

Abstract
Oxidative stress has been shown to underlie neuropathological aspects of Alzheimer's disease (AD). 4-Hydroxy-2-nonenal (HNE) is a highly reactive product of lipid peroxidation of unsaturated lipids. HNE-induced oxidative toxicity is a well-described model of oxidative stress-induced neurodegeneration. GSH plays a key role in antioxidant defense, and HNE exposure causes an initial depletion of GSH that leads to gradual toxic accumulation of reactive oxygen species. In the current study, we investigated whether pre-treatment of cortical neurons with acetyl-L-carnitine (ALCAR) and α-lipoic acid (LA) plays a protective role in cortical neuronal cells against HNE-mediated oxidative stress and neurotoxicity. Decreased cell survival of neurons treated with HNE correlated with increased protein oxidation (protein carbonyl, 3-nitrotyrosine) and lipid peroxidation (HNE) accumulation. Pretreatment of primary cortical neuronal cultures with ALCAR and LA significantly attenuated HNE-induced cytotoxicity, protein oxidation, lipid peroxidation and apoptosis in a dose-dependent manner. Additionally, pretreatment of ALCAR and LA also led to elevated cellular GSH and heat shock proteins (HSPs) levels compared to untreated control cells. We have also determined that pretreatment of neurons with ALCAR and LA leads to the activation of phosphoinositol-3 kinase (PI3K), PKG and ERK1/2 pathways, which play essential roles in neuronal cell survival. Thus, this study demonstrates a cross talk between the PI3K, PKG and ERK1/2 pathways in cortical neuronal cultures that contributes to ALCAR and LA-mediated pro-survival signaling mechanisms. This evidence supports the pharmacological potential of co-treatment of ALCAR and LA in the management of neurodegenerative disorders associated with HNE-induced oxidative stress and neurotoxicity, including AD.
Introduction
The Alzheimer's disease (AD) brain is under extensive oxidative stress, evidenced by significant protein oxidation, lipid peroxidation, and DNA oxidation (1-3). AD is characterized by deposition of amyloid β-peptide [Aβ (1-42)] (4). Aβ (1-42) induces lipid peroxidation in ways that are inhibited by free radical antioxidants (2,3). One of the reactive products of lipid peroxidation is 4-hydroxy-2-trans-nonenal (HNE), which covalently binds to cysteine, histidine, and lysine residues by Michael addition (5) and changes protein conformation and function. HNE binding to proteins, which introduces a carbonyl to the protein, makes the protein oxidatively modified as a consequence of lipid peroxidation (6). HNE has been found to be increased in AD brain (7,8).
Previous studies have shown that, though still an unsettled question, apoptosis may contribute among the mechanisms of neuronal loss in AD brain (9). The AD brain is under oxidative stress indexed by, among other markers, lipid peroxidation (3,6). A large body of evidence indicates that lipid peroxidation is directly responsible for the generation of the apoptotic phenotype (10). Many agents that induce apoptosis are also stimulators of cellular oxidative metabolism (11), and many inhibitors of apoptosis have antioxidant activities or enhance cellular antioxidant defenses (12,13).
Acetyl-L-carnitine (ALCAR) is present in high concentration in the brain and is involved in the production of acetylcholine (14). ALCAR has been shown to improve mitochondrial function and reverse age-related deficits (15). α-Lipoic acid (LA) is an essential cofactor in mitochondrial dehydrogenase reactions, functions as an antioxidant and reduces oxidative stress in aged animals (16). LA and its reduced form, dihydrolipoic acid (DHLA), protects neuronal cultures from oxidative stress, when treated with amyloid beta-peptide (Abeta 25-35) and iron/hydrogen peroxide [Fe/H2O2] (17). Therefore, in the present study, we evaluated the role of ALCAR and LA co-treatment, to demonstrate its possible protective effects against HNE-induced oxidative stress and neurotoxicity in cortical neuronal cells.
The brain is susceptible to oxidative stress compared to other tissues (18) and to combat this stress, the cells respond by induction of groups of protective proteins, one of which is heat shock proteins [Hsps] (19). Expression of the genes encoding Hsps has been found in neurons (20,21). Hsps serve as molecular chaperones and among the various Hsps, Hsp32 (also known as HO-1) and Hsp72 have been recently demonstrated to play neuroprotective role against oxidative stress (20). HO-1 is the inducible form of a group of enzymes that catalyze heme degradation to form the antioxidant biliverdin and CO (22). Overexpression of human HO-1 gene has been shown to result in a stable increase of HO activity associated with neuroprotection against oxidative injury (23). Hsp70 (also referred to as Hsp72) is an inducible form, which is induced in the nervous system in a variety of neurodegenerative and metabolic disorders (24,25).
The PI3K/Akt pathway has been found to consistently serve a pro-survival function in neurons exposed to various apoptosis-inducing stimuli (26). Akt (a serinethreonine kinase, also known as protein kinase B) is a key upstream component of anti-apoptosis processes associated with activated phosphoinositide 3-kinase (PI3K). PI3K phosphorylates membrane-localized phosphoinositides and leads to translocation of Akt from cytoplasm to the plasma membrane, the site of Akt phosphorylation (27). Phosphorylated Akt (phospho-Akt) acts both to stimulate anti-apoptotic factors and to inhibit pro-apoptotic factors [e.g., BAD and caspase-9] (27). The mitogen activated protein kinase (MAPK) family member, extracellular signal-regulated kinase (ERK), is activated by a wide range of stimuli that acts on a diverse range of cellular targets (28,29). In neurons, ERK can function to either support cell survival or promote cell death (30). In neurons, there is much evidence to support significant cross talk between PI3K and [ERK] (31,32) with the potential for PI3K to act as a required upstream activator of ERK.
In the present study we show that ALCAR and LA protect cortical neurons against HNE-induced protein oxidation, lipid-peroxidation, loss of mitochondria function, and DNA fragmentation by up-regulating GSH and Hsps levels and by triggering PI3K, PKG and ERK1/2 signaling pathways, thereby inducing the expression of anti-apoptotic and anti-oxidant proteins.
MATERIALS AND METHODS
Chemicals
All chemicals were of the highest purity and were obtained from Sigma (St. Louis, MO) unless otherwise noted. HNE was purchased from Cayman chemicals (Ann Arbor; MI). ALCAR (99.99% pure) was a generous gift from Sigma Tau Pharma. (Pomezia, Italy). Anti-HO-1, anti-Hsp72, anti-MnSOD, anti-phospho ERK1/2, anti-phospho-BAD, anti-Bcl2, anti-GAPDH and anti-iNOS were obtained from Stressgen Biotechnologies. The OxyBlot protein oxidation detection kit was purchased from Chemicon International (Temecula, CA). Anti-4-hydroxynonenal was purchased from Alpha Diagnostic International (San Antonio, TX). KT5823 (PKG inhibitor), LY294002 (PI3K inhibitor), PD98059 (ERK1/2 inhibitor), SB202190 (p38-SAPK inhibitor) and SP600125 (JNK inhibitor) were obtained from Calbiochem.
Cell Culture Experiments
Neuronal cultures were prepared from 18-day-old Sprauge-Dawley rat fetuses (33). HNE stocks were prepared in sterile phosphate buffer saline (PBS), pH 7.5 prior to addition to cultures. The final concentration of the HNE in the cell culture was 10 μM and its effects on the neuronal culture were measured after 24 h of exposure. These concentrations and time were chosen based on prior studies of HNE produced by Aβ (1-42) or HNE-only experiments (34,35). Inhibitors of HO-1, Hsp72 and iNOS proteins; Zinc protoporphyrin IX (ZnPP IX), quercitin, L-NMMA (a nonisoform-specific nitric oxide synthase inhibitor), respectively or KT5823 (the PKG inhibitor) or SB202190 (the p38-SAPK inhibitor) or SP600125 (the JNK inhibitor) or LY294002 (the PI3K inhibitor), or PD98059 (the ERK1/2 inhibitor) were added singly to the cell culture 1 h before addition of ALCAR+LA singly, which was added 2 hr prior to addition of 10 μM HNE. The concentrations of ALCAR used in this study were those used by Ishii et al., (36) and the concentration of LA according to Vincent et al. (37).
Sample Preparation
The samples were homogenized in a 10 mM HEPES buffer (pH 7.4) containing 137 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, 0.6 mM MgSO4, and protease inhibitors leupeptin (0.5 lg/ml), pepstatin (0.7 lg/ml), type II S soybean trypsin inhibitor (0.5 lg/ml), and phenylmethylsulfonyl fluoride (PMSF; 40 lg/ml). The protein concentration was estimated by using the Pierce BCA method.
Determination of cell viability
Mitochondrial function was evaluated by the 3-[4,5-dimethylthiazol-2-yl)-2,5-diphenyl] tetrazolium bromide (MTT) reduction assay. This cell proliferation assay was used as a quantitative colorimetric method for measurements of cellular cytotoxicity. Briefly, MTT was added to each well with a final concentration of 1.0 mg/ml and incubated for 1 h in CO2 incubator. The dark blue formazan crystals formed in intact cells were extracted with 250 μl of dimethyl sulfoxide, and the absorbance was read at 595 nm with a microtiter plate reader (Bio-Tek Instruments, Winooski, VT). Results were expressed as the percent MTT reduction of control cells (untreated cells).
Analysis of DNA fragmentation
The nuclear staining with Hoescht 332584 (1 μg/ml) followed by propidium iodide (PI) [5 μg/ml] staining provided morphological discrimination between normal and apoptotic cells as measured and detected by fluorescence microscopy (38). Cortical neuronal cells were treated with 10 μM HNE alone for 24 h or pretreated with ALCAR (75 μM) +LA (50 μM) or 75 μM ALCAR alone or 50 μM LA alone (for 2 h) followed by HNE (10 μM) and incubated for 24 h. Cultures were rinsed three times in PBS, fixed with 4% paraformaldehyde for 10 min at 37°C, rinsed, and stained with Hoechst 332584 or PI for 10 min at room temperature. Images were obtained sequentially with Hoechst 332584, and then PI. Random areas (approximately ten) in the cell culture dishes were selected, and the number of apoptotic cells was counted in neurons from control (untreated cells) and treated samples. The average of the apoptotic cells in each of the respective groups was determined as a percentage of the control (untreated cells).
Measurement of protein carbonyls
Protein carbonyls are an index of protein oxidation and were determined immunochemically. Samples (5 μl) were incubated for 20 min at room temperature with 5 μl of 12% sodium dodecyl sulfate (SDS) and 10 μl of 2,4-dinitrophenylhydrazine (DNPH) that was diluted 10 times with PBS (pH 7.5) from a 200 mM stock. The samples were neutralized with 7.5 μl of neutralization solution (2 M Tris in 30% glycerol). Derivatized protein samples were loaded onto nitrocellulose membrane (under vacuum pressure) with a slot blot apparatus (250 ng/well). The membrane was blocked with 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.2% (v/v) Tween 20 (wash blot) for 1 h and incubated with a 1:100 dilution of anti-DNP polyclonal antibody in wash blot for 1 h. After completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min each. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:8,000 in wash blot and added to the membrane for 1 h. The membrane was washed in wash blot three times for 5 min each and developed using Sigmafast Tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop (San Jose, CA), and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software. Controls in which carbonyls were reduced to alcohol by NaBH4 showed no detection of carbonyls by these immunochemical methods, demonstrating specificity of carbonyls and lack of non-specific binding by the primary or secondary antibodies (39).
Measurement of 4-hydroxy-2-trans-nonenal
Levels of HNE were quantified by slot-blot analysis as described previously (8). Samples (10 μl) were incubated with 10 μl of modified Laemmli buffer containing 0.125 M Tris base pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol. The resulting sample (250 ng) was loaded per well in the slot blot apparatus. Samples were loaded onto a nitrocellulose membrane under vacuum pressure. The membrane was blocked with 5% (w/v) BSA in wash blot for 1 h and incubated with a 1:5000 dilution of anti-4-hydroxynonenal (HNE) polyclonal antibody in wash blot for 90 min. Following completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min each. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:8000 in wash blot and added to the membrane for 90 min. The membrane was washed in wash blot three times for 5 min and developed using Sigma fast Tablets (BCIP/NBT substrate). The membrane was developed using Sigmafast tablets (BCIP/NBT substrate). The blot was dried, scanned with Adobe Photoshop, and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software.
Measurement of 3-nitrotyrosine (3NT)
The sample (10 μl) was incubated with 10 μl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol. The resulting samples were loaded onto nitrocellulose membrane (under vacuum pressure) with a slot blot apparatus (250 ng/well). The membrane was blocked with 5% (w/v) BSA in wash blot for 1 h and incubated with a 1:2000 dilution of 3-NT polyclonal antibody in wash blot for 90 min. Following completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min each. An anti-rabbit IgG alkaline phosphatase secondary antibody was diluted 1:8000 in wash blot and added to the membrane for 120 min. The membrane was washed in wash blot three times for 5 min and developed using Sigmafast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop, and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software.
Western blot analysis
Neuronal cultures were incubated with 10 μM HNE alone for 24 h or pretreated with ALCAR (75 μM) +LA (50 μM) or ALCAR (75 μM) alone or LA (50 μM) alone, 2 h prior to addition of HNE. Protein content of the cells was determined using the Bio-Rad protein assay reagent (BCA). Equal amounts of proteins were separated on 4-15% SDS-polyacrylamide gels, transferred to nitrocellulose membrane, and then probed with appropriate antibodies. HO-1, Hsp-72, iNOS, phospho-ERK1/2, phospho-BAD, Bcl2, MnSOD, thioredoxin and GAPDH were detected with specific primary antibodies. After incubation with the primary antibodies, the nitrocellulose membranes were incubated with a secondary alkaline phosphatase-conjugated antibody. Proteins were visualized by developing with Sigmafast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop, and quantitated with Scion software.
Statistical Analysis
Results were expressed as means ± SD from at least 5 independent samples. Each experiment was repeated at least three times to confirm the reproducibility of findings. Multiple groups were analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. P < 0.05 was considered significant.
RESULTS
ALCAR and LA inhibited HNE-induced protein oxidation and lipid-peroxidation
Protein carbonyls are elevated in vulnerable regions of AD brain (40-42). Accumulation of HNE (7,8) and elevated 3-NT (43,44) in AD brain has also been reported. In the present study, a concentration-dependent study (with various combinations of ALCAR and LA) was carried out (Table 1) in the cortical neurons. A 29% significant increase in carbonyl (p <0.02) or HNE (p < 0.05) or 3-NT (p < 0.05) levels, respectively, was observed in neurons following 24 h of treatment with 10 μM HNE (Table 1). Pretreatment of neurons with ALCAR and LA at different combinations of concentrations was shown to partially inhibit HNE-mediated protein oxidation and lipid-peroxidation. However, with ALCAR (75 μM) +LA (50 μM), there was a maximum significant decrease in the protein oxidation (p < 0.005) (Table 1) and lipid-peroxidation (p < 0.005) (Table 1). Pretreatment of cells with ZnPP IX or quercitin independently followed by ALCAR (75 μM) +LA (50 μM) and then 10 μM HNE produced less protection against HNE-induced protein carbonyl, HNE and 3-nitrotyrosine formation, compared to control than the protection produced by pretreatment of ALCAR (75 μM) +LA (50 μM) (p < 0.05) [data not shown]. In the presence of L-NMMA, there was less protection against HNE-induced 3-NT formation and a comparatively smaller effect on protein carbonyls or HNE formation [data not shown].
Table 1.
| PC±SD (P) | HNE±SD (P) | 3-NT±SD (P) | ||
|---|---|---|---|---|
| Control | 100±0.3 | 100±0.3 | 100±0.3 | |
| HNE | 129±1.4 (0.02) | 129±1.4 (0.05) | 129±4.1 (0.05) | |
| 75 μM | + 50 μM | 100±2.8 (0.05) | 101±3.4 (0.05) | 101±5.1 (0.05) |
| ALCAR | LA | |||
| HNE plus | HNE plus | |||
| ALCAR | LA | |||
| (μM) | (μM) | |||
| 50 | 50 | 126±1.9 (0.03) | 128±1.9 (0.05) | 125±5.4 (0.09) |
| 50 | 75 | 125±2.8 (0.05) | 125±3.4 (0.05) | 123±5.1 (0.10) |
| 50 | 100 | 123±3.9 (0.07) | 122±4.4 (0.10) | 120±7.2 (0.17) |
| 75 | 50 | 112±0.3 (0.005) | 111±0.7 (0.005) | 114±1.9 (0.005) |
| 75 | 75 | 111±1.1 (0.04) | 112±0.1 (0.005) | 114±2.6 (0.05) |
| 100 | 75 | 112±1.1 (0.04) | 112±0.1 (0.005) | 113±3.7 (0.12) |
| 100 | 100 | 113±3.5 (0.14) | 112±0.9 (0.05) | 114±4.1 (0.14) |
| 75 | – | 120±1.7 (0.02) | 121±1.4 (0.05) | 119±4.1 (0.05) |
| – | 50 | 121±1.2 (0.02) | 122±1.8 (0.05) | 121±2.1 (0.05) |
Protective effect of ALCAR+LA against HNE-induced oxidative stress. The treatment of cortical neuronal cultures is as described in “Materials and Methods” section. PC and P, refer to protein carbonyls and p value respectively. The data are the mean ± SD expressed as percentage of control values. Each experiment was repeated three times with five independent samples. Statistical comparison was by ANOVA (n=5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control.
ALCAR and LA protect mitochondria from HNE-mediated dysfunction
HNE was shown to decrease MTT reduction by 50% compared with untreated control (Fig. 1). Treatment of neurons with 75 μM ALCAR alone or 50 μM LA alone for 24 h did not show any change in MTT reduction compared to the control. Pretreatment of neurons with ALCAR and LA, at different combinations of concentrations, was shown to partially protect neurons against HNE-mediated mitochondrial dysfunction. However, pretreatment of ALCAR (75 μM) + LA (50 μM) [2 h prior] followed by 10 μM HNE showed maximum significant (p < 0.05) protection of mitochondrial function compared with that induced by 10 μM HNE alone, but still a significant decrease in MTT reduction compared with untreated control was observed (Fig.1). These results are consistent with the concentration of ALCAR+LA that protected neurons against HNE-mediated protein oxidation and lipid peroxidation (Table. 1). Since ALCAR (75 μM) + LA (50 μM) protected neurons against protein oxidation, lipid peroxidation and prevented loss of mitochondrial function, this concentration was used for all subsequent experiments.
Fig. 1.
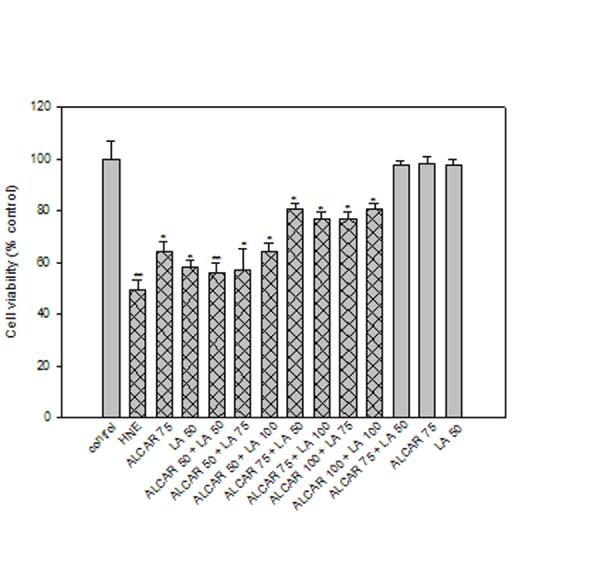
Protective effect of ALCAR+LA on cell viability against HNE-induced cytotoxicity in cortical neuronal culture. A dose-dependent effect of ALCAR+LA on neuronal cell culture was assessed by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reduction assay. The grid bars represent the presence of HNE and the plain bars represent studies without HNE. The data are the mean ± SD expressed as percentage of control values obtained for five independent preparations. Statistical comparison was by ANOVA (n=5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p < 0.05, **p < 0.005.
ALCAR and LA protects against HNE-mediated induction of neuronal apoptosis
Phase-contrast microscopy was used to examine the morphological changes in the neurons following treatment (Fig. 2). Neurons that were exposed to 10 μM HNE for 24 h (Fig. 2a) showed loss of their neuronal network, membrane blebbing and cell shrinkage, processes that are normally associated with apoptotic cell death. Conversely, neurons pretreated with ALCAR (75 μM) + LA (50 μM) 2 h prior to addition of HNE or ALCAR (75 μM) alone or LA (50 μM) alone (Fig. 2a), showed intact networks and cell bodies similar to those of control neurons (Fig. 2a). To further investigate and confirm the results obtained from the above MTT assay, neuronal apoptosis studies were carried out using Hoechst and PI staining. Neurons treated with 10 μM HNE (Fig. 2b, c) showed extensive apoptotic bodies by both stains, from which we conclude that late apoptotic and necrotic cells are found under the conditions of this experiment. In contrast, pretreatment of neurons with 75 μM ALCAR + 50 μM LA followed by addition of HNE (Fig. 2b, c) resulted in a significant (p < 0.005) reduction in apoptotic cells. ALCAR (75 μM) alone or LA (50 μM) alone treated neurons were similar to those of untreated control cells. The averages of late apoptotic cells were calculated and are reflected in the bar graph (Fig. 2d).
Fig. 2.
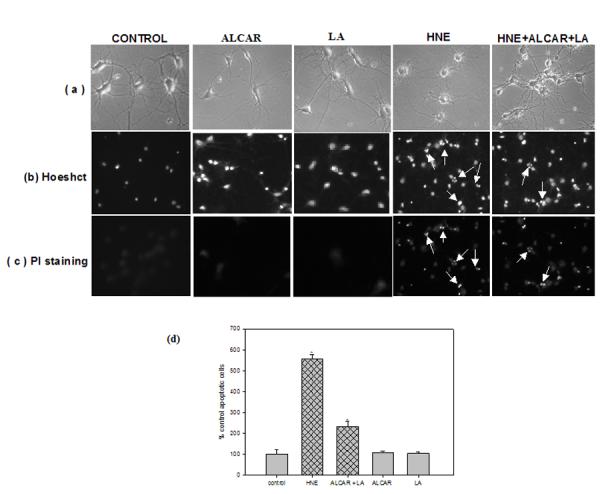
Protective effect of ALCAR+LA against DNA fragmentation. The cells were treated as described in the “Materials and Methods” section, following which cell morphology was visualized by phase contrast microscope (magnification 100X). The final concentration of 10μM HNE or 75μM ALCAR+50μM LA or 75μM ALCAR alone or 50μM LA alone were used in the study. (a) The images displayed here are those, which exhibit the maximum morphological protection at the respective concentration specified. (b) Hoechst staining (c) propidium iodide staining for DNA fragmentation. Arrows heads indicate apoptotic bodies. (d) The averages of late-stage apoptotic cells and necrotic cells were calculated and are reflected in this bar graph. The grid bars are in presence of HNE and the plain bars are without HNE. The results presented are the mean ± SD expressed as percentage of control values obtained for five independent preparations. Statistical comparison was by ANOVA (n =5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p < 0.005, compared with the untreated control.
ALCAR and LA increases endogenous GSH levels
An assay to measure the total GSH levels was conducted (Fig. 3) to determine whether ALCAR+LA protected neurons by up-regulating GSH. HNE treatment led to a significant reduction (p <0.05) in cellular GSH (50% compared to the untreated control cells). Treatment of cells with ALCAR (75 μM) + LA (50 μM) alone led to a mean 20% significant (p < 0.05) increase in GSH compared with control (untreated cells). However, ALCAR+LA pretreatment followed by addition of HNE raised total GSH levels significantly (p < 0.05) to more than 80% of control. That is, pretreatment of cortical neuronal cells with ALCAR (75 μM) + LA (50 μM) apparently led to elevated GSH levels. Indeed, compared to HNE-treated controls, ALCAR (75 μM) + LA (50 μM) pretreatment followed by HNE addition led to about 50% increase in brain GSH (p < 0.05).
Fig. 3.
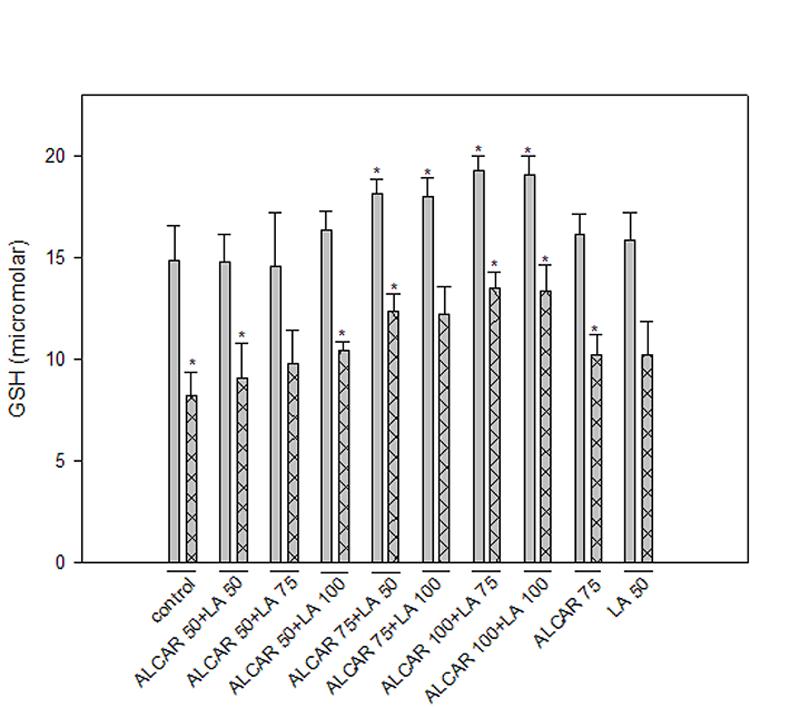
GSH assay. Cell cultures were pretreated with ALCAR+LA in a dose-dependant manner (as described in “Materials and Methods” section) 2 h prior to the addition of HNE following, which the cells were collected after 24 hr and assayed for total GSH. The grid bars are in presence of HNE and the plain bars are without HNE. The results are shown as mean ± SD expressed as percentage of control values obtained for five independent preparations. Statistical comparison was by ANOVA (n=5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p<0.005.
Protection of neurons against HNE-induced toxicity is associated with ALCAR and LA-mediated up-regulation of HO-1, Hsp72 and down-regulation of iNOS
Cortical neuronal cells treated with 10 μM HNE showed a significant 3-fold increase (p < 0.005) in HO-1 protein levels, consistent with oxidative tress-induced cellular response (21). Treatment of cells with 75 μM ALCAR + 50 μM LA alone, induced HO-1 by 4-fold (p < 0.005) compared to untreated control neurons (Fig. 4a, 4c). Pre-treatment of neurons with 75 μM ALCAR+50 μM LA or 75 μM ALCAR alone or 50 μM LA alone followed by addition of HNE (Fig. 4a, 4c) resulted in a significant (p < 0.005) 8- or 4-fold increase, respectively, in HO-1 induction, compared to the control. A significant (p < 0.005) 3-fold increase in Hsp72 protein (Figs 5a, 5c) levels was observed in HNE treated cells compared to untreated control neurons, and treatment of cells with 75 μM ALCAR + 50 μM LA alone significantly (p < 0.005) increased (4-fold) Hsp72 protein levels compared to untreated control neurons. Pretreatment of neurons with 75 μM ALCAR+50 μM LA or 75 μM ALCAR alone or 50 μM LA alone, in each case followed by treatment of HNE (Fig. 5a, 5c), resulted in a significant (p < 0.005) 7- or 4-fold increase, respectively, in Hsp72 induction, compared to the control. The large amount of NO produced by iNOS has been closely correlated with the pathophysiology in AD (44) and inflammation (45). As shown in Fig. 6, iNOS levels were significantly (p < 0.005) increased by more than 4-fold in HNE-treated primary neuronal cells. The iNOS levels in neurons treated with 75 μM ALCAR + 50 μM LA alone were similar compared to the control (untreated cells). However, pretreatment of neurons with 75 μM ALCAR+50 μM LA alone or 75 μM ALCAR alone or 50 μM LA alone, in each case followed by addition of HNE (Fig. 6a, 6c), resulted in a significant (p < 0.005) 4-fold decrease in iNOS levels, compared to the HNE-treated neurons.
Fig. 4.
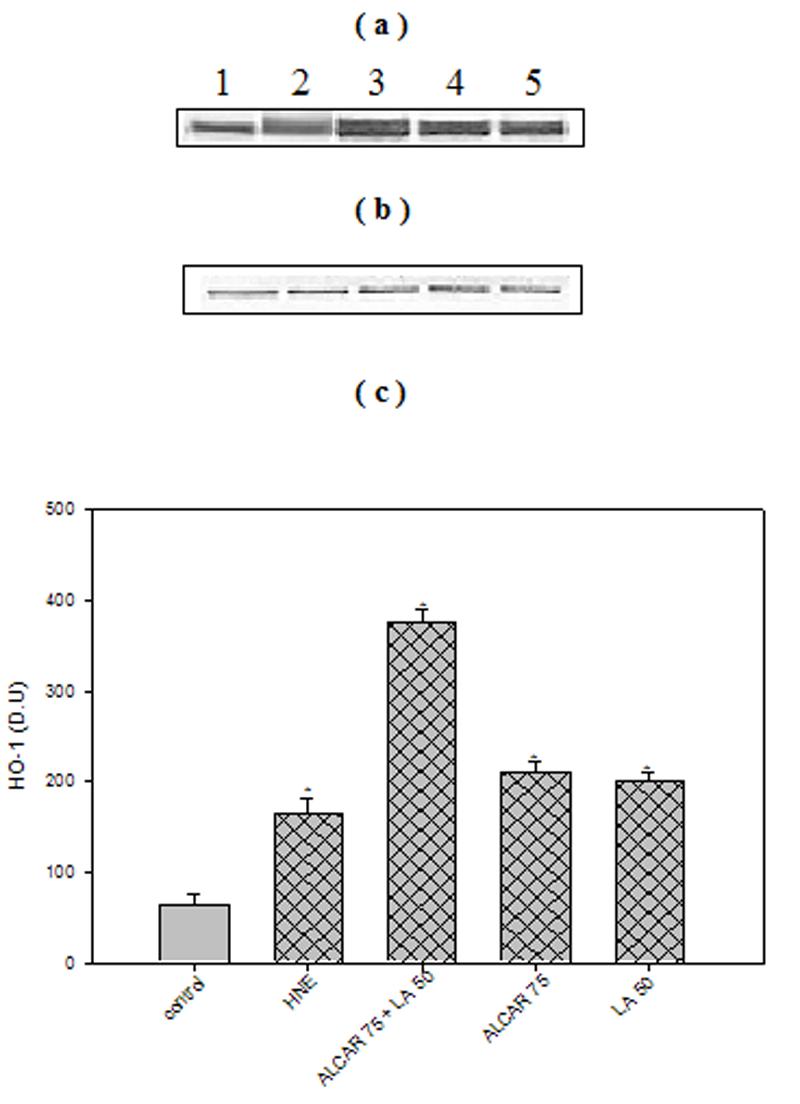
ALCAR+LA-mediated up-regulation of HO-1. (a) Representative Western immunoblot analysis of neuronal cells for HO-1 protein. 100μg of protein were analysed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis and immunoblotting using a mouse monoclonal anti-HO-1 antibody. The grid bars are in presence of HNE and the plain bars are without HNE. Lane 1: control (untreated cells); Lane 2: 10μM HNE; Lane 3: 10 μM HNE+75μM ALCAR+50μM LA; Lane 4: 75μM ALCAR+10μM LA; Lane 5: 10μM HNE+50 μΜ LA (b) Anti-GAPDH blot as control for equal protein loading. (c) Densitometric analysis from five independent experiments (mean ±SD of values expressed as relative units). Significant differences were assessed by ANOVA. Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p < 0.005.
Fig. 5.
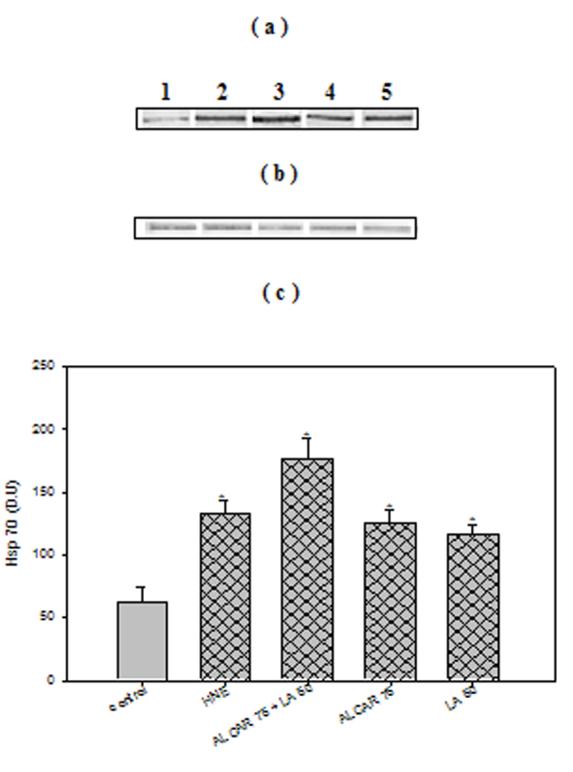
ALCAR+LA-mediated up-regulation of Hsp72. (a) Representative Western immunoblot analysis of neuronal cells for Hsp72. The treatments to the samples and method followed were the same as in Fig. 4(a), and immunoblotting was conducted using a mouse monoclonal anti-Hsp72 antibody. The grid bars are in presence of HNE and the plain bars are without HNE. (b) Anti-GAPDH blot as control for equal protein loading. (c) Densitometric analysis from five independent experiments (mean ±SD of values expressed as relative units). Significant differences were assessed by ANOVA. Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p < 0.005.
Fig. 6.
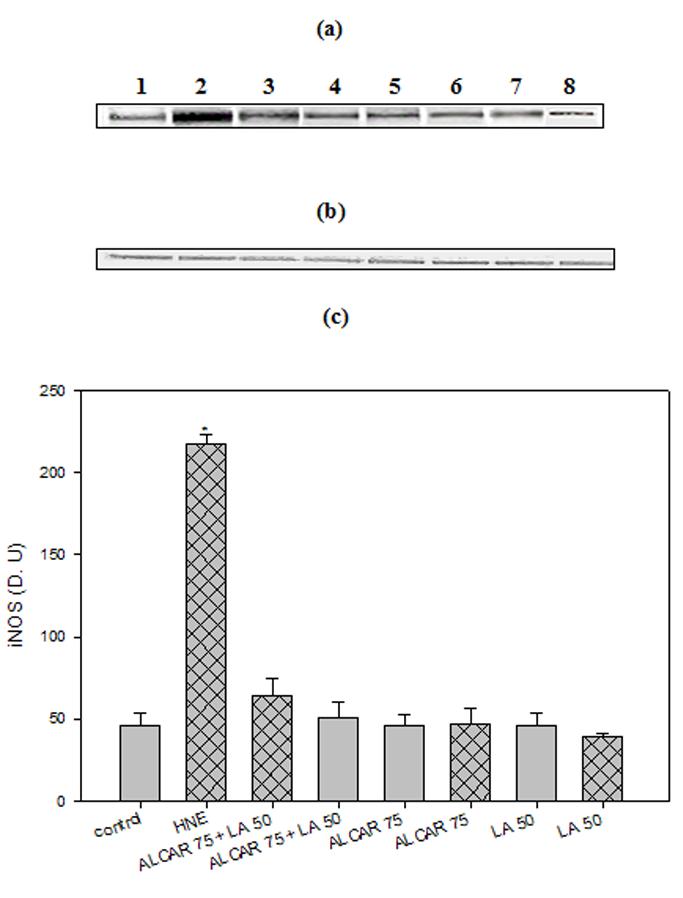
ALCAR+LA-mediated down-regulation of iNOS. (a) Representative western immunoblot analysis of neuronal cells for inducible nitric oxide synthase (iNOS) protein. The treatments to the samples and method followed were the same as in fig. 4(a) and immunoblotting was conducted using a mouse monoclonal anti-iNOS antibody. The grid bars are in presence of HNE and the plain bars are without HNE. (b) Anti-GAPDH blot as control for equal protein loading. (c) Densitometric analysis from five independent experiments (mean ±SD of values expressed as relative units). Significant differences were assessed by ANOVA. Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p < 0.005.
ALCAR and LA-mediated neuroprotection against HNE toxicity involves activation of multiple signaling pathways
To explore the molecular mechanisms underlying ALCAR+LA-mediated protective effects against HNE neurotoxicity, we screened five different pharmacological inhibitors each targeted at a specific signaling pathway to test which pathway may be capable of modulating the ALCAR+LA neuroprotective effect. The results suggest that (Fig. 7a. 7b, 7c), ALCAR+LA-mediated neuroprotection against HNE-induced toxicity could be significantly (p< 0.05) antagonized, either completely or partially, by KT5823 (PKG inhibitor), LY294002 (PI3K inhibitor), and PD98059 (ERK1/2 inhibitor), but not by SB202190 (p38-SAPK inhibitor) or SP600125 (JNK inhibitor), suggesting that PKG, PI3K and ERK1/2 pathways may be involved in mediating ALCAR+LA neuroprotective action. The treatments with the inhibitors noted above had a minimal effect on control cells when added alone.
Fig. 7.
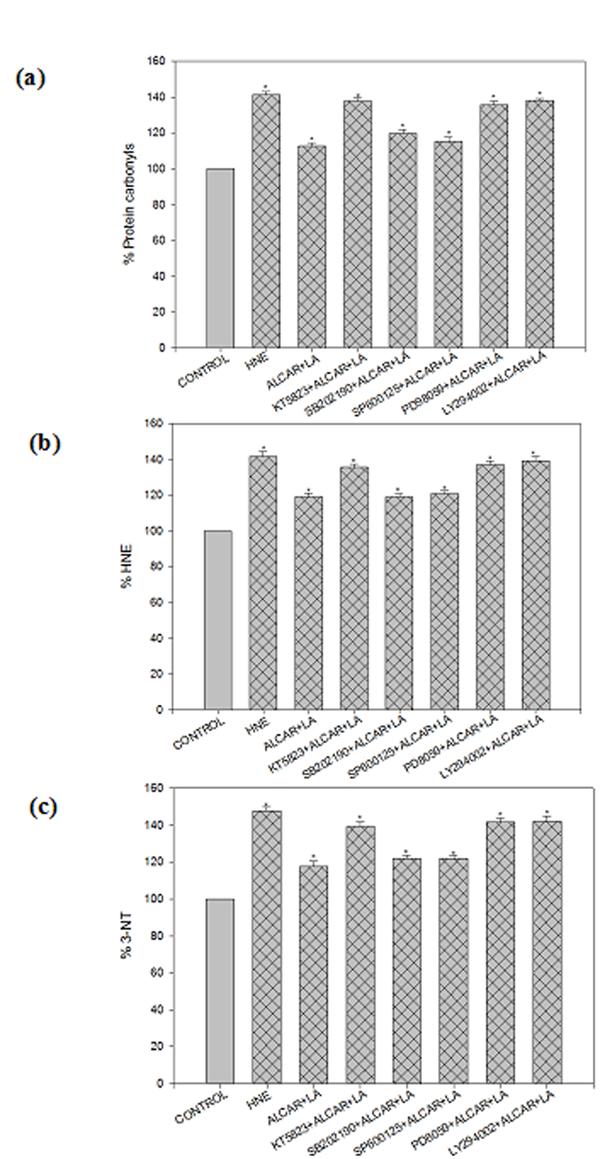
ALCAR and LA-mediated neuroprotection via activation of multiple signaling pathwaysagainst HNE toxicity. KT5823 (the PKG inhibitor), SB202190 (the p38-SAPK inhibitor), SP600125 (the JNK inhibitor), LY294002 (the PI3K inhibitor), PD98059 (the ERK1/2 inhibitor) were added singly to the cell culture 1 h before addition of ALCAR+LA singly, which was added 2 hr prior to addition of 10 μM HNE. The grid bars are in presence of HNE and the plain bars are without HNE. The results are shown as mean ± SD expressed as percentage of control values obtained for five independent preparations. Statistical comparison was by ANOVA (n=5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p<0.005.
Role of PKG pathway in ALCAR+LA-mediated neuroprotection
Guanylyl cyclase activation by nitric oxide (NO) results in elevation of cellular cGMP levels, and subsequent activation of PKG is a pro-survival signaling pathway in the nervous system (46). The PKG inhibitor (KT5823) significantly (p< 0.05) suppressed ALCAR+LA-mediated neuroprotection against HNE (Fig. 7a. 7b, 7c), suggesting the involvement of the PKG pathway in mediating ALCAR+LA neuroprotective action. We therefore tested whether direct activation of PKG is capable of mimicking ALCAR+LA-mediated neuroprotection. To test this hypothesis 8-Br-cGMP (a NO-independent stimulator of PKG) was used in this study. The cortical neurons were pretreated with 8-Br-cGMP (2 h prior) followed by the addition of 10 μM HNE, incubated for 24h following which oxidative stress parameters were studied. The results shown (Fig. 8a, 8b, 8c) demonstrate that 15μM 8-Br-cGMP protected neurons against HNE-induced oxidative stress (protein-oxidation and lipid-peroxidation), indicative of a neuroprotective role of the PKG signaling cascade against HNE toxicity in cortical neurons.
Fig. 8.
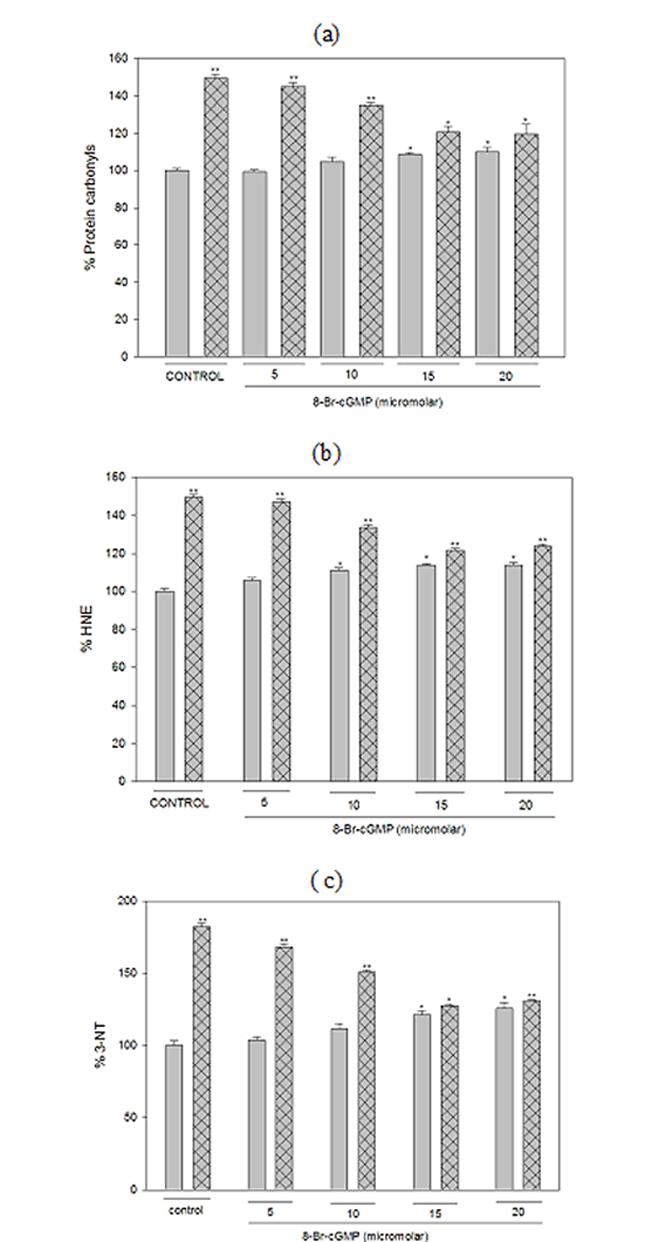
8-Br-cGMP mimics ALCAR and LA-mediated neuroprotective action. The treatments to the samples and method followed are same as described in “Results”. The grid bars are in presence of HNE and the plain bars are without HNE. The results are shown as mean ± SD of three independent measurements obtained for five independent preparations. Statistical comparison was by ANOVA (n =5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p<0.05.
Phosphorylation of ERK1/2
Active PKG may promote the phosphorylation of ERK1/2, thereby inducing the expression of antiapoptotic and antioxidative genes (47). Since ALCAR+LA-mediated neuroprotective effects are abrogated by PD98059, the ERK1/2 inhibitor (Fig. 7a. 7b, 7c), we therefore analyzed the phosphorylation status of ERK1/2. The results (Fig. 9) suggest that HNE reduced the cellular contents of phospho-ERK1/2 in cortical cultures; in contrast, pretreatment of neurons with 75 μM ALCAR+50 μM LA slightly enhanced ERK1/2 phosphorylation (Fig. 9). Treatment of 75μM ALCAR+50μM LA alone increased 2-fold ERK1/2 phosphorylation, compared to the untreated control cells (Fig. 9).
Fig. 9.
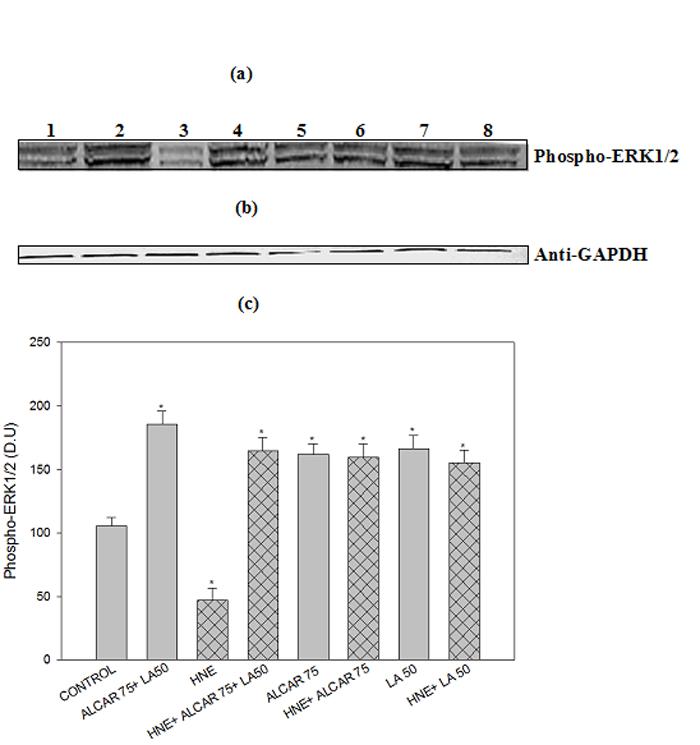
ALCAR and LA-mediated phosphorylation of ERK1/2. (a) Representative Western immunoblot analysis of neuronal cells for phospho-ERK1/2 protein levels. The grid bars are in presence of HNE and the plain bars are without HNE. Lane 1: control (untreated cells); Lane 2: 75μM ALCAR+50μM LA; Lane 3: 10μM HNE; Lane 4: 10 μM HNE+75μM ALCAR+50μM LA; Lane 5: 75μM ALCAR; Lane 6: 10μM HNE+75 μM ALCAR; Lane 7: 50 μM LA; Lane 8: 10μM HNE+50 μM LA (b) Anti-GAPDH blot as control for equal protein loading. Each experiment was repeated five times with independent samples.
ALCAR and LA-mediated PKG-dependent induction of anti-apoptotic and antioxidant proteins
Recently it has been shown that cGMP-dependent expression of thioredoxin plays a pivotal role in neuroprotection against oxidative stress (48). We therefore explored the possible contribution of thioredoxin in the observed ALCAR+LA -mediated neuroprotection against HNE toxicity. HNE significantly attenuated thioredoxin levels compared to the control (untreated cells), whereas treatment of ALCAR+LA alone enhanced thioredoxin levels (partially) compared to HNE treated neuronal cells (Fig. 10a). However, pretreatment of neurons with 75μM ALCAR+50μM LA or 75 μM ALCAR alone or 50 μM LA alone followed by addition of HNE enhanced thioredoxin protein levels (Fig. 10a), which was down-regulated by HNE, suggesting a neuroprotective effect of ALCAR+LA against HNE-induced down-regulation of thioredoxin (Fig. 10a).
Fig. 10.
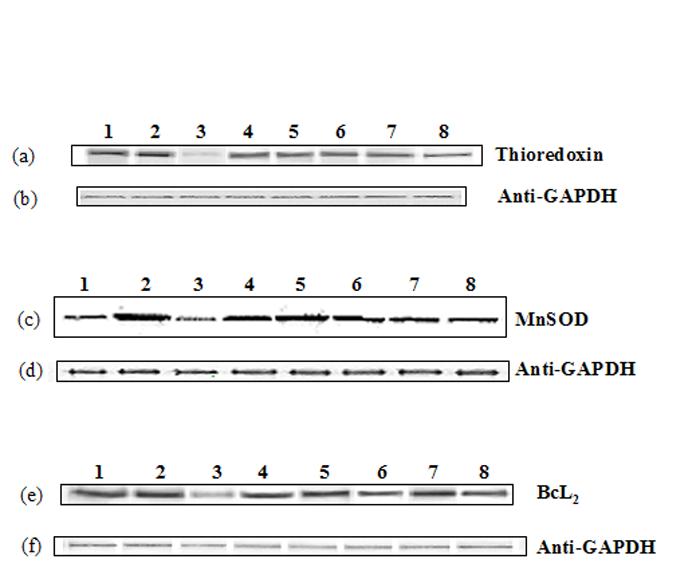
ALCAR and LA-mediated enhanced expression of thioredoxin, MnSOD and Bcl2. The treatments to the samples and method followed were the same as in Fig. 9. Each experiment was repeated five times with independent samples.
(a) Representative Western immunoblot analysis of neuronal cells for thioredoxin protein (probed with mouse polyclonal anti-thioredoxin antibody). (b) Anti-GAPDH blot of 10a (c) Representative western immunoblot analysis of neuronal cells for MnSOD protein (probed with mouse monoclonal anti-MnSOD antibody). (d) Anti-GAPDH blot of 10c. (e) Representative western immunoblot analysis of neuronal cells for BcL2 protein (probed with mouse polyclonal anti- BcL2 antibody. (f) Anti-GAPDH blot of 10e.
It has recently been demonstrated that cGMP and PKG are intermediate effectors for NO-dependent induction of the antiapoptotic Bcl2 protein in cerebellar granule neuronal cultures (49). Additionally, the NO-cGMP-PKG pathway mediated the preconditioning-induced upregulation of antiapoptotic protein Bcl2 and MnSOD in SHSY5Y neuroblastoma cells (50). We therefore examined the possible contribution of MnSOD and Bcl2 in the observed ALCAR+LA -mediated neuroprotection against HNE toxicity. The results suggest that ALCAR+LA enhanced MnSOD (Fig. 10c) and Bcl2 (Fig. 10e) expression that was down-regulated by HNE in cortical neurons.
PI3K/Akt pathway involvement in ALCAR+LA -mediated neuroprotection against HNE toxicity
Our data in this study also indicated that LY294002 (a PI3K inhibitor) abolished ALCAR+LA-mediated neuroprotective effects against HNE toxicity (Fig. 7a, 7b, 7c). PI3K-mediated activation of Akt leads to BAD phosphorylation (26). We therefore examined the effects of ALCAR+LA and HNE on the phosphorylation status of BAD protein. Western blot analysis using an antibody specifically recognizing phospho-BAD revealed that HNE decreased the extent of BAD-phosphorylation, which was reversed by ALCAR+LA pretreatment (Fig. 11a). Pretreatment of neurons with 75 μM ALCAR alone or 50 μM LA alone followed by the addition of HNE, partially reversed the effect of HNE (Fig. 11a).
Fig. 11.
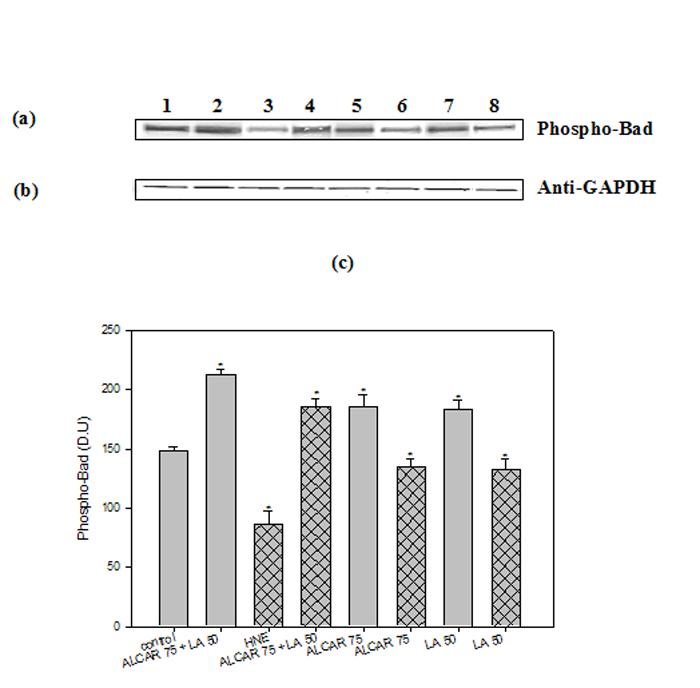
ALCAR and LA-mediated phosphorylation of Bad. The treatments to the samples and method followed were the same as in Fig. 9. Each experiment was repeated five times with independent samples. (a) Representative Western immunoblot analysis of neuronal cells for phospho-BAD (probed with mouse monoclonal anti-phospho-BAD antibody). (b) Anti-GAPDH blot as control for equal protein loading. (c) Densitometric analysis from five independent experiments (mean ±SD of values expressed as relative units). Significant differences were assessed by ANOVA. (n=5). Post hoc analysis was via Student-Newman-Keuls test, and the P values given are compared with the control. *p<0.005.
Discussion
We previously have shown that ALCAR protected neurons against Aβ (1-42)-induced oxidative stress, mitochondrial dysfunction and neurotoxicity, in primary cortical cultures (51). In the present study, we show that ALCAR+LA pretreatment, protected against HNE-induced oxidative stress (protein oxidation and lipid-peroxidation), mitochondrial dysfunction and neurotoxicity in cortical cultures. Furthermore, HNE-mediated neuronal death can be antagonized effectively by application of ALCAR+LA, via activation of multiple pro-survival signaling pathways including PI3K/Akt and cGMP/PKG.
LA functions as an antioxidant and reduces oxidative stress in aged animals (16) and its reduced form, dihydrolipoic acid (DHLA) protects neuronal cultures from Aβ (25-35) or iron/hydrogen peroxide-induced oxidative stress (17). ALCAR is the L-carnitine ester of acetic acid and is synthesized in the brain, kidney and liver by the enzyme acetyl carnitine transferase. ALCAR is suggested to enhance learning capacity in aging animals (52) and acts a source of acetyl groups available for synthesis for acetylcholine (53). ALCAR crosses the blood-brain barrier, improves neuronal energetic and repair mechanism, decreases the level of lipid peroxidation in the aged rat brain (54), is involved in mitochondrial metabolism (15) and may have anti-oxidant properties (54,55). However, the precise mechanism of action by which ALCAR may be neuroprotective in aging and neurodegeneration, is not known. HNE is an aldehydic molecule generated endogenously during the process of lipid peroxidation and is causally involved in many of the pathophysiological effects associated with oxidative stress in cells and tissues (56). HNE is believed to be largely responsible for cytopathological effects observed during oxidative stress in vivo and is one of the most studied cytotoxic products of lipid peroxidation (56). As noted, HNE-adducts are been reported in AD brain (7,8,57).
The exact role of HNE in neurodegenerative disorders has yet to be established, but it is acknowledged that HNE is a highly toxic compound capable of causing neuronal cell death (58). We postulated that pretreatment of ALCAR+LA might protect neurons against HNE-mediated oxidative stress. Pretreatment of ALCAR+LA partially reduced the neurotoxic effects of HNE, with the maximum significant protective dosage at ALCAR (75μM) +LA (50μM) (Fig. 1). Consistent with this result, pretreatment of ALCAR (75μM) +LA (50μM) also reduced cell death in cortical cultures caused by HNE (Fig. 1). It was shown that Aβ peptide induces apoptosis in mouse neuronal cultures (59). Earlier studies reported (60) evidence for DNA fragmentation in neurons from subjects with AD. Mitochondria are particularly vulnerable to oxidative damage and mitochondrial dysfunction has been observed in AD brain (61). In the present study, we studied the cytotoxic effects (by MTT assay) and morphological changes by examining the dynamic neuronal-network processing in the HNE or ALCAR+LA treated cortical neurons. The results suggest that there is an extensive loss of neuronal connections as well as the presence of dying cells in the HNE treated cells (Fig. 2a, 2b, 2c). Pretreatment of neurons with ALCAR (75μM) +LA (50μM) alone prevented the change in neuronal morphology, resulting in morphology almost similar to that of control cells (without treatment) consistent with a protective role for ALCAR and LA.
To further confirm our results, Hoechst or PI staining were used to detect the presence of apoptotic bodies as an indication of apoptotic cells, and pretreatment of ALCAR (75μM) +LA (50μM) showed an anti-apoptotic effect on HNE-treated neuronal cells by reducing the formation of apoptotic bodies. ALCAR (1 – 100μM) promoted neuronal survival and mitochondrial activity in a concentration-dependent manner attenuating DNA fragmentation and nuclear condensation in cultured neurons (36). Prolonged pretreatment with LA protects cultured neurons against hypoxic, glutamate, or iron or Aβ-induced injury (17,62). Our results are consonant with the above studies that suggest the antiapoptotic action of ALCAR and LA contributes to their neuroprotective effect.
HNE treatment of the cells (rat liver epithelial RL34) resulted in depletion of intracellular glutathione (GSH) and in the formation of protein-bound HNE in plasma membrane. HNE normally is detoxified by conjugation with GSH (63). Aβ (1-42) has been shown to deplete GSH levels in astrocytes (64), leaving neurons vulnerable to ROS attack (65) leading to a significant loss of neurons in vitro. In the present study, HNE significantly depleted the cellular GSH level in the cortical neuronal cells. Moreover, treatment of neurons with 75 μM ALCAR+50 μM ALCAR alone was shown to elevate GSH levels (Fig. 3) 24 h after administration. Upon pretreatment with 75 μM ALCAR+50 μM ALCAR followed by 10 μM HNE, GSH levels ameliorated compared to HNE treated cells. As such, increasing GSH levels in the mitochondria may prove to be an important therapeutic approach to prevent cell death in oxidative stress-linked, age-dependent neurodegenerative disorders (66). Taken together, these results suggest ALCAR+LA induces up-regulation of GSH, which, in turn, protects neurons against protein oxidation and neurotoxicity.
Increasing evidence now underscores the role of heme-oxygenase (HO) in cell protection against oxidative stress, such as free radicals and certain degenerative diseases, as well as the ageing process in which enhanced HO activity seems to have an antioxidant effect (21). A number of in vitro studies show that both heat shock and Hsp overproduction protect CNS cells against glutamate-mediated toxicity or lethal acidosis (67,68). In addition, Hsp70 has been demonstrated to inhibit caspase-3 activation caused by ceramide (69). This insight has opened new perspectives in medicine and pharmacology, as possible candidates for novel cytoprotective strategies (21,25,70,71). In this study, we tested the hypothesis that ALCAR+LA protects primary neuronal cell cultures against HNE-mediated oxidative stress and neurotoxicity by up- regulating Hsp's (Fig. 4a, 4c, 5a, 5c). Our results also suggested that inhibition of HO-1, Hsp72, or iNOS proteins by the inhibitors ZnPP IX, quercitin, or L-NMMA, respectively, abrogated the protective effect of 75 μM ALCAR+50 μM LA (data not shown), demonstrating that HO-1 or Hsp72 or iNOS are likely involved in the ALCAR+LA-mediated cytoprotection against HNE-induced oxidative stress. It has been recently demonstrated that LA induces HO-1 expression in THP-1 monocytic cells via Nrf2 and p38 (72). Our results are consonant with the recent studies, which shows that ALCAR is cytoprotective against inflammatory and oxidative insults in astrocytes in part by being able to up-regulate cytoprotective cellular stress responses, particularly induction of HO-1, while inhibiting induction of iNOS (73).
ALCAR+LA -mediated attenuation of HNE toxicity, as shown in the present study, appears to involve several signaling pathways including PKG, ERK1/2 and PI3K. ERK is implicated in neuronal cell survival and many studies have confirmed its contribution to neuronal cell survival (74). ERK1/2 is an endogenous negative regulator of gamma-secretase activity (75). The mechanisms that underlie such varied effects of ERK are unclear but could be based on differences in both the temporal and spatial pattern of ERK activation induced by the various treatments (76). Our results on PKG-mediated elevated levels of phospho-ERK1/2 by ALCAR+LA (Fig. 9) are consistent with the studies that demonstrate active PKG promotes the elevation of phosphorylated ERK1/2 (47). Activation of PKG pathway results in the induction of antioxidative and antiapoptotic proteins (47). Recently, cGMP-dependent expression of thioredoxin, the redox protein with potent antioxidative properties, has been shown to play a pivotal role in neuroprotection against oxidative stress mediated by estrogen (48). Interestingly, a decrease in thioredoxin protein level was observed in AD brains as compared to normal controls (77). Since thioredoxin-II resides in mitochondria, the results in Fig. 10(a) may be related to the mitochondrial protection induced by ALCAR and LA (Fig. 1). A brief preconditioning stress by serum deprivation for 2 h also induced neuronal nitric oxide synthase (nNOS) that was accompanied by up-regulation of Bcl2 via activation of PKG (47). In addition to Bcl2, cGMP-dependent expression of thioredoxin was also involved in estrogen-induced neuroprotection against oxidative stress caused by serum deprivation (48). We therefore investigated the possible contribution of thioredoxin, MnSOD and Bcl2 in the observed ALCAR+LA-mediated protection against HNE toxicity. Our results are in agreement with the above studies, as thioredoxin, MnSOD and Bcl2 protein levels were down-regulated by HNE (Fig. 10a, 10c, 10e), but pretreatment of neurons with 75 μM ALCAR+50 μM LA enhanced thioredoxin, MnSOD and Bcl2 levels (Fig. 10a, 10c, 10e).
HNE may affect cellular NO levels by causing impairment in activity of constitutive NOS upon HNE treatment, thereby modulating NO-mediated intrinsic neuroprotective mechanisms (78). Pharmacological blockade of NO production by a nNOS inhibitor down-regulated the Akt pathway, causing apoptosis in cerebellar granule cell cultures (49). The PI3K/Akt pathway was identified as another potentially important signaling pathway downstream of ALCAR+LA in protecting cortical cultures against HNE-induced oxidative stress and neurotoxicity. Alterations in the PI3K/Akt/BAD phosphorylation pathway have recently been implicated in AD pathogenesis. BAD phosphorylation occurs secondary to PI3K-mediated activation of Akt (26). Akt is activated in response to Aβ25-35 in a PI3K-dependent manner and protects PC12 cells against apoptosis (79). Additionally, down-regulation of Akt also paralleled intracellular Abeta accumulation in vivo in the Tg2576 AD mouse model, and overexpression of constitutively active Akt reversed the toxic effects of Abeta through a mechanism involving the induction of Hsp70 (80). Since Aβ(1-42) leads to HNE formation (8,35), HNE might initiate a cascade of biochemical alterations in primary cortical neurons, including dephosphorylation, and hence inactivation of Akt followed by dephosphorylation of BAD, mitochondrial depolarization and permeabilization leading to neurotoxicity and cell death. In our studies, ALCAR+LA enhanced BAD phosphorylation that was attenuated by HNE (Fig.11). These observations are consistent with the notion that ALCAR+LA is capable of modulating HNE-mediated down-regulation of the PI3K/Akt/Bad phosphorylation, thereby exerting neuroprotective action.
NO inhibits apoptosis through its ability to bind to guanylate cyclase, which in turn activates PKG. Together, these results support the notion that ALCAR+LA may cause PKG activation, ERK1/2 phosphorylation, induction of antiapoptotic and antioxidative genes, and up-regulation of PI3K pathway-mediated enhanced BAD phosphorylation.
In conclusion, we have demonstrated that ALCAR+LA protected rat cortical neurons from HNE-mediated oxidative stress and neurotoxicity. We further identified several signal pathways that may play pivotal roles in mediating ALCAR+LA neuroprotective action, which include, PKG activation, ERK1/2 phosphorylation, induction of thioredoxin/MnSOD/Bcl2, and PI3K-induced BAD phosphorylation and up-regulation of HO-1/Hsp 70 and down-regulating iNOS (Fig. 12). Activation of these signaling pathways by ALCAR+LA protects cortical neurons from HNE-mediated oxidative stress and neurotoxicity. ALCAR+LA-mediated induction of cytoprotective and antiapoptotic proteins may provide a new combination therapeutic approach for protection of neurons against oxidative stress in AD and other oxidative stress-related neurodegenerative disorders.
Fig. 12.

Schematic representation of mechanisms of action of ALCAR+LA-mediated neuroprotection against HNE-induced oxidative stress and neurotoxicity.
Acknowledgements
We thank Dr. Edgardo Dimayuga for technical advice and for providing some materials used in this study. We thank Sigma-Tau Pharma for the generous gift of ALCAR. This work was supported in part by grants from NIH [AG-05119; AG-10836].
Abbreviations
- PI3K
phosphatidyl inositol 3-kinase
- iNOS
inducible nitric oxide synthase
- 3-NT
3-nitrotyrosine
- Q
quercitin
- ZnPP
Zn protoporphyrin IX
- L-NMMA
N (G)-monomethyl-l-arginine
Footnotes
Address correspondence to: Professor D. Allan Butterfield, Dept. of Chemistry, Center of Membrane Sciences, and Sanders-Brown Center on Aging, University of Kentucky, Lexington KY 40506 Tel: 859 257-3184; Fax 859-257-5876; E-mail: dabcns@uky.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Butterfield DA, Boyd-Kimball D, Castegna A. Proteomics in Alzheimer's disease: insights into potential mechanisms of neurodegeneration. J Neurochem. 2003;86:1313–1327. doi: 10.1046/j.1471-4159.2003.01948.x. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 3.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 4.Katzman R, Saitoh T. Advances in Alzheimer's disease. Faseb J. 1991;5:278–286. [PubMed] [Google Scholar]
- 5.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 6.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 7.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 8.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 9.Shimohama S. Apoptosis in Alzheimer's disease--an update. Apoptosis. 2000;5:9–16. doi: 10.1023/a:1009625323388. [DOI] [PubMed] [Google Scholar]
- 10.Kagan VE, Fabisiak JP, Shvedova AA, Tyurina YY, Tyurin VA, Schor NF, Kawai K. Oxidative signaling pathway for externalization of plasma membrane phosphatidylserine during apoptosis. FEBS Lett. 2000;477:1–7. doi: 10.1016/s0014-5793(00)01707-5. [DOI] [PubMed] [Google Scholar]
- 11.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 12.Aluigi MG, De Flora S, D'Agostini F, Albini A, Fassina G. Antiapoptotic and antigenotoxic effects of N-acetylcysteine in human cells of endothelial origin. Anticancer Res. 2000;20:3183–3187. [PubMed] [Google Scholar]
- 13.Park SA, Choi KS, Bang JH, Huh K, Kim SU. Cisplatin-induced apoptotic cell death in mouse hybrid neurons is blocked by antioxidants through suppression of cisplatin-mediated accumulation of p53 but not of Fas/Fas ligand. J Neurochem. 2000;75:946–953. doi: 10.1046/j.1471-4159.2000.0750946.x. [DOI] [PubMed] [Google Scholar]
- 14.Dolezal V, Tucek S. Utilization of citrate, acetylcarnitine, acetate, pyruvate and glucose for the synthesis of acetylcholine in rat brain slices. J Neurochem. 1981;36:1323–1330. doi: 10.1111/j.1471-4159.1981.tb00569.x. [DOI] [PubMed] [Google Scholar]
- 15.Hagen TM, Ingersoll RT, Wehr CM, Lykkesfeldt J, Vinarsky V, Bartholomew JC, Song MH, Ames BN. Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci U S A. 1998;95:9562–9566. doi: 10.1073/pnas.95.16.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poon HF, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Proteomic analysis of specific brain proteins in aged SAMP8 mice treated with alpha-lipoic acid: implications for aging and age-related neurodegenerative disorders. Neurochem Int. 2005;46:159–168. doi: 10.1016/j.neuint.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Lovell MA, Xie C, Xiong S, Markesbery WR. Protection against amyloid beta peptide and iron/hydrogen peroxide toxicity by alpha lipoic acid. J Alzheimers Dis. 2003;5:229–239. doi: 10.3233/jad-2003-5306. [DOI] [PubMed] [Google Scholar]
- 18.Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol. 1999;56:1449–1452. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 19.Santoro MG. Heat shock factors and the control of the stress response. Biochem Pharmacol. 2000;59:55–63. doi: 10.1016/s0006-2952(99)00299-3. [DOI] [PubMed] [Google Scholar]
- 20.Kravets A, Hu Z, Miralem T, Torno MD, Maines MD. Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J Biol Chem. 2004;279:19916–19923. doi: 10.1074/jbc.M314251200. [DOI] [PubMed] [Google Scholar]
- 21.Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci. 2004;59:478–493. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- 22.Takeda A, Perry G, Abraham NG, Dwyer BE, Kutty RK, Laitinen JT, Petersen RB, Smith MA. Overexpression of heme oxygenase in neuronal cells, the possible interaction with Tau. J Biol Chem. 2000;275:5395–5399. doi: 10.1074/jbc.275.8.5395. [DOI] [PubMed] [Google Scholar]
- 23.Abraham NG, Jiang S, Yang L, Zand BA, Laniado-Schwartzman M, Marji J, Drummond GS, Kappas A. Adenoviral vector-mediated transfer of human heme oxygenase in rats decreases renal heme-dependent arachidonic acid epoxygenase activity. J Pharmacol Exp Ther. 2000;293:494–500. [PubMed] [Google Scholar]
- 24.Calabrese V, Scapagnini G, Colombrita C, Ravagna A, Pennisi G, Giuffrida Stella AM, Galli F, Butterfield DA. Redox regulation of heat shock protein expression in aging and neurodegenerative disorders associated with oxidative stress: a nutritional approach. Amino Acids. 2003;25:437–444. doi: 10.1007/s00726-003-0048-2. [DOI] [PubMed] [Google Scholar]
- 25.Calabrese V, Renis M, Calderone A, Russo A, Reale S, Barcellona ML, Rizza V. Stress proteins and SH-groups in oxidant-induced cellular injury after chronic ethanol administration in rat. Free Radic Biol Med. 1998;24:1159–1167. doi: 10.1016/s0891-5849(97)00441-3. [DOI] [PubMed] [Google Scholar]
- 26.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimoto T, Uchino H, He QP, Li PA, Siesjo BK. Cyclosporin A, but not FK506, prevents the downregulation of phosphorylated Akt after transient focal ischemia in the rat. Brain Res. 2001;899:148–158. doi: 10.1016/s0006-8993(01)02220-x. [DOI] [PubMed] [Google Scholar]
- 28.Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 29.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 30.Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 31.Perkinton MS, Sihra TS, Williams RJ. Ca(2+)-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci. 1999;19:5861–5874. doi: 10.1523/JNEUROSCI.19-14-05861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- 33.Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta- peptide(1-42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem. 2005;92:749–758. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- 34.Yatin SM, Yatin M, Aulick T, Ain KB, Butterfield DA. Alzheimer's amyloid beta-peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: protective effect of vitamin E. Neurosci Lett. 1999;263:17–20. doi: 10.1016/s0304-3940(99)00101-9. [DOI] [PubMed] [Google Scholar]
- 35.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 36.Ishii T, Shimpo Y, Matsuoka Y, Kinoshita K. Anti-apoptotic effect of acetyl-l-carnitine and I-carnitine in primary cultured neurons. Jpn J Pharmacol. 2000;83:119–124. doi: 10.1254/jjp.83.119. [DOI] [PubMed] [Google Scholar]
- 37.Vincent AM, McLean LL, Backus C, Feldman EL. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. Faseb J. 2005;19:638–640. doi: 10.1096/fj.04-2513fje. [DOI] [PubMed] [Google Scholar]
- 38.Darzynkiewicz Z, Li X, Gong J. Assays of cell viability: discrimination of cells dying by apoptosis. Methods Cell Biol. 1994;41:15–38. doi: 10.1016/s0091-679x(08)61707-0. [DOI] [PubMed] [Google Scholar]
- 39.Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 40.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 41.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 42.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 43.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 44.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 46.Fiscus RR. Involvement of cyclic GMP and protein kinase G in the regulation of apoptosis and survival in neural cells. Neurosignals. 2002;11:175–190. doi: 10.1159/000065431. [DOI] [PubMed] [Google Scholar]
- 47.Andoh T, Chiueh CC, Chock PB. Cyclic GMP-dependent protein kinase regulates the expression of thioredoxin and thioredoxin peroxidase-1 during hormesis in response to oxidative stress-induced apoptosis. J Biol Chem. 2003;278:885–890. doi: 10.1074/jbc.M209914200. [DOI] [PubMed] [Google Scholar]
- 48.Lee SY, Andoh T, Murphy DL, Chiueh CC. 17beta-estradiol activates ICI 182,780-sensitive estrogen receptors and cyclic GMP-dependent thioredoxin expression for neuroprotection. Faseb J. 2003;17:947–948. doi: 10.1096/fj.02-0807fje. [DOI] [PubMed] [Google Scholar]
- 49.Ciani E, Guidi S, Bartesaghi R, Contestabile A. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem. 2002;82:1282–1289. doi: 10.1046/j.1471-4159.2002.01080.x. [DOI] [PubMed] [Google Scholar]
- 50.Andoh T, Chock PB, Chiueh CC. Preconditioning-mediated neuroprotection: role of nitric oxide, cGMP, and new protein expression. Ann N Y Acad Sci. 2002;962:1–7. doi: 10.1111/j.1749-6632.2002.tb04051.x. [DOI] [PubMed] [Google Scholar]
- 51.Abdul HM, Calabrese V, Calvani M, Butterfield DA. Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1-42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer's disease. J Neurosci Res. 2006 doi: 10.1002/jnr.20877. [DOI] [PubMed] [Google Scholar]
- 52.Ando S, Tadenuma T, Tanaka Y, Fukui F, Kobayashi S, Ohashi Y, Kawabata T. Enhancement of learning capacity and cholinergic synaptic function by carnitine in aging rats. J Neurosci Res. 2001;66:266–271. doi: 10.1002/jnr.1220. [DOI] [PubMed] [Google Scholar]
- 53.Kuratsune H, Watanabe Y, Yamaguti K, Jacobsson G, Takahashi M, Machii T, Onoe H, Onoe K, Matsumura K, Valind S, Kitani T, Langstrom B. High uptake of [2-11C]acetyl-L-carnitine into the brain: a PET study. Biochem Biophys Res Commun. 1997;231:488–493. doi: 10.1006/bbrc.1996.5919. [DOI] [PubMed] [Google Scholar]
- 54.Kaur J, Sharma D, Singh R. Acetyl-L-carnitine enhances Na(+), K(+)-ATPase glutathione-S-transferase and multiple unit activity and reduces lipid peroxidation and lipofuscin concentration in aged rat brain regions. Neurosci Lett. 2001;301:1–4. doi: 10.1016/s0304-3940(01)01576-2. [DOI] [PubMed] [Google Scholar]
- 55.Poon HF, Calabrese V, Calvani M, Butterfield DA. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid Redox Signal. 2006;8:381–394. doi: 10.1089/ars.2006.8.381. [DOI] [PubMed] [Google Scholar]
- 56.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 57.Calingasan NY, Uchida K, Gibson GE. Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer's disease. J Neurochem. 1999;72:751–756. doi: 10.1046/j.1471-4159.1999.0720751.x. [DOI] [PubMed] [Google Scholar]
- 58.Shibata E, Ejima K, Nanri H, Toki N, Koyama C, Ikeda M, Kashimura M. Enhanced protein levels of protein thiol/disulphide oxidoreductases in placentae from pre-eclamptic subjects. Placenta. 2001;22:566–572. doi: 10.1053/plac.2001.0693. [DOI] [PubMed] [Google Scholar]
- 59.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U S A. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer's disease. Neuroreport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 61.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muller U, Krieglstein J. Prolonged pretreatment with alpha-lipoic acid protects cultured neurons against hypoxic, glutamate-, or iron-induced injury. J Cereb Blood Flow Metab. 1995;15:624–630. doi: 10.1038/jcbfm.1995.77. [DOI] [PubMed] [Google Scholar]
- 63.Ullrich O, Grune T, Henke W, Esterbauer H, Siems WG. Identification of metabolic pathways of the lipid peroxidation product 4-hydroxynonenal by mitochondria isolated from rat kidney cortex. FEBS Lett. 1994;352:84–86. doi: 10.1016/0014-5793(94)00922-8. [DOI] [PubMed] [Google Scholar]
- 64.Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci. 2003;23:5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu R, Choi J. Age-associated decline in gamma-glutamylcysteine synthetase gene expression in rats. Free Radic Biol Med. 2000;28:566–574. doi: 10.1016/s0891-5849(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 66.Gegg ME, Beltran B, Salas-Pino S, Bolanos JP, Clark JB, Moncada S, Heales SJ. Differential effect of nitric oxide on glutathione metabolism and mitochondrial function in astrocytes and neurones: implications for neuroprotection/neurodegeneration? J Neurochem. 2003;86:228–237. doi: 10.1046/j.1471-4159.2003.01821.x. [DOI] [PubMed] [Google Scholar]
- 67.Narasimhan P, Swanson RA, Sagar SM, Sharp FR. Astrocyte survival and HSP70 heat shock protein induction following heat shock and acidosis. Glia. 1996;17:147–159. doi: 10.1002/(SICI)1098-1136(199606)17:2<147::AID-GLIA6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 68.Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK. The neuroprotective potential of heat shock protein 70 (HSP70) Mol Med Today. 1999;5:525–531. doi: 10.1016/s1357-4310(99)01599-3. [DOI] [PubMed] [Google Scholar]
- 69.Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol. 1997;17:5317–5327. doi: 10.1128/mcb.17.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calabrese V, Scapagnini G, Catalano C, Bates TE, Dinotta F, Micali G, Giuffrida Stella AM. Induction of heat shock protein synthesis in human skin fibroblasts in response to oxidative stress: regulation by a natural antioxidant from rosemary extract. Int J Tissue React. 2001;23:51–58. [PubMed] [Google Scholar]
- 71.Ropeleski MJ, Tang J, Walsh-Reitz MM, Musch MW, Chang EB. Interleukin-11-induced heat shock protein 25 confers intestinal epithelial-specific cytoprotection from oxidant stress. Gastroenterology. 2003;124:1358–1368. doi: 10.1016/s0016-5085(03)00282-8. [DOI] [PubMed] [Google Scholar]
- 72.Ogborne RM, Rushworth SA, O'Connell MA. Alpha-lipoic acid-induced heme oxygenase-1 expression is mediated by nuclear factor erythroid 2-related factor 2 and p38 mitogen-activated protein kinase in human monocytic cells. Arterioscler Thromb Vasc Biol. 2005;25:2100–2105. doi: 10.1161/01.ATV.0000183745.37161.6e. [DOI] [PubMed] [Google Scholar]
- 73.Calabrese V, Ravagna A, Colombrita C, Scapagnini G, Guagliano E, Calvani M, Butterfield DA, Giuffrida Stella AM. Acetylcarnitine induces heme oxygenase in rat astrocytes and protects against oxidative stress: involvement of the transcription factor Nrf2. J Neurosci Res. 2005;79:509–521. doi: 10.1002/jnr.20386. [DOI] [PubMed] [Google Scholar]
- 74.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim SK, Park HJ, Hong HS, Baik EJ, Jung MW, Mook-Jung I. ERK1/2 is an endogenous negative regulator of the gamma-secretase activity. Faseb J. 2006;20:157–159. doi: 10.1096/fj.05-4055fje. [DOI] [PubMed] [Google Scholar]
- 76.Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- 77.Lovell MA, Xie C, Gabbita SP, Markesbery WR. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer's disease brain. Free Radic Biol Med. 2000;28:418–427. doi: 10.1016/s0891-5849(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 78.Venturini G, Colasanti M, Persichini T, Fioravanti E, Ascenzi P, Palomba L, Cantoni O, Musci G. Beta-amyloid inhibits NOS activity by subtracting NADPH availability. Faseb J. 2002;16:1970–1972. doi: 10.1096/fj.02-0186fje. [DOI] [PubMed] [Google Scholar]
- 79.Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78:1000–1008. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 80.Magrane J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci. 2005;25:10960–10969. doi: 10.1523/JNEUROSCI.1723-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]