

Abstract
The concept that during an immune challenge the release of glucocorticoids (GC) provides feedback inhibition on evolving immune responses has been drawn primarily from studies of autoimmune and/or inflammatory processes in animal models. The epidemic form of haemolytic uraemic syndrome (HUS) occurs secondary to infection with Gram-negative bacteria that produce Shiga toxin (Stx). Although Stx binding to the specific receptors present on renal tissue is the primary pathogenic mechanism, inflammatory or immune interactions are necessary for the development of the complete form of HUS. The aim of this study was to investigate the influence of endogenous GC on Stx-toxicity in a mouse model. Stx2 was injected into GC-deprived mice and survival rate, renal damage and serum urea levels were evaluated. Plasma corticosterone and cytosolic GC receptor (GR) concentration were also determined at multiple intervals post-Stx2 treatment. Higher sensitivity to Stx2 was observed in mice lacking endogenous GC, evidenced by an increase in mortality rates, circulating urea levels and renal histological damage. Moreover, Stx2 injection was associated with a transient but significant rise in corticosterone secretion. Interestingly, 24 h after Stx inoculation significant increases in total GR were detected in circulating neutrophils. These results indicate that interactions between the neuroendocrine and immune systems can modulate the level of damage significantly during a bacterial infection.
Keywords: glucocorticoid receptors, glucocorticoids, HUS, neutrophils, Stx2
Introduction
Haemolytic uraemic syndrome (HUS) is characterized by the triad of microangiopathic haemolytic anaemia, thrombocytopenia and nephropathy. Typically, HUS develops in young children as a vascular disease several days after the occurrence of diarrhoea and bloody gastroenteritis [1]. It is caused by Gram-negative bacteria such as Shigella dysenteriae 1 [2] or by particular serotypes of Escherichia coli which produce significant amounts of Shiga toxins (Stx) [3,4], also referred to as verotoxins or Shiga-like toxins. Two antigenically different Stx types, Stx1 or Stx2, are the primary pathogenic factors [5,6]. However, epidemiological and experimental studies have suggested that Stx2 is clinically more significant than Stx1 [6,7]. Stx exert their cytotoxic effects by binding to their specific cell receptor, a neutral glycolipid known as globotriaosylceramide (Gb3) [5], present on renal epithelial, endothelial and glomerular mesangial cells [6–8]. However, compelling evidence has indicated that inhibition of protein synthesis after Stx-receptor interaction is not sufficient to induce tissue injury and that other pathogenic factors are necessary to develop HUS. Several animal models have been used to study the contribution of the inflammatory response to Stx pathogenicity [9–12]. These results, together with clinical evidence, have suggested that inflammatory cytokines [13–15] and neutrophils play a central role in the development of HUS [9, 16, 17]. In contrast, little is known about the physiological factors that can protect against Stx toxicity.
It is well known that the hypothalamo–pituitary–adrenal axis (HPA) is activated during infectious diseases and after the injection of an inflammatory agent [18–21]. The concept that glucocorticoids (GC) released under physiological conditions contribute to an endogenous inflammatory control system is now well accepted. For example, the limitation of severity and duration of inflammatory disease models, including adjuvant arthritis, and experimental allergic encephalomyelitis (EAE) has been demonstrated to be dependent on an adequate physiological glucocorticoid response [22,23]. Moreover, the protective role of endogenous corticosteroids during streptococcal cell wall-induced arthritis in Lewis rats and during sepsis has been well evidenced [24,25]. However, their role during the development of HUS has not been investigated previously.
The aim of this study was to analyse in vivo the protective role of endogenous glucocorticoids on Stx type 2 (Stx2) toxicity.
Materials And Methods
Antibodies and reagents
Fluorescein isothiocyanate (FITC)-conjugated mouse monoclonal antibody (MoAb) 5E4 (IgG1) against GC-receptor (GR) was produced as described previously [26]. FITC-conjugated isotype controls of mouse IgG1 were obtained from Immunotech (Marseille, France). Fix & Perm Cell Permeabilization Kit were obtained from Caltag Laboratories (Burlingame, CA, USA).
Mice
BALB/c mice were bred in the animal facility of the Department of Experimental Medicine, Academia Nacional de Medicina, Buenos Aires. Male mice aged 9–16 weeks and weighing 20–25 g were used throughout the experiments. They were maintained under a 12-h light–dark cycle at 22 ± 2°C and fed with standard diet and water ad libitum. The experiments performed herein were conducted according to principles set forth in the Guide for the Care and Use of Laboratory Animals (National Institute of Health, 1985).
Stx2 preparation
Stx2 was kindly provided by Dr Sugiyama Junichi from Denka Seiken Co. Ltd. (Nigata, Japan). Purity was analysed by the supplier, which showed only one peak in HPLC. Stx2 preparation was checked for endotoxin contamination by the Limulus amoebocyte lysate assay, given that 1 IU/ml is equal to 0·1 ng/ml of United States Pharmacopea standard E. coli endotoxin [27]. Stx2 preparation contained less than 40 pg LPS/µg of Shiga toxin protein. Stx2 was tested for cytotoxic activity on Vero cells, as described previously [4] in the Instituto Nacional de Enfermedades Infecciosas, ANLIS, Dr C. G. Malbran (Buenos Aires, Argentina). Briefly, Vero cells were grown in Eagle's minimum essential medium with Earle salts and non-essential amino acids (Gibco Diagnostics, Madison, WI, USA) supplemented with 7% fetal calf serum (Sigma Chemical Co., St Louis, MO, USA), 0·03 m glutamine, 50 µg/ml gentamicin and 2·5 µg/ml fungizone in microtitre plates (Nunc, Intermed, Roskilde, Denmark). Aliquots (50 µl) of serial twofold dilutions of the samples containing Stx2 were added to each well (25 000 Vero cells) and incubated for 3 days at 37°C in 5% CO2. Cells were washed with normal saline, stained with crystal violet dye for 30 min at 37°C, and after washing thoroughly remaining cell-stain was solved with a 30% acetic acid solution and read on a Microwell System Reader 230S (Organon, Teknika, USA) with a 550-nm filter. The 50% cytotoxic dose (CD50) corresponded to the dilution required to kill 50% of the Vero cells: CD50 was ∼ 0·063 pg.
Stx2 treatment
The same batch of Stx2 preparation was used throughout the experiments. In a previous manuscript [15], in vivo lethality of Stx2 was evaluated by i.v. injection in the retro-orbital plexus of serial dilutions in pyrogen-free saline, selecting a dose of 12·5 ng/kg (approx. 250 pg/mouse), which induced a mortality of ≈ 50% between 3 and 4 days after injection.
Surgical procedures
Bilateral adrenalectomy (surgical removal of adrenal gland, ADX) and sham (SHAM) operations were performed between 0900 and 1100 h as described previously [21]. Before surgery, mice were placed in a clean cage for at least 30 min and then transferred to the operation room. Surgical recovery lasted a total of 5 days and ADX mice received 0·9% saline as drinking solution.
In vivo treatment with dexamethasone or the antagonist of type II steroid receptor Ru486
Mice were injected daily i.p. with 600 µg/mice of the glucocorticoid receptor antagonist Ru486[17-hydroxy-11-(4-dimethylaminophenyl) 17-(1-propynyl) estra-4,9-diene-3-one] (Sigma) starting 12 h before Stx2 until the end of the experiment. This dose has been proved to block GC action completely [28]. Dexamethasone (DEX) administration to ADX mice was performed by adding 300 µg/ml (Sigma, St Louis, MO, USA) to the drinking water starting 6 h before Stx2 injection until the end of the experiment. Appropriate amounts of stock DEX (10 mg/ml) in 100% ethanol were added to the drinking water. The final concentration of ethanol in the drinking water of all mice was standardized at 0·078%w/v. When normal mice were treated with DEX, doses of 2·5 mg/kg were i.v. injected into the retro-orbital plexus as described previously [29].
Neutrophil purification and flow cytometry
Neutrophils from peripheral blood of individual mice were obtained by layering a Ficoll-Hypaque density gradient, followed by dextran sedimentation as previously described [30]. Intracellular staining of the GR in murine isolated neutrophils was performed on ice, using the specific mouse monoclonal antibody 5E4 [26] following the instructions of Fix & Perm Cell Permeabilization Kit. Briefly, cells were fixed in suspension, washed and then permeabilized in the presence of specific conjugated antibody. The non-specific binding sites were blocked with 10% normal mouse serum containing permeabilization buffer. This procedure gives antibodies access to intracellular structures and leaves the morphological scatter characteristics of the cells intact [31]. Control of isotype-matched antibody was assayed in parallel. Then, cells were washed with cold PBS supplemented with 1% FCS and suspended in 0·3 ml of ISOFLOW. Fluorescence was measured with a Becton Dickinson FACScan equipment. Forward and side scatter gating excluded dead cells. Separate gates were set on neutrophils and mononuclear cells. The analysis was carried out on 20 000 events on each sample by using the Cell Quest program. The anti-GR-FITC staining was always compared (by overlaying the histogram plots) to the autofluorescent and isotype-FITC control samples.
Corticosterone assays
To minimize stress induced by handling, the day before the experiments animals were housed one per cage. Mice were injected intravenously with saline and Stx2, and intraperitoneally (i.p.) with LPS. Blood samples were collected at different times post-treatment in another room; controls were always killed last in the sequence. Blood samples were obtained by puncture of the retro-orbital plexus and collected in heparin plastic centrifuge tubes. Plasma was immediately separated and stored at −20°C until assayed. Plasma corticosterone concentration was determined by radioimmunoassay (RIA) using the commercial 125I-RIA Kit ImmunoChem Double antibody (ICN Biomedicals, Costa Mesa, CA, USA). The detection limit was 25 ng/ml. Intra- and interassay coefficients of variation were 4% and 7%, respectively.
Histological studies
For histological studies both kidneys from each mouse were obtained 72 h post-Stx2 or saline injection. Kidneys were bisected longitudinally, fixed in 10% neutral formalin and embedded in paraffin. Then, the tissue were stained with haematoxilin and eosin (H&E) or periodic acid Schiff (PAS) and examined by light microscopy. A mean number of five sections of both kidneys from each mouse were examined for glomerular and tubular histological appearance and epithelial damage.
Serum determinations of urea and TNF-αand IL-1β
Blood was obtained by puncture of the retro-orbital plexus. Biochemical determinations of urea in mouse sera were performed in an autoanalyser CCX Spectrum (Abbott Diagnostics Systems, Buenos Aires, Argentina) following standardized instructions. Aliquots of the sera were stored at −20°C until cytokine measurement. TNF-α and IL-1β were quantified by the sandwich enzyme-linked immunosorbent assay (ELISA) Kits (Endogene, MA, USA) and TNF-α biological activity was determined by a cytotoxic assay using L929 cells in the presence of actinomycin D.
Statistical analysis
All data correspond to the mean ± s.e.m. of individual mice. Statistical differences were determined using the one-way analysis of variance (anova), and P < 0·05 was considered significant. Individual groups were compared using the unpaired Student's t-test. The χ2 test, Fisher's exact test, was used for class variables in two independent samples.
Results
Stx2 toxicity in adrenalectomized (ADX) or Ru486-treated mice
Although it has been demonstrated that pretreatment of mice with a therapeutic dose of dexamethasone (DEX) diminishes Stx2 lethality [29], the role of endogenous glucocorticoids (GC) has not been analysed previously. Hence, we evaluated Stx2 toxicity under two experimental conditions: adrenalectomized (ADX) mice, and mice injected daily with Ru486, which prevents translocation of ligated GR to the nucleus by inhibiting dissociation of GR from heat shock protein 90. In both groups, mice were observed regularly to determine the survival rate from inoculation at 0 h to 144 h. As shown in Fig. 1a, ADX mice were significantly more susceptible to Stx2 toxicity. Additionally, the simultaneous treatment with DEX reversed the increase observed in the mortality rates. In order to confirm that this increase in mortality rates in ADX mice was a consequence of GC-deprivation (and not due to mineralocorticoid absence), we evaluated Stx2 toxicity in Ru486-treated mice. The animals were injected daily with 600 µg of Ru486, beginning 24 h before Stx2 inoculation (0 h) until the end of the experiment. As shown in Fig. 1b, a significant increase in the lethal effect was observed in Ru486-treated group. In spite of variations in the mortality rates of control mice after Stx2 treatment, both experimental protocols confirm that endogenous GC are able to attenuate Stx2-induced toxicity.
Fig 1.
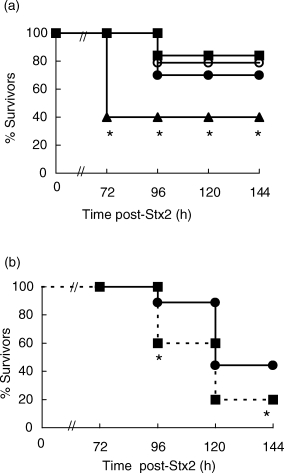
Role for GC in protection against Stx2 toxicity. (a) ADX mice and SHAM operated mice were injected with Stx2 (250 pg/mice) 5 days after surgery. DEX (300 µg/ml) was provided from 6 h prior to Stx2 in the drinking water until the end of the experiment. •, Stx2; ○, SHAM + Stx2; ▴, ADX + Stx2; ▪, ADX + DEX + Stx2. (b) Ru486 (600 µg/mice, i.p.) was administered daily from 12 h prior to Stx2 injection. One experiment representative of three (n = 12 mice per group); *P < 0·05, significantly different compared with all control groups by Fisher's exact test. •, Stx2; ▪, Stx2 + Ru486.
Effect of Ru486 treatment at different intervals of time
In order to determine when GC must be present to exert their protection against Stx2 toxicity, different protocols of Ru486-treatment were evaluated. Mice were injected once a day with Ru486: (1) every day since the Stx2 injection up to 96 h, (2) during the first 48 h or (3) from 48 h to 96 h post-Stx2 injection. Percentage of survivors was recorded at 120 h post-Stx2. Figure 2a shows that mice injected with Ru486 every day or during the first 48 h have an increased susceptibility to Stx2 toxicity. On the contrary, mice injected with Ru486 after 48 h post-Stx2, show higher percentage of survivors than control mice (inoculated with vehicle). These results suggest that GC have a beneficial role during only the first stage of HUS development. Since the early absence of GC-action turns mice more susceptible to the toxin, we evaluated the effects of DEX administration following the same time schedule than with Ru486 treatment. As shown in Fig. 2b, treatment of mice with DEX from Stx2 injection up to 96 h diminishes Stx2 lethality. On the contrary, mice treated with DEX from 48 h to 96 h post-Stx2 show a significant increase in mortality rates. These results are complementary to those observed in Ru486- treated mice (Fig. 2a).
Fig 2.
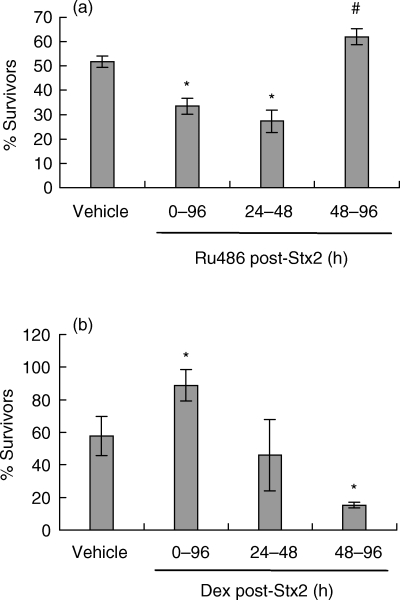
Effect of Ru486 and DEX treatment at different time intervals on the modulation of Stx2 toxicity. (a) Mice were injected with vehicle (control) or Ru486 (600 µg/mouse, i.p.) and (b) vehicle (control) or DEX (50 µg/mouse, i.v.) during the time intervals indicated in the figure after Stx2 injection. Bars represent the percentage of survivors (a) n= 18 mice/group; (b) n= 19 mice/group) at 120 h post-Stx2-injection. P < 0·001 and #P < 0·05, significantly different from vehicle-treated group, by Student's t-test.
Stx2-induced renal damage
We have demonstrated previously in the mouse model that urea and creatinine serum levels are good indicators of Stx2-induced renal damage [29]. In this context, we measured levels of urea in sera from ADX and Ru486 treated mice 72 h after Stx2 inoculation. Urea levels were increased significantly in both groups compared to Stx2-treated control mice (Table 1), which correlates with higher mortality rates.
Table 1.
Serum urea, measured 72 h post-Stx2 injection
| Treatment1 | Mice (n) | Plasma urea (mg%) X ± s.e.m. |
|---|---|---|
| SHAM + saline | 6 | 47 ± 12 |
| SHAM + Stx2 | 12 | 92 ± 7a |
| SHAM + DEX + Stx2 | 6 | 67 ± 3 |
| ADX + saline | 6 | 50·5 ± 5 |
| ADX + Stx2 | 6 | 140 ± 9b |
| ADX + DEX + Stx2 | 6 | 68 ± 15 |
| Ru486 + Stx2 | 6 | 150 ± 22c |
| Ru486 + DEX + Stx2 | 6 | 58 ± 3 |
Mouse serum was obtained 72 h post-Stx2 injection. Dexamethasone (DEX) (300 µg/ml) was provided in the drinking water to adrenalectomized (ADX) mice and injected i.p. (50 µg/mice) to Ru486-treated mice (600 µg/mice).
aP < 0·01 compared with SHAM-operated mice treated with saline or DEX.
bP < 0·05 compared with SHAM injected with Stx2 and ADX treated with saline, and P < 0·005 compared with ADX mice treated with DEX.
cP < 0·005 compared with SHAM-operated mice treated with Stx2, saline or Ru486 + DEX-treated mice.
Histological study
To confirm whether the enhancement on Stx2 toxicity in survival rate and urea levels, due to the absence of GC, was conveyed by an enhanced renal damage, we evaluated the histological lesions. All experimental groups were sacrificed 72 h after Stx2 injection in order to remove the kidneys that were fixed immediately in paraffin wax and then stained with haematoxylin and eosin. Figure 3 shows representative histological images of kidneys from mice either sham-operated or adrenalectomized (ADX) and treated with or without Stx2. Mice given saline, either sham or ADX, showed no damage (Fig. 3a). The kidneys from sham-operated mice treated with Stx2 showed focal cortical necrosis involving detached tubular epithelial cells, together with intraluminal deposits of amorphous material (Fig. 3b). Some glomeruli contained extensive capillary congestion and acidophilic material consistent with thrombosis (Fig. 3b, insert). The thrombi were confirmed by deposits of PAS positive material in the lumen of glomerular capillaries (not shown). Kidneys from ADX mice treated with Stx2 displayed diffuse bilateral cortical necrosis with loss of tubular epithelium, nuclear pyknosis and the interstitium contained deposits of haemosiderin pigments (Fig. 3c). The extent of necrosis observed included the glomeruli (Fig. 3c, insert). Mice treated with Ru486 and Stx2 developed similar lesions (not shown). Renal lesions were consistent within sections from the same kidney, in kidneys from the same mouse and in kidneys of all the mice in each treatment group. Thus, the absence of glucocorticoids enhances Stx2 toxicity in the kidneys.
Fig 3.
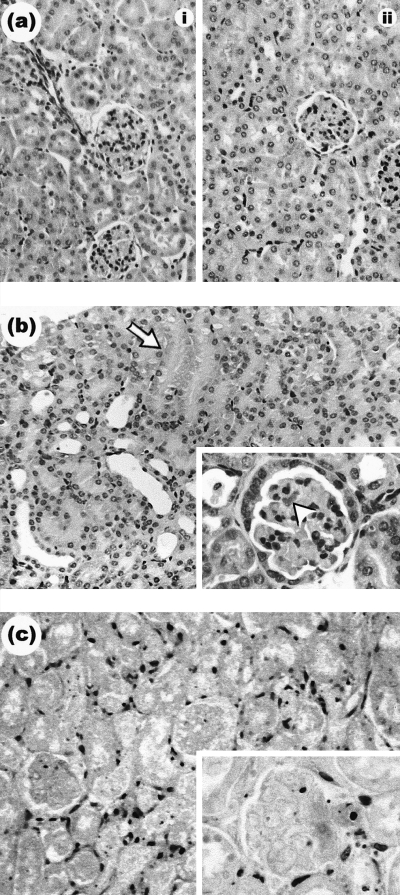
Representative haematoxylin and eosin staining of paraffin wax sections from murine renal sections (glomeruli and tubules) of ADX or sham-operated mice, treated with or without saline or Stx2. (a) The renal cortexes of the control groups, either (i) sham or (ii) ADX, were histologically normal. (b) Renal cortex of sham-operated mice treated with Stx2. This group developed focal necrosis of tubules as indicated by arrowhead. Glomeruli were histologically altered (b, insert). Acidophilic material deposits is indicated by arrowhead. (c) ADX mice treated with Stx2. Note the extent of diffuse cortical tubular and glomerular necrosis with severe dilation of tubule lumens and extensive loss of epithelial cells. Necrotic glomeruli (c, insert). Magnification, × 200. The magnification of all inserts is ×400.
Corticosteroid induction after Stx2 inoculation
To address whether Stx2 is able to induce GC secretion mice were injected with Stx2 on day 0, and corticosterone was measured after 2, 6, 24 and 48 h post-Stx2 injection, always in the morning to avoid diurnal variations on the baseline of endogenous GC level. As shown in Fig. 4, Stx2 inoculation induced a significant increase in plasmatic corticosterone 2 h post-treatment. Basal levels were recovered gradually between 24 and 48 h post-Stx2. Since LPS is a bacterial product, which stimulates HPA strongly, we bled mice 2 h after i.p. injection of 25 µg/mouse LPS as a positive control. As can be seen in Fig. 4, this group of mice showed a marked corticosterone response to LPS compared to the control.
Fig 4.
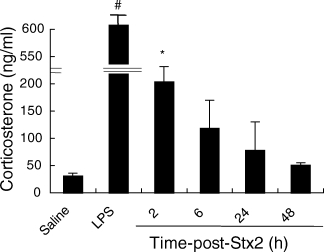
Plasma corticosterone concentration in mice treated with Stx2. Plasma corticosterone determinations were made at multiple time points (2, 6, 24, 48 h) post-Stx2 i.v. injection (250 pg/mice), or 2 h after saline and LPS i.p. injection (25 µg/mice), always in the morning (1000–1200 h), using a commercial kit as described in Materials and methods. Values are expressed as the mean (± s.e.m.) of six mice per time-point. *P < 0·05, significantly different from the control value and #P < 0·0001, compared with all other groups, using Student's t-test.
Inflammatory cytokines after Stx2 inoculation
To determine whether increased susceptibility to Stx2 in Ru486-treated mice could be ascribed to an overproduction of inflammatory cytokines, we assayed TNF-α and IL-1β at different times after Stx2 injection. No increase was observed in serum concentration of these cytokines neither after Stx2 alone nor in Ru486-pretreated mice, as assayed by an ELISA KIT and, in the case of TNF-α, by its biological activity on L929 cell line (data not shown).
Intracellular glucocorticoid receptors (GR) in neutrophils
GC action depends not only on GC levels but also on the number and distribution of the specific receptors [32]. Taking into account that several studies have reported neutrophil involvement in HUS pathogenesis [9, 16, 17], we evaluated whether Stx2 caused variations in GR expression in peripheral neutrophils. For this purpose, flow cytometric detection of intracellular (cytoplasmic and nuclear) GR with a specific monoclonal antibody was used, because it provides an alternative and sensitive method for intracellular GR antigen detection [26]. Mice were injected with Stx2 or saline and blood samples were obtained after different intervals. The positive control was evaluated on neutrophils from DEX-treated mice [26]. As depicted in Fig. 5, a significant increase in GR expression was detected in neutrophils as soon as 24 h after Stx2 injection.
Fig 5.
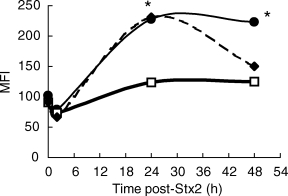
Induction of GR expression by Stx2 in neutrophils. PMN were isolated from mice at 0, 2, 24 and 48 h post-treatment with saline (squares), DEX (50 µg/mice i.p.) (diamonds) or Stx2 (250 pg/mice) (circles). Then, the cells were centrifuged, washed and stained with specific MoAb anti-GR for cytometric studies as described in Materials and methods. Data represent the mean ± s.e.m. of mean fluorescence intensity (MFI) from six mice per group. *P < 0·05, significantly different from saline treated group using Student's t-test.
Discussion
This study demonstrates the capacity of endogenous glucocorticoids (GC) to modulate Stx2 toxicity. They were shown to protect against Stx2-induced lethality, as mice rendered glucocorticoid deficient by adrenalectomy (ADX) or by blocking the glucocorticoid receptors (GR) with Ru486, had increased sensitivity to Stx2 and because the normal susceptibility was restored by corticosterone replacement. Glucocorticoid effects on Stx sensitivity were evaluated by lethality rate, biochemical determination of urea and histological observation of the kidneys. Temporal studies with Ru486 treatment indicated that GC play a protective role during the first stage of HUS development, suggesting that GC are necessary as soon as an inflammatory reaction is triggered, probably as a consequence of Stx2 endothelial damage. A similar conclusion was reached through the administration of DEX at different times after Stx2. Although these results could have important therapeutic implications, it is difficult to extrapolate to humans the ‘window time’ for GC treatment, mainly for two reasons. First, disease days are usually counted from the onset of diarrhoea in humans, the lack of diarrhoea in the mouse model makes it difficult to compare the time courses of the diseases between these species. Secondly, it is not known when Stx enters the bloodstream and induces irreversible damage to target organs in humans.
Corticosteroids, the final product of activation of HPA, are well known for their profound effects on virtually every aspect of the immune response [18]. In fact, GC are known to down-regulate macrophage function [33], impairing cytokine production and action [34–36] and to inhibit several neutrophil responses [37–39]. One or a combination of more than one of these effects could be acting in concert to diminish Stx2 toxicity. In particular, we have demonstrated previously that Stx2 toxicity was significantly reduced in mice depleted of macrophages [15], suggesting that its effects on these cells play an important role in mediating tissue damage. Moreover, macrophages stimulated in vitro with Stx can release TNF-α and IL-1β[40], two inflammatory cytokines involved in Stx2 damage. Thus, the increased sensitivity to Stx2 toxicity in the absence of GC could be mediated by the up-regulation of inflammatory cytokine secretion and/or responsiveness. However, plasmatic TNF-α and IL-1β levels were not elevated significantly in Ru486-treated mice at any time after Stx2 injection. In spite of this, it is possible that GC deficiency results in additional effects promoting cytokine-mediated sensitivity or in a higher local production within certain vital organs, such as the kidneys. In this regard, renal mRNA TNF-α induction by Stx2 has been demonstrated in mice [14]. Therefore, increases in local cytokine production in different tissues may have a significant contribution to lethality although not proportionally reflected in the circulation. Moreover, ADX has been demonstrated to increase sensitivity to exogenously administered TNF-α (and IL-1β) [41,42], and there are several pathways by which GC might modulate TNF-α responsiveness.
Corticosteroid action is a function of two major factors: (a) the relative availability of the hormone, which is determined by its level in the circulation, and (b) the number and function of the hormone receptors [43,44]. We have demonstrated that Stx2 induces transient significant increases in the secretion of corticosterone, the principal corticosteroid in mice, as well as in the expression of its receptor in neutrophils. Increase in GR number and/or affinity might provide a mechanism whereby cells could regulate their gene expression and ultimately their glucocorticoid sensitivity, thereby promoting feedback inhibition [45]. Results from our study indicate that the number of GR in neutrophils begins to increase as the concentration of Stx2-induced GC present in the serum begins to wane, perhaps providing a mechanism for increasing GC sensitivity. Thus, the increased availability of neutrophil GR induced by Stx2 may contribute to the protective capacity of glucocorticoids in regulating the pathogenic potential of this population. In this regard, HUS neutrophils have been found to adhere avidly to endothelium and to damage the endothelial cell [16], and we have found that peripheral neutrophils from Stx-treated mice had increased expression of CD11b, higher adhesive properties in lung vessels and increased cytotoxic capacity [9]. All these results suggest that activation of PMN may play an important role in HUS pathogenesis. At present, it is not known whether increased GR number in neutrophils would indeed increase glucocorticoid sensitivity. However, several reports have suggested a direct relationship between GR number and biologic responses [44–46]. Glucocorticoid action on neutrophil function includes reduction of the binding capacity of β2-integrins [47] and l-selectins [48]. Both effects, together with the inhibition of endothelial expression of adhesion molecules for neutrophils [49] may account for a minor neutrophil rolling and recruitment of PMN to inflamed areas [49,50].
It can be argued that the induction of glucocorticoids and their receptors in PMN could be a direct effect of Stx2, or could be the consequence of the complex interaction between platelet activation, endothelial injury, and/or could be mediated indirectly by the secretion of inflammatory mediators, such as TNF-α, IL-1 or IL-6. All these factors play an important role not only in HPA stimulation but also in GR-induction in different cells [51].
In conclusion, we can say that endogenous glucocorticoids protect against Stx2-toxicity, and these results reinforce the concept that endocrine–immune interactions could be beneficial in the control of bacterial-induced damage.
Acknowledgments
This work was supported by grants from CONICET (Consejo Nacional de Investigaciones Científicas y Tecnológicas), Fundación Alberto J. Roemmers and Agencia Nacional de Promoción Científica y Tecnológica, Argentina. The authors thank Juan Portaluppi, Antonio Morales, Marta Felippo, Nora Galassi and Norma Riera for their excellent technical assistance and Christiane Dosne Pasqualini for reviewing the manuscript. The authors also thank Fundación de la Hemofilia for the use of the FACScan flow cytometer.
References
- 1.Remuzzi G, Ruggenenti P. The hemolytic uremic syndrome. Kidney Int. 1995;47:2–19. doi: 10.1038/ki.1995.261. [DOI] [PubMed] [Google Scholar]
- 2.Koster F, Boonpucknavig V, Sujaho S, Gilman R, Rahaman M. Renal histopathology in the hemolytic uremic syndrome following shigellosis. Clin Nephrol. 1984;21:126–33. [PubMed] [Google Scholar]
- 3.Arbus GS. Association of verotoxin-producing E. coli and verotoxin with hemolytic uremic syndrome. Kidney Int. 1997;58:S91–6. [PubMed] [Google Scholar]
- 4.Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. The association between idiopathic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis. 1985;151:775–82. doi: 10.1093/infdis/151.5.775. [DOI] [PubMed] [Google Scholar]
- 5.Keusch GT, Acheson DWK. Thrombotic thrombocytopenic purpura associated with shiga toxins. Semin Hematol. 1997;34:106–16. [PubMed] [Google Scholar]
- 6.Paton JC, Paton AW. Pathogenesis and diagnosis of shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev. 1998;11:450–79. doi: 10.1128/cmr.11.3.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcato P, Mulvey G, Read RJ, Vander Helm K, Nation PN, Armstrong GD. Immunoprophylactic potential of cloned Shiga toxin 2 B subunit. J Infect Dis. 2001;183:435–43. doi: 10.1086/318080. [DOI] [PubMed] [Google Scholar]
- 8.Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Maloney L, Daniel TO. Endothelial heterogeneity in shiga toxin receptors and responses. J Biol Chem. 1993;268:15484–8. [PubMed] [Google Scholar]
- 9.Fernández GC, Rubel C, Dran G, Gomez S, Isturiz MA, Palermo MS. Shiga toxin-2 induces neutrophilia and neutrophil activation in a murine model of hemolytic uremic syndrome. Clin Immunol. 2000;95:227–34. doi: 10.1006/clim.2000.4862. [DOI] [PubMed] [Google Scholar]
- 10.Taylor FB, Jr, Tesh VL, DeBault L, et al. Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to Stx-1. Am J Pathol. 1999;154:1285–99. doi: 10.1016/S0002-9440(10)65380-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wadolkowski EA, Burris JA, O'Brien AD. Mouse model for colonization and disease caused by enterohemorragic Escherichia coli O157:H7. Infect Immun. 1990;58:2438–45. doi: 10.1128/iai.58.8.2438-2445.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis DH, Moxley RA, Andraos CY. Edema disease-like brain lesions in gnotobiotic piglets infected with Escherichia coli serotype O157:H7. Infect Immun. 1989;57:1339–42. doi: 10.1128/iai.57.4.1339-1342.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barret TJ, Potter ME, Wachsmuth K. Bacterial endotoxin both enhances and inhibits the toxicity of Shiga-like toxin II in rabbits and mice. Infect Immun. 1989;57:3434–7. doi: 10.1128/iai.57.11.3434-3437.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harel Y, Silva M, Giroir B, Weinberg A, Cleary TB, Beutler B. A reporter transgene indicates renal-specific induction of tumor necrosis factor (TNF) by shiga-like toxin. J Clin Invest. 1993;92:2110–6. doi: 10.1172/JCI116811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palermo MS, Alves Rosa MF, Van Rooijen N, Isturiz MA. Depletion of liver and splenic macrophages reduces the lethality of Shiga toxin-2 in a mouse model. Clin Exp Immunol. 1999;116:462–7. doi: 10.1046/j.1365-2249.1999.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forsyth KD, Simpson AC, Fitzpatrick MM, Barratt TM, Levinsky RJ. Neutrophil mediated endothelial injury in haemolytic uraemic syndrome. Lancet. 1989;2:411–4. doi: 10.1016/s0140-6736(89)90591-6. [DOI] [PubMed] [Google Scholar]
- 17.Te Loo DMWM, Monnens LAH, van der Velden TJAM, et al. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood. 2000;95:3396–402. [PubMed] [Google Scholar]
- 18.Cupps TR, Fauci AS. Corticosteroid-mediated immunoregulation in man. Immunol Rev. 1982;65:133–53. doi: 10.1111/j.1600-065x.1982.tb00431.x. [DOI] [PubMed] [Google Scholar]
- 19.Kapcala LP, Chautard T, Reskay RL. The protective role of the hypothalamic–pituitary–adrenal axis against lethality produced by immune, infectious and inflammatory stress. Ann NY Acad Sci. 1995;771:419–37. doi: 10.1111/j.1749-6632.1995.tb44699.x. [DOI] [PubMed] [Google Scholar]
- 20.Blalock JE. The syntax of immune-neuroendocrine communication. Immunol Today. 1985;15:504–11. doi: 10.1016/0167-5699(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 21.Miller AH, Spencer RL, Pearce BD, et al. Effects of viral infection on corticosterone secretion and glucocorticoid receptor binding in immune tissues. Psychoneuroendocrinology. 1997;22:455–74. doi: 10.1016/s0306-4530(97)00028-0. [DOI] [PubMed] [Google Scholar]
- 22.Del Rey A, Klusman I, Besedovsky HO. Cytokines mediate protective stimulation of glucocorticoid output during autoimmunity: involvement of IL-1. Am J Physiol. 1998;275:R1146–51. doi: 10.1152/ajpregu.1998.275.4.R1146. [DOI] [PubMed] [Google Scholar]
- 23.MacPhee IAM, Antoni FA, Mason WD. Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on regulation of the immune system by endogenous adrenal corticosteroids. J Exp Med. 1989;169:431–45. doi: 10.1084/jem.169.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sternberg EM, Hill JM, Chrousos GP, et al. Inflammatory mediator-induced hypothalamic–pituitary–adrenal axis activation is defective in streptococcal cell wall arthritis susceptible Lewis rats. Proc Natl Acad Sci USA. 1989;86:2374–8. doi: 10.1073/pnas.86.7.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middelveld RJ, Wanecek M, Bergman D, Weitzberg E, Alving K. Effect of cortisol-synthesis inhibition on endotoxin-induced porcine acute lung injury, shock and nitric oxide production. Shock. 1999;12:382–90. doi: 10.1097/00024382-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Berki T, Kumánovics GK, Kumánovics A, Falus A, Újhelyi E, Németh P. Production and flow cytometric application of a monoclonal anti-glucocorticoid receptor antibody. J Immunol Meth. 1998;214:19–27. doi: 10.1016/s0022-1759(98)00037-4. [DOI] [PubMed] [Google Scholar]
- 27.Duner KI. A new kinetic single stage Limulus amoebocyte lysate method for the detection of endotoxin in water and plasma. J Biochem Biophys Meth. 1993;26:131–42. doi: 10.1016/0165-022x(93)90043-n. [DOI] [PubMed] [Google Scholar]
- 28.Vinson RB, Carroll JL, Pret SB. Mechanism of suppressed neutrophil mobilization in a mouse model for binge drinking: role of glucocorticoids. Am J Physiol. 1998;275:R1049–57. doi: 10.1152/ajpregu.1998.275.4.R1049. [DOI] [PubMed] [Google Scholar]
- 29.Palermo MS, Rubel C, Alves Rosa F, et al. Pretreatment of mice with LPS or IL-1β exerts opposite effects on Shiga-toxin-2 lethality. Clin Exp Immunol. 1999;119:77–83. doi: 10.1046/j.1365-2249.2000.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coligan J, Knisblek A, Margulies D, Shevach E, Warren S. Coligan J, Knisblek A, Margulies D, et al., editors. In vitro assays for mouse lymphocyte function. Current protocols in immunology. National Institute of Health. USA: Wiley & Sons Inc. 1994. pp. 3.20.3–4.
- 31.Knapp W, Majdic O, Strobl H. Flow cytometric analysis of cell surface and intracellular antigens in leukemia diagnosis. Cytometry. 1994;18:187–98. doi: 10.1002/cyto.990180402. [DOI] [PubMed] [Google Scholar]
- 32.Miller AH, Spencer RL, Pearce BD, et al. Glucocorticoid receptors are differentially expressed in the cells and tissues of the immune system. Cell Immunol. 1998;186:45–54. doi: 10.1006/cimm.1998.1293. [DOI] [PubMed] [Google Scholar]
- 33.Baybutt HN, Holsboer NJ. Inhibition of macrophage differentiation and function by cortisol. Endocrinology. 1990;127:476–80. doi: 10.1210/endo-127-1-476. [DOI] [PubMed] [Google Scholar]
- 34.Zanker B, Walz G, Wleder J, Strom TB. Evidence that glucocorticoids block expression of the human interleukin-6 gene by accessory cells. Transplantation. 1990;49:183–5. doi: 10.1097/00007890-199001000-00040. [DOI] [PubMed] [Google Scholar]
- 35.Snyder DS, Unanue ER. Corticosteroids inhibit murine macrophage Ia expression and interleukin 1 production. J Immunol. 1982;129:1803–5. [PubMed] [Google Scholar]
- 36.Fantuzi G, Di Santo E, Sacco S, Benigni F, Ghezzi P. Role of hypothalamus-pituitary-adrenal axis in the regulation of TNF production in mice. J Immunol. 1995;155:3552–5. [PubMed] [Google Scholar]
- 37.Youssef P, Roberts-Thomson P, Ahern M, Smith M. Pulse methylprednisolone in rheumatoid arthritis: effects on peripheral blood and synovial fluid neutrophil surface phenotype. J Rheumatol. 1995;22:2065–71. [PubMed] [Google Scholar]
- 38.Goulding NJ, Euzger HS, Butt SK, Perretti M. Novel pathways for glucocorticoid effects on neutrophils in chronic inflammation. Inflamm Res. 1998;47:S158–65. doi: 10.1007/s000110050310. [DOI] [PubMed] [Google Scholar]
- 39.Yazawa H, Kato T, Nakata T, Sendo F. Glucocorticoid hormone suppression of human neutrophil-mediated tumor cell cytostasis. Int J Cancer. 1999;81:74–80. doi: 10.1002/(sici)1097-0215(19990331)81:1<74::aid-ijc14>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 40.Foster GH, Armstrong CS, Sakiri R, Tesh VL. Shiga toxin-induced tumor necrosis factor alpha expression: requirement for toxin enzymatic activity and monocyte protein kinase C and protein tyrosine kinases. Infect Immun. 2000;68:5183–9. doi: 10.1128/iai.68.9.5183-5189.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1708–12. doi: 10.1084/jem.167.5.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butler LD, Layman NK, Riedl PE, et al. Neuroendocrine regulation of in vivo cytokine production and effects: I. In vivo regulatory networks involving the neuroendocrine system, interleukin-1 and tumor necrosis factor-alpha. J Neuroimmunol. 1989;24:143–53. doi: 10.1016/0165-5728(89)90108-2. [DOI] [PubMed] [Google Scholar]
- 43.McEwen BS, Biron CA, Brunson KW, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- 44.Vanderbilt JN, Miesfield R, Maler BA, Yamamoto KR. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol. 1987;1:68–74. doi: 10.1210/mend-1-1-68. [DOI] [PubMed] [Google Scholar]
- 45.Salkowski AA, Vogel SN. Lipopolysaccharide increases glucocorticoid receptor expression in murine macrophages. A posible mechanism for glucocorticoid-mediated suppression of endotoxicity. J Immunol. 1992;149:4041–7. [PubMed] [Google Scholar]
- 46.Danielsen M, Stallcup MR. Down-regulation of glucocorticoid receptors in mouse lymphoma cell variants. Mol Cell Biol. 1984;4:449–53. doi: 10.1128/mcb.4.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torsteinsdottir I, Arvidson NG, Hallgren R, Hakansson L. Enhanced expression of integrins and CD66b on peripheral blood neutrophils and eosinophils in patients with rheumatoid arthritis, and the effects of glucocorticoids. Scand J Immunol. 1999;50:433–9. doi: 10.1046/j.1365-3083.1999.00602.x. [DOI] [PubMed] [Google Scholar]
- 48.Filep JG, Delalandre A, Payette Y, Foldes-Filep E. Glucocorticoid receptor regulates expression of l-selectin and CD11/CD18 on human neutrophils. Circulation. 1997;97:2279–81. doi: 10.1161/01.cir.96.1.295. [DOI] [PubMed] [Google Scholar]
- 49.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weismann G. A mechanism for the antiinflamatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1992;89:9991–5. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leech M, Hutchinson P, Holsworth SR, Morand EF. Endogenous glucocorticoids modulate neutrophil migration and synovial P-selectin but not neutrophil phagocytic or oxidative function in experimental arthritis. Clin Exp Immunol. 1998;112:383–8. doi: 10.1046/j.1365-2249.1998.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Besedovsky HO, Del Rey A. Immune-neuro-endocrine interaction: facts and hypotheses. Endocr Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]