAV11. Bands were visualized by chemiluminescence using HRP-conjugated secondary antibodies (Amersham) and captured on X-ray film.
Mass spectroscopy and N-terminal sequencing
For mass spectroscopy and N-terminal sequencing, DOC/CsCl/heparin purified rAAV2-GFP virions were digested with trypsin (0.02% trypsin for 24 hours at 37° C) and heated to 100°C for 5 minutes in the presence of 0.6M dithiothreitol (DTT). The common C- terminal trypsin digestion product of VP1, VP2, and VP3 (designated as the VP1,2,3T fragment) was isolated by HPLC on a Vydac C4 5μm 150mm 2.1mm ID column eluted using a 15% A to 80% B buffer gradient (Buffers: A is 0.1% Tri Fluoro Acetic Acid (TFA) in H2O and B is 0.1% TFA in Acetonitrite (CH3CN)) over 30min at a flow rate of 200μl/min. Detection was at 215 nm and column peak #3 of 5 peaks (~0.5ml fractions) contained VP1,2,3T as verified by Western blotting with polyclonal and B1 antisera. For matrix assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF), the VP1,2,3T fragment was reconstituted in 50% acetonitrile/0.1% acetic acid and evaluated using an Applied Biosystems QSTAR with electrospray ionization and the Bayesian Protein Reconstruction algorithm was used to deconvolute the mass. N-terminal sequence of the VP1,2,3T fraction was obtained using an Applied Biosystems 494/HT PROCISE Protein Sequencing System with standard liquid blot cycles.
Semi-native PAGE
Trypsin-treated (0.02% trypsin at 37°C) and untreated rAAV2-GFP virions were subjected to semi-native gel electrophoresis. Loading dye (5% glycerol, 50mM Tris-Cl pH 6.8, and bromophenol blue) was added to the samples, and they were electrophoresed on a 5% native polyacrylamide gel in Tris/glycine buffer containing 0.1% SDS. Following electrophoresis, the gel was either stained with Gel Code Blue (Pierce) or proteins were transferred to nitrocellulose in 25mM Tris/192mM Glycine/0.1% (w/v) SDS/20%MeOH for 2hours at 0.5 Amp in a Criterion-transblot apparatus (Bio Rad) that reached 45°C, and probed with the B1 antibody at a dilution of 1:2000.
Electron microscopy (EM)
Purified rAAV2-GFP virions were digested with 0.02% trypsin for 24 hours at 37°C. 3–5μl of treated and untreated samples (at approximately 0.05mg/ml) were loaded onto 400 mesh carbon-coated formar copper grids (Ted Pella, Inc., Redding, CA), and negatively stained with 2% uranyl acetate. The grids were viewed on a Hitachi H-7000 transmission electron microscope at 30,000X and 70,000X magnification.
Immuno dot-blot
The immuno dot-blot procedure was essentially as described in Bleker, et al. [42]. Whatman filter paper No. 3 (Whatman International, Ltd., Maidstone, England) and supported nitrocellulose, 0.2 μm pore size, (Bio-Rad, Hercules, CA) were soaked briefly in TBS (50 mM Tris, 100 mM NaCl, pH 8.0) prior to assembling the dot-blot manifold (Schleicher and Schuell). After proteolysis, 25μl samples were directly applied to the wells of the dot-blot manifold and allowed to adsorb to the membrane. In some cases, the trypsinized virions were treated with 0.1% SDS and 20% MeOH for 2h at 37°C or 45°C and then applied. Excess fluid was drawn through the membrane by vacuum filtration. Each well was washed with 100ul of TBS. The membrane was removed from the manifold and blocked with TBS/0.05% Tween 20 (TTBS) + 5% milk for 1 hour. Membranes were then probed with antisera to AAV2 for 1 hour, in TTBS+5% milk at antibody dilutions of 1:2000, washed 3 times for 5 minutes each with TTBS + 0.05% milk, and were probed with secondary antibody, HRP-linked anti-mouse antibody diluted 1:5000 in TTBS+5% milk for 1 hour. Membranes were washed 3 times for 5 minutes each with TTBS + 0.05% milk prior to detection using Pierce SuperSignal West Pico Chemiluminescent Substrate.
3D-structure analysis
Potential protease cleavage sites were analyzed using the protein sequences of AAV1 (NP049542), AAV2 (AAC03780), AAV3 (NP043941), AAV4 (NP044927), AAV5 (YP068409), AAV6 (NP045758), AAV7 (YP077178), AAV8 (YP077180), AAV9 (AAS99264), AAV10 (AAT46337), AAV11 (AAT46339), and AAAV (AAT48613). Structure analysis to map potential tryptic cleavage sites on the AAV2 and AAV1 capsids utilized available coordinates for the AAV2 VP3 monomer [24] (PBD Accession No. 1LP3) and homologous models were generated for AAV1 VP3 and AAV5 VP3 based on a structure-guided sequence alignment with AAV2 [25]. VIPER was used to apply icosahedral symmetry operators to the VP3 coordinates to generate 3D models [50]. The coordinates were visualized using the program PyMOL (http://www.pymol.org, DeLano Scientific, San Carlos, CA).
Acknowledgments
We thank Nicholas Muzyczka and Philippe Moullier for helpful comments; Scott McClung and Stanley Stevens of the Interdisciplinary Center for Biotechnology Research for mass spectroscopy and protein sequencing; Lynda Schneider, Karen Kelley, and Gregory Erdos of the Electron Microscopy Core Laboratory; the Powell Gene Therapy Center Vector Core; and Lakshmanan Govindasamy for help with 3D structure modeling. RS is an inventor on patents related to recombinant AAV technology. RS owns equity in a gene therapy company that is commercializing AAV for gene therapy applications. To the extent that the work in this manuscript increases the value of these commercial holdings, RS has a conflict of interest. This project was funded, in part, by UF College of Medicine start-up funds to RS, and NIH projects P01 HL59412 and P01 HL51811 to MA-M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warrington KA, Jr, Herzog RW. Treatment of human disease by adeno-associated viral gene transfer. Hum Genet. 2006;119:571–603. doi: 10.1007/s00439-006-0165-6. [DOI] [PubMed] [Google Scholar]
- 2.Stedman H, Wilson JM, Finke R, Kleckner AL, Mendell J. Phase I clinical trial utilizing gene therapy for limb girdle muscular dystrophy: alpha-, beta-, gamma-, or delta-sarcoglycan gene delivered with intramuscular instillations of adeno-associated vectors. Hum Gene Ther. 2000;11:777–790. doi: 10.1089/10430340050015671. [DOI] [PubMed] [Google Scholar]
- 3.During MJ, Kaplitt MG, Stern MB, Eidelberg D. Subthalamic GAD gene transfer in Parkinson disease patients who are candidates for deep brain stimulation. Hum Gene Ther. 2001;12:1589–1591. [PubMed] [Google Scholar]
- 4.Crystal RG, Sondhi D, Hackett NR, Kaminsky SM, Worgall S, Stieg P, Souweidane M, Hosain S, Heier L, Ballon D, Dinner M, Wisniewski K, Kaplitt M, Greenwald BM, Howell JD, Strybing K, Dyke J, Voss H. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum Gene Ther. 2004;15:1131–1154. doi: 10.1089/hum.2004.15.1131. [DOI] [PubMed] [Google Scholar]
- 5.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko J, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 6.Aitken ML, Moss RB, Waltz DA, Dovey ME, Tonelli MR, McNamara SC, Gibson RL, Ramsey BW, Carter BJ, Reynolds TC. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum Gene Ther. 2001;12:1907–1916. doi: 10.1089/104303401753153956. [DOI] [PubMed] [Google Scholar]
- 7.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt M, Grot E, Cervenka P, Wainer S, Buck C, Chiorini JA. Identification and characterization of novel adeno-associated virus isolates in ATCC virus stocks. J Virol. 2006;80:5082–5085. doi: 10.1128/JVI.80.10.5082-5085.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CL, Jensen RL, Schnepp BC, Connell MJ, Shell R, Sferra TJ, Bartlett JS, Clark KR, Johnson PR. Molecular characterization of adeno-associated viruses infecting children. J Virol. 2005;79:14781–14792. doi: 10.1128/JVI.79.23.14781-14792.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rutledge EA, Halbert CL, Russell DW. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J Virol. 1998;72:309–319. doi: 10.1128/jvi.72.1.309-319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chao H, Liu Y, Rabinowitz J, Li C, Samulski RJ, Walsh CE. Several Log Increase in Therapeutic Transgene Delivery by Distinct Adeno-Associated Viral Serotype Vectors. Mol Ther. 2000;2:619–623. doi: 10.1006/mthe.2000.0219. [DOI] [PubMed] [Google Scholar]
- 12.Grimm D, Kay MA, Kleinschmidt JA. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol Ther. 2003;7:839–850. doi: 10.1016/s1525-0016(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 13.Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. Recombinant AAV Viral Vectors Pseudotyped with Viral Capsids from Serotypes 1, 2, and 5 Display Differential Efficiency and Cell Tropism after Delivery to Different Regions of the Central Nervous System. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 14.Girod A, Wobus CE, Zadori Z, Ried M, Leike K, Tijssen P, Kleinschmidt JA, Hallek M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol. 2002;83:973–978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- 15.Hoque M, Ishizu K, Matsumoto A, Han SI, Arisaka F, Takayama M, Suzuki K, Kato K, Kanda T, Watanabe H, Handa H. Nuclear transport of the major capsid protein is essential for adeno-associated virus capsid formation. J Virol. 1999;73:7912–7915. doi: 10.1128/jvi.73.9.7912-7915.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruffing M, Zentgraf H, Kleinschmidt JA. Assembly of viruslike particles by recombinant structural proteins of adeno-associated virus type 2 in insect cells. J Virol. 1992;66:6922–6930. doi: 10.1128/jvi.66.12.6922-6930.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warrington KH, Jr, Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J Virol. 2004;78:6595–6609. doi: 10.1128/JVI.78.12.6595-6609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wistuba A, Kern A, Weger S, Grimm D, Kleinschmidt JA. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J Virol. 1997;71:1341–1352. doi: 10.1128/jvi.71.2.1341-1352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter BJ. AAV DNA Replication, Integration, and Genetics. In: Tijssen P, editor. Handbook of Parvoviruses. Vol. 1. CRC Press; Boca Raton: 1990. pp. 169–226. [Google Scholar]
- 20.Agbandje M, McKenna R, Rossmann MG, Strassheim ML, Parrish CR. Structure determination of feline panleukopenia virus empty particles. Proteins. 1993;16:155–171. doi: 10.1002/prot.340160204. [DOI] [PubMed] [Google Scholar]
- 21.Tsao J, Chapman MS, Agbandje M, Keller W, Smith K, Wu H, Luo M, Smith TJ, Rossmann MG, Compans RW, et al. The three-dimensional structure of canine parvovirus and its functional implications. Science. 1991;251:1456–1464. doi: 10.1126/science.2006420. [DOI] [PubMed] [Google Scholar]
- 22.Xie Q, Chapman MS. Canine parvovirus capsid structure, analyzed at 2.9 A resolution. J Mol Biol. 1996;264:497–520. doi: 10.1006/jmbi.1996.0657. [DOI] [PubMed] [Google Scholar]
- 23.Kronenberg S, Kleinschmidt JA, Bottcher B. Electron cryo-microscopy and image reconstruction of adeno-associated virus type 2 empty capsids. EMBO Rep. 2001;2:997–1002. doi: 10.1093/embo-reports/kve234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:10405–10410. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Padron E, Bowman V, Kaludov N, Govindasamy L, Levy H, Nick P, McKenna R, Muzyczka N, Chiorini JA, Baker TS, Agbandje-McKenna M. Structure of adeno-associated virus type 4. J Virol. 2005;79:5047–5058. doi: 10.1128/JVI.79.8.5047-5058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walters RW, Agbandje-McKenna M, Bowman VD, Moninger TO, Olson NH, Seiler M, Chiorini JA, Baker TS, Zabner J. Structure of adeno-associated virus serotype 5. J Virol. 2004;78:3361–3371. doi: 10.1128/JVI.78.7.3361-3371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kronenberg S, Bottcher B, von der Lieth CW, Bleker S, Kleinschmidt JA. A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J Virol. 2005;79:5296–5303. doi: 10.1128/JVI.79.9.5296-5303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman MS, Agbandje-McKenna M. Atomic structure of viral particles. In: Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, editors. Parvoviruses. Edward Arnold, Ltd; New York: 2006. pp. 107–123. [Google Scholar]
- 29.Agbandje-McKenna M, Chapman MS. Correlating structure with function in the viral capsid. In: Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, editors. Parvoviruses. Edward Arnold, Ltd; New York: 2006. pp. 125–139. [Google Scholar]
- 30.Kern A, Schmidt K, Leder C, Muller OJ, Wobus CE, Bettinger K, Von der Lieth CW, King JA, Kleinschmidt JA. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Opie SR, Warrington KH, Jr, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Handa A, Muramatsu S, Qiu J, Mizukami H, Brown KE. Adeno-associated virus AAV-3-based vectors transduce haematopoietic cells not susceptible to transduction with AAV-2-based vectors. J Gen Virol. 2000;81(Pt 8):2077–2084. doi: 10.1099/0022-1317-81-8-2077. [DOI] [PubMed] [Google Scholar]
- 33.Walters RW, Yi SM, Keshavjee S, Brown KE, Welsh MJ, Chiorini JA, Zabner J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J Biol Chem. 2001;276:20610–20616. doi: 10.1074/jbc.M101559200. [DOI] [PubMed] [Google Scholar]
- 34.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S, Kapturczak M, Loiler SA, Zolotukhin S, Glushakova OY, Madsen KM, Samulski RJ, Hauswirth WW, Campbell-Thompson M, Berns KI, Flotte TR, Atkinson MA, Tisher CC, Agarwal A. Efficient transduction of vascular endothelial cells with recombinant adeno-associated virus serotype 1 and 5 vectors. Hum Gene Ther. 2005;16:235–247. doi: 10.1089/hum.2005.16.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grieger JC, Snowdy S, Samulski RJ. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J Virol. 2006;80:5199–5210. doi: 10.1128/JVI.02723-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berns KI, Rose JA. Evidence for a single-stranded adenovirus-associated virus genome: isolation and separation of complementary single strands. J Virol. 1970;5:693–699. doi: 10.1128/jvi.5.6.693-699.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Q, Hare J, Turnigan J, Chapman MS. Large-scale production, purification, and crystallization of wild-type adeno-associated virus-2. J Virol Methods. 2004;122:17–27. doi: 10.1016/j.jviromet.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 40.Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tattersall P. Parvovirus Protein Structur e and Virion Maturation. In: Ward DCaT P., editor. Replication of Mammalian Parvoviruses. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1978. pp. 53–72. [Google Scholar]
- 42.Bleker S, Sonntag F, Kleinschmidt JA. Mutational analysis of narrow pores at the fivefold symmetry axes of adeno-associated virus type 2 capsids reveals a dual role in genome packaging and activation of phospholipase A2 activity. J Virol. 2005;79:2528–2540. doi: 10.1128/JVI.79.4.2528-2540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sommer JN, Smith PH, Parthasarathy S, Isaacs J, Vijay S, Kieran J, Powell SK, McClelland A, Wright JF. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol Ther. 2003;7:122–128. doi: 10.1016/s1525-0016(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 44.Snyder RO, Francis J. Adeno-associated viral vectors for clinical gene transfer studies. Curr Gene Ther. 2005;5:311–321. doi: 10.2174/1566523054065066. [DOI] [PubMed] [Google Scholar]
- 45.Muzyczka N, Warrington KH., Jr Custom adeno-associated virus capsids: The next generation of recombinant vectors with novel tropism. Hum Gene Ther. 2005;16:408–416. doi: 10.1089/hum.2005.16.408. [DOI] [PubMed] [Google Scholar]
- 46.Douar AM, Poulard K, Danos O. Deleterious effect of peptide insertions in a permissive site of the AAV2 capsid. Virology. 2003;309:203–208. doi: 10.1016/s0042-6822(03)00186-7. [DOI] [PubMed] [Google Scholar]
- 47.Girod A, Ried M, Wobus C, Lahm H, Leike K, Kleinschmidt J, Deleage G, Hallek M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2 [published erratum appears in Nat Med 1999 Dec;5(12):1438] Nat Med. 1999;5:1052–1056. doi: 10.1038/71021. [DOI] [PubMed] [Google Scholar]
- 48.Snyder RO, Xiao X, Samulski RJ. Production of Recombinan t Adeno-Associated Viral Vectors. In: Dracopoli N, Haines J, Krof B, Moir D, Morton C, Seidman C, Seidman J, Smith D, editors. Current Protocols in Human Genetics. John Wiley and Sons; New York: 1996. pp. 12.1.1–12.1.24. [Google Scholar]
- 49.Mohiuddin I, Loiler S, Zolotukhin I, Byrne BJ, Flotte TR, Snyder RO. Herpesvirus-based infectious titering of recombinant adeno-associated viral vectors. Mol Ther. 2005;11:320–6. doi: 10.1016/j.ymthe.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 50.Reddy VS, Natarajan P, Okerberg B, Li K, Damodaran KV, Morton RT, Brooks CL, 3rd, Johnson JE. Virus Particle Explorer (VIPER), a website for virus capsid structures and their computational analysis. J Virol. 2001;75:11943–11947. doi: 10.1128/JVI.75.24.11943-11947.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]