

Abstract
Nucleated cells employ several strategies to evade killing by homologous complement. We studied complement resistance in the human carcinoma cell lines (CA) T47D (mammary), SKOV3 (ovarian), and PC-3 (prostate) with emphasis on the following mechanisms of defense: 1. Expression and shedding of the membrane complement regulatory proteins (mCRP) CD46, CD55 and CD59; 2. Resistance based on protein phosphorylation; 3. Cell surface expression of sialic acid residues; 4. Desensitization to complement upon exposure to sublytic complement doses. Anti-mCRP antibody blocking experiments demonstrated that CD59 is the main mCRP protecting these CA from complement. Soluble CD59 was also found in supernates of PC-3> SKOV3 > T47D cells. Second, inhibitors of PKC, PKA and MEK sensitized the CA to lysis, thus implicating these protein kinases in CA complement resistance. Third, removal of sialic acid residues with neuraminidase also sensitized CA to lysis. Finally, exposure of CA to sublytic doses of complement conferred on them enhanced resistance to lytic complement doses in a PKC-dependent process. Combined treatment of CA with anti-CD59 antibodies, PD98059 (a MEK inhibitor) and neuraminidase produced a large enhancement in CA sensitivity to complement. Our results show that CD59 and sialic acid residues present on the cell surface, and intracellular processes involving protein phosphorylation act additively to secure CA resistance to complement-mediated lysis. Therefore, the effectiveness of antibody- and complement-based cancer immunotherapy will markedly improve by suppression of the various complement resistance mechanisms.
Keywords: complement resistance, regulators, CD59, sialic acid, tumour immunology
Introduction
Resistance of cancer cells to lysis mediated by homologous complement is one of their immune escape strategies that will be a major obstacle to development of immunotherapy based on complement-fixing anti-tumour antibodies [1,2]. To date, several complement protective molecules and mechanisms have been described in malignant and normal cells (reviewed in 3). Best studied is the family of membrane complement regulatory proteins (mCRP: CD46, CD55 and CD59) that provide normal and cancer cells with a first line defense from complement attack. Membrane cofactor protein (MCP or CD46) and decay accelerating factor (DAF or CD55) block the complement cascade at the C3 activation/amplification stage [4]. In contrast, CD59 interacts with the C8 and C9 complement components and inhibits assembly of the membrane attack complex (MAC) [5,6]. Larger amounts of mCRP have been found on certain tumour cells relative to the corresponding normal cells [7–10]. Production and binding of the soluble complement inhibitor factor H has been demonstrated in glioblastoma cells [11]. Surface membrane proteases have also been shown to confer complement resistance on certain tumour cells, probably by digestion and removal of cell bound complement components [12,13]. Another cell surface component that has been implicated in cell resistance to complement is sialic acid [14,15]. Other mechanisms of complement resistance involve intracellular protein phosphorylation by protein kinase C (PKC) [3, 16, 17] and by the extracellularly regulated protein kinase (ERK) [18] and protein synthesis [19]. The exact mode of protection conferred by PKC and ERK is still not known, and one possibility is that they participate in the process of MAC removal by vesiculation or endocytosis [20,21].
Basal cell resistance to complement may be modified by various treatments. Thus, blocking of RNA and protein synthesis sensitized tumour cells to complement-mediated lysis [19]. On the other hand, K562 erythroleukaemic and other cells treated with ionomycin, A23187, cAMP, forskolin or phorbol myristate acetate gained, within several minutes, enhanced resistance to complement damage [16,22]. Treatment with sublytic doses of antibody and complement desensitized leukaemic cells to lytic complement doses [23]. Similarly, the pore-formers perforin, streptolysin O and mellitin induced in K562 cells enhanced resistance to complement [22]. The protective effect of sublytic complement depends upon a signalling process involving calcium ion influx, PKC and ERK activation and protein synthesis induced by the complement MAC [17, 18, 24].
Until recently, each of the complement resistance mechanisms had been studied separately from the others. It has become now important to assess the relative impact of the various evasion strategies in the same target cell population. Cooperation among mCRPs has been demonstrated [25,26], however, the possible cooperation between the mCRPs and the other protective mechanisms mentioned above has been barely examined. In a previous study, we showed that K562 eythroleukaemic cells are equipped with at least three distinct protective strategies acting together to promote basal cell refractoriness to complement-mediated lysis [27]. These are the mCRP (especially CD59 and CD55), a surface expressed serine protease and a PKC-dependent intracellular mechanism. In this study, the complementary action of three distinct complement protective mechanisms has been studied in human carcinoma cell lines representing breast, prostate and ovarian carcinoma. As shown here, the 3 carcinoma cell types protect themselves from the lytic action of complement by using extracellular mCRP and sialic acid residues and intracellular defense mechanisms dependent on protein phosphorylation.
Materials And Methods
Cell cultures
PC-3, a human prostate carcinoma cell line was cultured in RPMI-1640 supplemented with 10% (v/v) heat inactivated-fetal bovine serum (Gibco Laboratories, Grand Island, NY, USA), 1% glutamine, 2% pyruvate and antibiotics mixture (Bio-Lab, Jerusalem, Israel) at 37°C and 5% CO2. T47D, a human breast carcinoma cell line and SKOV3, a human ovarian carcinoma cell line, were cultured in DMEM supplemented with the same ingredients as RPMI-1640.
Antibodies and reagents
Polyclonal antibodies directed to T47D, SKOV3 and PC-3 cells were prepared in rabbits by 3 biweekly s.c. injections of 20 × 106 cells and used after heat inactivation (30 min, 56°C). The following mAb directed to membrane complement regulatory proteins were used: anti-CD46 (clone GB24; kindly provided by Dr J. Atkinson, St. Louis, MO, USA), anti-CD55 (clone BRIC110), anti-CD55 (clone BRIC216) and anti-CD59 (clone BRIC229) (International Blood Group Reference Laboratory, IBGRL, Birmingham, UK). In addition, mouse anti-human CD55 monoclonal antibodies were purchased from PharMingen (clone IA10, San Diego, CA, USA) and mouse anti-human CD59 monoclonal antibodies (MEM-43) were purchased from Serotec (Oxford, UK). Rabbit anti-human CD59 antibodies were kindly provided by Dr P. Lachmann (Cambridge, UK) and Dr M. Daha (Leiden, the Netherlands) and mouse anti-human mannose-binding lectin (MBL) monoclonal antibodies 3F8 and 1C10 were a kind gift from Dr G. Stahl (Boston, MA, USA). FITC-conjugated goat anti-mouse IgG was from Sigma Chemical Co. (St. Louis, MO, USA), and FITC-conjugated goat anti-rabbit IgG was from Jackson ImmunoResearch laboratories (West Grove, PA, USA). d-mannose and PMA were purchased from Sigma; calphostin C, PD98059, recombinant α2–3,6,8,9 neuraminidase, GF109203X and H89 were from Calbiochem (San Diego, CA, USA).
Sera
Normal human serum (NHS) used as a source of complement was freshly prepared from healthy donors. Heat inactivation of complement in NHS (HI-NHS) was performed for 30 min at 56°C. Factor B-depleted human serum was purchased from Advanced Research Technologies (San Diego, CA, USA). Factor B was purified from NHS as described by Gotze & Muller-Eberhard [28]. All sera and proteins were kept frozen at −70°C.
Cell lysis and induced protection assays
Cytotoxicity assays were performed using trypan blue exclusion as described before [24]. Briefly, cells were incubated with diluted rabbit anti-carcinomas antiserum for 30 min at 4°C and then with NHS or HI-NHS (final dilution: 1 : 2) for 60 min at 37°C. In some experiments, tumour cells were preincubated in addition with noncomplement fixing neutralizing mAb against CD46 (GB24), CD55 (BRIC110 and BRIC216) each at 2·5 or 10 µg/ml and/or CD59 (BRIC229) at 2·5–10 µg/ml. Alternatively, complement-mediated cytotoxicity was analysed by using a novel nonradioactive cytotoxicity assay based on time-resolved fluorometry [27,29] (Fig. 2). Briefly, 1 × 106 cells were washed two times in culture medium and incubated with 20 µm of the fluorescence enhancing ligand bis(acetoxymethyl)2,2′:6′,2′-terpyridine-6,6′′-dicarboxylic acid (BATDA, Wallac, Turku, Finland) in 1 ml culture medium for 25 min at 37°C. The labelled cells were washed 5 times and resuspended in test buffer and adjusted to 105 cells/ml. 50 µl BATDA-labelled target cells per well of a round bottom 96-well plate (Greiner, Frickenhausen, Germany) were incubated for 30 min at 37°C with anti-cell antibody (50 µl). NHS (10%) as a source of complement or heat-inactivated NHS as control (100 µl) were then added for additional 60 min incubation at 37°C. Spontaneous release was determined by incubating the target cells with test buffer instead of antiserum and NHS and maximal release was evaluated by incubating the cells with 160 µl test buffer containing 40 µl digitonin stock solution (1 mg/ml). After centrifugation (5 min, 500 g), 20 µl of the supernatant from each well were transferred to wells of flat-bottom 96-well plates. 200 µl of the Europium solution (Wallac) were added to each well and the plates were shaken for 5 min. Fluorescence of EuTDA chelates was measured in a time-resolved fluorometer (VICTOR, Wallac). Specific release (%) was calculated according to the formula:
Fig. 2.
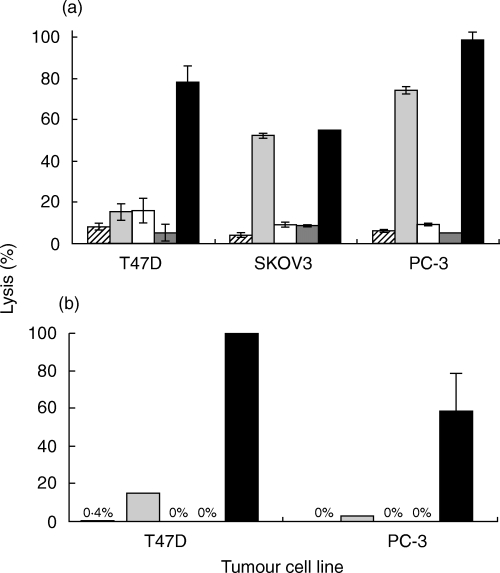
Neutralization of mCRP augments susceptibility of carcinoma cells to complement-mediated lysis. Cells were incubated with neutralizing mAb directed to CD59, CD55 or CD46 or a mixture of these antibodies or with buffer alone. (a) 10 µg/ml mAb (an optimal concentration) (b) 2·5 µg/ml mAb (a suboptimal concentration). Next, the cells were subjected to lysis by rabbit anti-CA antibodies and NHS. Cells in Fig. 2b also received a lower concentration of anti-CA antibodies (1 : 500 antiserum dilution) than in Fig. 2a (1 : 300 dilution). Percentage lysis was determined by released-TDA fluorometry (see Methods). 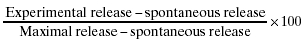 w/o Ab;
w/o Ab;  anti-CD59 Ab; □ anti-CD55 Ab;
anti-CD59 Ab; □ anti-CD55 Ab;  anti-CD46 Ab; ▪ Abs mixture.
anti-CD46 Ab; ▪ Abs mixture.
In the induced protection experiments, the cells were first treated with sublytic doses of antibody and NHS, washed and then incubated with lytic doses of antibody and complement [23]. To study the effect of PKC inhibitors on complement-induced protection, cells were pretreated with calphostin C for 30 min at 37°C.
Percentage induced protection was calculated as follows:
Flow-cytometry analysis
Cells were removed from the culture plates by gentle scraping with a Cell Scraper (Costar, NY, USA), washed and resuspended in PBS. Tumour cells (0·5 × 106) in 50 µl PBS were incubated for 30 min on ice with monoclonal anti-human CD55 (clone IA10, 2 µg/ml), CD46 (clone GB24, 44 µg/ml) or CD59 (clone MEM43, 5 µg/ml) antibodies or with polyclonal anti-human CD59 antiserum (1 : 200). The cells were washed three times with PBS and incubated for 30 min on ice with FITC-conjugated goat anti-mouse IgG (8·8 µg/ml) or with FITC-conjugated goat anti-rabbit IgG (20 µg/ml). The cells were washed three times with PBS and analysed in a Becton Dickinson FACScan.
Detection of sCD59 in cell supernatants by ELISA
The amount of soluble CD59 (sCD59) in cell culture supernates was determined by ELISA. In brief: 96-well microtitre plates (NUNC-MaxiSorb, Roskilde, Denmark) were coated with monoclonal anti-CD59 (BRIC229) in 50 mm Na2CO3/NaHCO3, pH 9·6 for 16 h at 4°C. After blocking remaining unspecific binding sites with 1% gelatin/PBS, 50 µl per well of each sample was added in duplicates for 1 h at room temperature. The plates were washed three times and then incubated with rabbit anti-human CD59 antiserum for 1 h at room temperature, followed by peroxidase-conjugated mouse anti-rabbit IgG (Dianova, Hamburg, Germany) for 1 h at room temperature. The assay was developed using ABTS (Sigma)/H2O2 as substrate. After stopping the reaction with 0·2 m oxalic acid, the microtitre plates were analysed at λ= 405 nm/492 nm in an ELISA reader (EAR 340, SLT Labinstruments, Overath, Germany). Purified sCD59 (kindly provided by Dr P. Morgan, Cardiff, UK) was used as a standard. sCD59 thus obtained from cell supernatants was shown to be active by its capacity to bind to human complement C9 on nitrocellulose membrane (not shown).
Statistical analysis
Student's unpaired and paired t-tests were used to determine statistical significance of differences between various data sets. Results in Figs 2,3,4,5,6 are expressed as mean ± SE. Statistical significance was assumed when P < 0·05.
Results
Expression of CD59, CD55 and CD46 on CA
The level of expression of CD59, CD55 and CD46 in breast (T47D), ovarian (SKOV3) and prostate (PC-3) carcinoma cell lines (CA) was determined by flow cytometry. The 3 carcinoma cell types were found to express the three membrane complement regulatory proteins (mCRP) (Fig. 1). The amount of soluble CD59 (sCD59) secreted spontaneously from CA, was next tested. The concentration of sCD59 found in CA supernatant, after 72 h of cell culture, is shown in Table 1. PC-3 cells secreted more sCD59 than SKOV3 and T47D cells.
Fig. 1.
Expression of CD55, CD46 and CD59 on T47D, SKOV3 and PC-3 cells. T47D, SKOV3 and PC-3 cells (0·5 × 106) were treated for 30 min on ice with mAb anti-human CD59, CD55 or CD46 (□) or without antibody (▪) and washed. Then, the cells were treated for 30 min on ice with FITC-conjugated goat anti-mouse IgG, washed and analysed by flow cytometry.
Table 1.
Secretion of soluble CD59 by carcinoma cells
| Carcinoma Cell line | sCD59 concentration* (pg/106 cells ± SE) |
|---|---|
| T47D | 384 ± 41 |
| SKOV3 | 811 ± 95 |
| PC-3 | 2516 ± 268 |
Soluble CD59 (sCD59) was collected from supernatants of CA cells and quantified in an ELISA as described under Methods.
To determine which of the mCRP has a major impact on complement resistance, the mCRP activity was blocked with specific antibodies. This has been shown to lead to significant sensitization of tumour cells to complement-mediated lysis [27,30]. Neutralization of CD59 had a large effect on resistance of SKOV3 and PC-3 cells to lysis (Fig. 2). In contrast, anti-CD55 antibodies had only a small effect on these cells. Anti–CD46 antibodies were ineffective and induced some lysis only in SKOV3 cells. In T47D cells, anti-CD59 and anti-CD55 antibodies produced practically the same rather low level of increased cell lysis by antibody and complement. A mixture of the three inhibitory antibodies produced a pronounced effect on the breast (T47D) and prostate (PC-3) carcinoma cells (Fig. 2). This was evident at optimal (Fig. 2a) and suboptimal (Fig. 2b) dilutions of the blocking antibodies, suggesting a synergism in the activity of the mCRP.
Contribution of PKC, PKA and ERK to CA resistance to complement
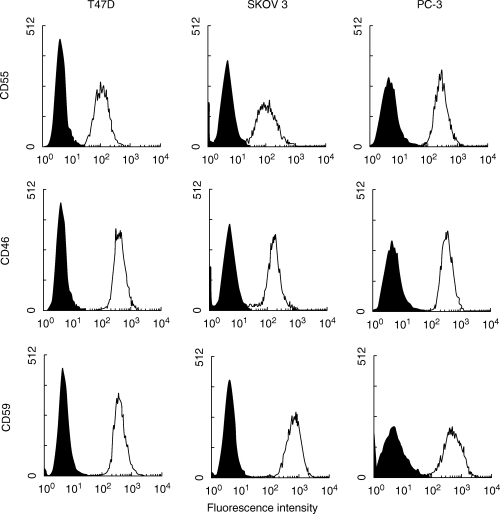
Our earlier results [17] demonstrated that PKC supports resistance of the K562 human erythroleukaemia cells to complement-mediated lysis. To analyse the significance of PKC activity in complement resistance of the carcinoma cell lines, we employed GF109203X [31], a selective PKC inhibitor. The optimal, nontoxic dose of each of the inhibitors described below was determined in a separate dose–response titration (not shown). Pre-incubation of tumour cells with GF109203X significantly increased their sensitivity to complement, in comparison with control cells incubated with DMSO (Fig. 3). The differences were all highly statistically significant (T47D, SKOV3: P < 0·001; PC-3: P < 0·0001).
Fig. 3.
Inhibitors of PKC, PKA and MEK increase cell sensitivity to lysis by antibody and complement. Carcinoma cells (0·5 × 106) were pretreated with GF109203X, H89 or PD98059, each at 1 µm, or with combinations of these inhibitors, for 30 min at 37°C. Control cells were pretreated with DMSO, 0·2%. Then, the cells were treated with rabbit anti-carcinoma antiserum for 30 min on ice followed by NHS, 60 min at 37°C. Results shown in Figs 3, 1, 2, 3, 4, 5 are each representative of 3 independent experiments and are expressed as the mean percentage of cell lysis (trypan blue inclusion) ± SE.
The involvement of the cAMP-dependent kinase PKA in tumour cell protection was next studied by using the PKA specific inhibitor H89 [32,33]. Pre-incubation of tumour cells with H89 increased complement-mediated lysis relative to control cells (statistically significant: for T47D and SKOV3, P < 0·001 and for PC-3, P < 0·0001) (Fig. 3). Another protein kinase that plays a role in the process of cell protection from complement is the extracellular-regulated protein kinase ERK [18]. PD98059 is an inhibitor of MEK, the kinase activating ERK. T47D, SKOV3 and PC-3 cells pretreated with PD98059 were more sensitive (P < 0·01; P < 0·005; P < 0·0001, respectively) to lysis by antibody and complement than untreated cells (Fig. 3).
The effect of various combinations of protein kinase inhibitors was also studied (Fig. 3). The combined action of two kinase inhibitors was, in most cases, significantly more effective than each inhibitor alone, and the combined action of the 3 inhibitors was significantly more effective than combination of two inhibitors (P < 0·05 and below). However, the overall increase was rather small (10–20%) and could not support a claim of an additive effect in the combined action of the protein kinases tested. Under the assay conditions used in the experiments described above, the protein kinase inhibitors themselves (in the absence of antibody and complement), separately or combined, did not cause cell death (necrotic or apoptotic) even at 48 h post-treatment.
Restriction of lysis by sialic acid
Removal of sialic acid from the cell surface with neuraminidase (NA) has been shown to confer on certain cell types increased sensitivity to complement-mediated lysis. Here we tested whether or not surface sialic acid residues contribute to the resistance of carcinoma cells to complement lysis. T47D, SKOV3 and PC-3 cells were treated with neuraminidase under condition known to remove most of the sialic acid residues from the cell surface. As shown in Fig. 4, these cells became more sensitive to lysis by antibody and complement. Flow cytometry analyses showed that NA-treated cells express the same amount of mCRPs as control cells, but bind a slightly higher (10–15%) amount of the rabbit anti-tumour cell antibodies. To identify the pathway of complement activated by NA-treated cells, we first subjected them to lysis by factor B-depleted human serum. In absence of factor B, lysis of NA-treated cells was reduced by 65–69% and lysis of control cell was reduced by 38–49% (Tables 2 and 3). Upon addition of purified factor B to factor B-depleted serum, cell lysis of CA was comparable to that obtained with NHS. Upon Ca2+ chelation with MgEGTA, lysis of control and NA-treated T47D, SKOV3 and PC-3 cells was reduced by 30–38%, and the factor of enhancement of lysis by NA was similar in absence or presence of MgEGTA (results not shown). The effect of the inhibitory 3F8 anti-MBL mAb and of the isotype control 1C10 anti-MBL mAb [34] on lysis of NA-treated and control cells was tested. Lysis of NA-treated cells was not inhibited by 3F8 and 1C10, whereas lysis of control cells was slightly reduced (12–18%) by 3F8 and not by 1C10 (results not shown). The relative percentage inhibition of lysis by fB removal, MgEGTA or 3F8 mAb is shown in Table 3.
Fig. 4.
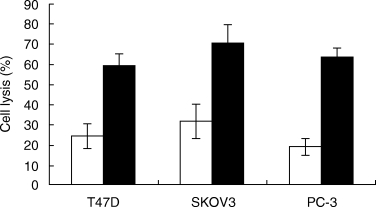
Treatment with neuraminidase is decreasing tumour cell resistance to complement-mediated lysis. CA cells (0·15 × 106) were incubated for 60 min at room temperature with 10 µU of recombinant α2–3,6,8,9 neuraminidase in PBS (▪). Control cells were incubated with PBS (□). Cells were washed and subjected to lysis by antibody (T47D, 1 : 64; SKOV3, 1 : 16; PC-3, 1 : 16) and NHS (1 : 2).
Table 2.
Involvement of the alternative pathway in lysis of neuraminidase-treated carcinoma cells by complement
| Cells | Treatment* | Serum† | Lysis (%) |
|---|---|---|---|
| T47D | Control | NHS | 38·0 ± 1·4 |
| NA | NHS | 71·0 ± 15·1 | |
| Control | RB | 18·3 ± 3·6 | |
| NA | RB | 20·3 ± 6·7 | |
| Control | RB + fB | 33·0 ± 3·4 | |
| NA | RB + fB | 65·5 ± 14·0 | |
| SKOV3 | Control | NHS | 41·8 ± 6·1 |
| NA | NHS | 72·3 ± 8·2 | |
| Control | RB | 22·5 ± 3·0 | |
| NA | RB | 27·8 ± 1·0 | |
| Control | RB + fB | 44·3 ± 5·7 | |
| NA | RB + fB | 78·8 ± 5·1 | |
| PC-3 | Control | NHS | 36·8 ± 7·9 |
| NA | NHS | 70·5 ± 11·1 | |
| Control | RB | 19·5 ± 2·4 | |
| NA | RB | 24·8 ± 3·3 | |
| Control | RB + fB | 31·5 ± 6·6 | |
| NA | RB + fB | 76·5 ± 10·0 |
Cells were treated first with neuraminidase (NA) or PBS (control) and then lysed by antibody and serum. Percentage lysis was determined by trypan blue inclusion.
NHS, normal human serum; RB, human serum depleted of factor B (fB); RB + fB, RB supplemented with purified fB.
Table 3.
Analysis of the complement pathways involved in lysis of CA cells by antibody and human serum
| Inhibition of lysis (%)† | |||
|---|---|---|---|
| Cells* | RB | MgEGTA-NHS | Anti-MBL (3F8)-NHS |
| T47D | 49 | 30 | 12 |
| SKOV3 | 38 | 38 | 15 |
| PC-3 | 45 | 34 | 18 |
| NA-T47D | 65 | 39 | ‡ns |
| NA-SKOV3 | 68 | 37 | ‡ns |
| NA-PC-3 | 69 | 35 | ns |
Cells were treated first with neuraminidase (NA) or PBS (control) and then lysed by antibody and serum. Percentage lysis was determined by trypan blue inclusion.
Percentages presented indicate a significant (P < 0·05) inhibition of cell lysis relative to control NHS. NHS, normal human serum; RB, human serum depleted of factor B; MgEGTA, 2·5 mm MgCl2 and 10 mm EGTA in NHS; 3F8, 67 µg/ml 3F8 mAb in NHS.
No significant inhibition.
Increase of CA cell lysis by mixtures of inhibitors of complement resistance mechanisms
Neutralization of CD59 (Fig. 2), inhibition of MEK/ERK (Fig. 3) and removal of sialic acid residues (Fig. 4), each leads to some reduction in CA protection from complement. To optimize complement-mediated killing of CA, we applied combinations of anti-CD59 antibody, neuraminidase and PD98059 (Fig. 5). The combined treatment of CA with any two of these agents significantly enhanced CA lysis in comparison with CA treated with anti-CD59 antibody, neuraminidase or PD98059 alone. Maximal killing was observed in CA treated with the 3 inhibitors. For the three cell types, the combined effect of the three inhibitory protocols was statistically more effective (P < 0·05 and below) than any combination of 2 inhibitory treatments. None of the ‘sensitizers’, the anti-CD59 antibodies (BRIC 229), PD98059 and neuraminidase, induced complement-mediated tumour cell killing in the absence of the rabbit anti-tumour cells antibodies. To rule out a possible interference between the complement activating polyclonal antibodies and the anti-CD59 antibodies, the carcinoma cells were first treated with the polyclonal rabbit anti-tumour antibodies, washed and then treated with the anti-CD59 antibodies. Binding of the anti-CD59 antibodies was detected by using FITC-labelled rabbit anti-mouse IgG antibodies. Analysis by flow cytometry showed that the binding of the anti-CD59 antibodies was not affected by the binding of the anti-tumour cells antibodies.
Fig. 5.
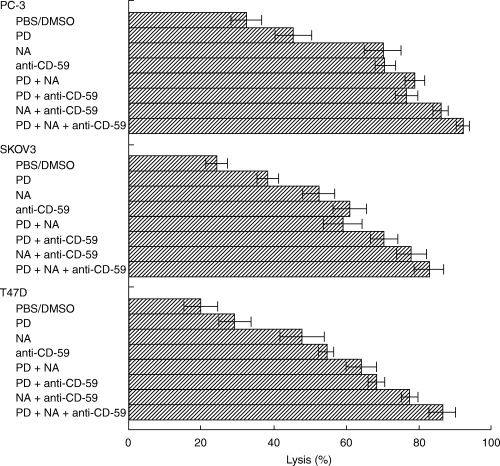
Mixtures of antibody anti-CD59, neuraminidase and MEK inhibitor have additive effects on complement-mediated lysis of carcinoma cells. CA cells (0·15 × 106) were incubated with PD98059 (1 µm) (PD) or DMSO (0·001%) for 60 min at 37°C and then with (or without) 10 µU neuraminidase (NA) for 60 min at room temperature. Control cells were incubated with PBS. Washed cells were incubated with anti-carcinomas antibodies mixed with or without anti-CD59 antibody (clone BRIC229) for 30 min on ice. Next, NHS was added and the cells were further incubated for 60 min at 37°C. Percentage cell lysis was determined by trypan blue exclusion.
Complement-induced protection and PKC
Leukaemic cells treated with sublytic doses of antibody and complement become resistant to higher, lytic doses of complement [23]. As shown here (Fig. 6), breast, ovarian and prostate CA cells responded similarly to sublytic complement doses. Within 1 h after treatment with sublytic complement, sensitivity of the CA cells to complement-mediated lysis decreased by 35–45%. Pre-treatment of the carcinoma cells with calphostin C, a selective PKC inhibitor [35], significantly impaired their capacity to respond to the complement-induced protection signal (Fig. 6). Apparently, PKC activation is induced in carcinoma cells in response to sublytic complement and is essential to the enhancement of complement resistance.
Fig. 6.
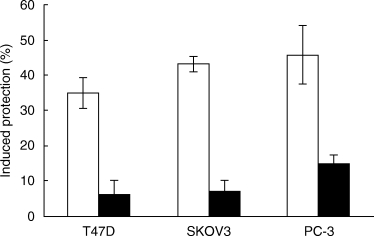
A sublytic dose of antibody and complement protects carcinoma cells from lytic doses of complement; involvement of PKC. T47D, SKOV3 and PC-3 cells were pretreated under light with calphostin C (1 µm) (▪) or with DMSO (0·03%) as control (□), for 30 min at 37°C. Next, the cells were incubated with rabbit anti-CA antibodies and with NHS or HI-NHS at a dilution producing a sublytic effect of complement (further details in Methods). Finally, the cells were washed and incubated with lytic doses of antibody and complement (NHS). Percentage cell lysis was determined by trypan blue exclusion and percentage protection was calculated as explained under Methods.
Discussion
Earlier studies with normal and malignant tissues have disclosed the existence of several mechanisms of cell protection from complement-mediated lysis. Those studies suggested that each cell type is equipped with a composite of resistance mechanisms. This assumption was tested here for the first time with human carcinoma cell lines. As shown in this comparative analysis, breast, ovarian and prostate carcinoma cells are equipped with three distinct mechanisms conferring resistance against antibody-dependent complement-mediated cell lysis. These cells utilize surface mCRP and sialic acid residues and intracellular protein phosphorylation cascades to resist a complement attack (Figs 2–5). In addition, in response to extracellular signals (e.g. by PMA and sublytic MAC), they can enhance their basal resistance to complement. It is reasonable to assume that these cells contain additional protective mechanisms, yet undiscovered. A similar study performed recently on K562 erythroleukaemic cells [27] also demonstrated a cooperation of several complement defense mechanisms: extracellular mCRP, soluble complement regulatory proteins and serine proteases and intracellular PKC-dependent protein phosphorylation processes.
The three carcinoma cell types studied here were found to express on their surface CD46, CD55 and CD59 (Fig. 1). mCRP have been described in K562 [36,37], lung [9], melanoma [38], ovarian [39], colon [40], breast [41], prostate [42] and other tumours. Blocking anti-mCRP monoclonal antibodies have been used to demonstrate that mCRP contribute to cell resistance to complement. As shown here, anti-CD46 mAb alone has almost no effect on basal resistance of the carcinoma cells [30] (Fig. 2) or K562 cells [27] to complement-mediated lysis. Anti-CD55 mAb has a significant lysis promoting effect only on breast carcinoma cells [30] (Fig. 2) and on K562 cells [27]. On the other hand, blocking of CD59 with mAb sensitizes breast, ovarian and prostate carcinoma (Fig. 2) as well as K562 cells [27] to complement. The variation among cell types in the significance of the mCRP to complement resistance, could perhaps reflect differences in their cell surface topology, for example, the composition and thickness of their glycocalyx [43] that surrounds the mCRP. Surface bound C3b and C3 convertases may also show variable accessibility to CD46 and CD55 depending on their surface location and possible association with other molecules. Effective MAC must form at the plane of the plasma membrane and its accessibility to CD59 may be affected by other surface structures. Analysis of mCRP in frozen sections of carcinomas demonstrated variable expression of CD46, CD55 and CD59 on tumours cells but also occurrence of large quantities of CD55 and CD59, probably in a soluble form, in tumour stroma [7]. As demonstrated also in melanoma cells [44], K562 cells [27] and the CA lines tested here (Table 1), a soluble form of CD59 is secreted and possibly even amplifies the protective capacity of membrane CD59. The antibody blocking results suggest that in the cell types studied, CD59 executes a more complete inhibition of the complement cascade than CD46 and CD55. Alternatively, the observed inefficiency of anti-CD55 and anti-CD46 mAb may be simply due to a lower inhibitory capacity relative to that of the anti-CD59 mAb. A recent study with renal CA suggested that CD46 is the major in vivo complement protector of these cells [45]. Clearly, cooperation and synergism among the three mCRP may amplify their individual protective capacity [25] (Fig. 2b).
Carcinoma cells utilize also sialic acid residues on their surface for protection from complement (Fig. 4). To avoid the possibility that some of the activity detected is due to contaminating proteases, this study was performed with a highly purified recombinant neuraminidase. Our findings are not surprising, as earlier studies have demonstrated that removal of sialic acid from red blood cells [14,46], rabbit lymph node cells [47], murine sarcoma cells [48], human bladder CA [15], Trypanosoma cruzi trypomastigotes [49], Neisseria meningitides[50] and N. gonorrhoeae[51] sensitized them to lysis by complement; high sialic acid expression correlates with lower complement activation. Inactivation of C3b by Factors H and I occurs more efficiently on surfaces of cells that are rich in sialic acid [46, 52, 53]. Therefore, it is reasonable to assume that following removal of sialic acid with neuraminidase, the carcinoma cells deposit more C3b and this, in turn, leads to formation of larger numbers of MAC. Following sialic acid removal, binding of the rabbit anti-tumour cell antibodies to the cells slightly increased. It is not clear yet, whether this increased binding represents an enhancement in antigenic exposure or increased nonspecific adherence of the rabbit IgG to the NA-treated cells. The fact that lysis of NA-treated cells depends more on factor B (65–69%) than lysis of control cells (38–49%) (Tables 2 and 3) indicates that NA-treated cells activate better the alternative complement pathway. Involvement of the lectin complement pathway in lysis of NA-treated cells was ruled out by data showing no significant effect of anti-MBL antibodies on cell lysis (Table 3). Interestingly, lysis of control carcinoma cells in presence of MgEGTA or of the 3F8 anti-MBL mAb was reduced by 30–40% or 15%, respectively (Table 3). Hence, it appears that the alternative, lectin and classical initiation pathways cooperate in lysis of CA cells by antibody and NHS. However, upon removal of sialic acid, the CA cells appear to activate primarily the alternative and classical complement pathways. It is possible that the capacity of MBL to bind to or activate the lectin pathway on CA cells is controlled in a negative or positive manner by sialic acid residues. Lysis of K562 erythroleukaemia cells by antibody and NHS is also mostly dependent on activation of both alternative and classical complement pathways [54].
Cells are triggered by the activated complement proteins generated upon complement attack, in particular by the MAC deposited on their surface. Perhaps the earliest response induced by the MAC is the increase in intracellular Ca2+ occurring within a minute post MAC insertion [55]. A wide range of physiological responses has been reported in cells stressed by the complement MAC. Our goal was to identify those intracellular responses that contribute to resistance of human carcinoma cells to the lytic action of the MAC. As shown here (Figs 3 and 5), carcinoma cells utilize three protein kinases, PKC, PKA and ERK, in their defense against MAC action. To determine whether these kinases belong to the same or different signalling cascades, the combined effect of kinase inhibitors was tested. We found that the mixture of kinase inhibitors produced in CA cells only a small increase in their sensitivity to complement as compared with the effect of each of the kinase inhibitors alone. This suggests that PKC, PKA and ERK act in protection of these CA cells from complement damage as components of the same signalling pathway. Recent analysis of the signalling cascade induced by MAC in K562 cells showed that activation of PKC precedes and is required for MEK/ERK activation [18].
Basal resistance to complement may be up-regulated by treatment of the CA cells with PMA (data not shown) or with sublytic MAC doses (Fig. 6). Both mechanisms involve intracellular protein phosphorylation and are inhibited by kinase inhibitors. This confirms earlier findings obtained with K562 cells [16,23] and breast carcinoma cells [30]. The protection conferred by long treatment with PMA can be explained by the enhanced expression of mCRP [30,37]. Similar effects of other pro-inflammatory agents such as TNFα, IL-1α, IL-1β and IL-4 on mCRP expression have been reported [9, 56, 57]. The phenotypic conversions induced by the latter cytokines usually occur only after several hours of treatment. However, sublytic MAC doses and PMA are known to induce within minutes in K562 cells enhanced resistance to complement-mediated lysis without an effect on the level of mCRP expression [3, 23, 58]. The protective mechanism activated within 15–45 min is still under investigation. It may involve members of the heat shock proteins family [59]. The protein kinases activated by sublytic MAC may also facilitate removal of the MAC from the cell surface and/or repair of the damage caused by the MAC.
Future approaches aimed at utilizing complement-based immunotherapeutic reagents against cancer cells will have to overcome complement resistance of these cells. Each tumour probably contains a different composite of evasion strategies. Besides the expression of mCRP and sialic acid, carcinoma cells may perhaps bear on their surface other protective molecules such as proteases [27,60] or kinases [61] or secrete soluble complement inhibitors [27,44]. Attention should also be paid to the possibility that certain therapeutical reagents may actually confer on the cancer cells resistance to immunotherapy. Better insights into the intracellular pathways and mechanisms involved in complement resistance are required to enable development of efficient counter-acting reagents. It is conceivable that a concerted action against the extracellular and intracellular complement protective mechanisms will be required to achieve efficient antibody- and complement-mediated cancer immunotherapy.
Acknowledgments
This work was supported in part by the Cooperation Program in Cancer Research of the Deutsches Krebsforschungszentrum (DKFZ) and Israeli Ministry of Science (MOS), by the Israel Cancer Research Foundation and by a CapCURE award.
References
- 1.Green MC, Murray JL, Hortobagyi GN. Monoclonal antibody therapy for solid tumors. Cancer Treat Rev. 2000;26:269–86. doi: 10.1053/ctrv.2000.0176. [DOI] [PubMed] [Google Scholar]
- 2.Vose JM. Antibody-targeted therapy for low-grade lymphoma. Semin Hematol. 1999;36:15–20. [PubMed] [Google Scholar]
- 3.Jurianz K, Ziegler S, Garcia-Schuler H, et al. Complement resistance of tumor cells: basal and induced mechanisms. Mol Immunol. 1999;36:929–39. doi: 10.1016/s0161-5890(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 4.Hourcade D, Liszewski MK, Krych-Goldberg M, et al. Functional domains, structural variations and pathogen interactions of MCP, DAF and CR1. Immunopharmacology. 2000;49:103–16. doi: 10.1016/s0162-3109(00)80296-9. [DOI] [PubMed] [Google Scholar]
- 5.Davies A, Lachmann PJ. Membrane defence against complement lysis. the structure and biological properties of CD59. Immunol Res. 1993;12:258–75. doi: 10.1007/BF02918257. [DOI] [PubMed] [Google Scholar]
- 6.Meri S, Morgan BP, Davies A, et al. Human protectin (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- 7.Niehans GA, Cherwitz DL, Staley NA, et al. Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin) Am J Pathol. 1996;149:129–42. [PMC free article] [PubMed] [Google Scholar]
- 8.Simpson KL, Jones A, Norman S, et al. Expression of the complement regulatory proteins decay accelerating factor (DAF, CD55), membrane cofactor protein (MCP, CD46) and CD59 in the normal human uterine cervix and in premalignant and malignant cervical disease. Am J Pathol. 1997;151:1455–67. [PMC free article] [PubMed] [Google Scholar]
- 9.Varsano S, Rashkovsky L, Shapiro H, et al. Human lung cancer cell lines express cell membrane complement inhibitory proteins and are extremely resistant to complement-mediated lysis; a comparison with normal human respiratory epithelium in vitro, and an insight into mechanism (s) of resistance. Clin Exp Immunol. 1998;113:173–82. doi: 10.1046/j.1365-2249.1998.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Spendlove I, Morgan J, et al. CD55 is over-expressed in the tumour environment. Br J Cancer. 2001;84:80–6. doi: 10.1054/bjoc.2000.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junnikkala S, Jokiranta TS, Friese MA, et al. Exceptional resistance of human H2 glioblastoma cells to complement-mediated killing by expression and utilization of factor H and factor H-like protein 1. J Immunol. 2000;164:6075–81. doi: 10.4049/jimmunol.164.11.6075. [DOI] [PubMed] [Google Scholar]
- 12.Charriaut-Marlangue C, Barel M, Frade R. Identification of P-57, a serine proteinase, from human erythrocyte membranes, which cleaves both chains of human third component (C3) of complement. Biochem Biophys Res Commun. 1986;140:1113–20. doi: 10.1016/0006-291x(86)90750-3. [DOI] [PubMed] [Google Scholar]
- 13.Ollert MW, Frade R, Fiandino A, et al. C3-cleaving membrane proteinase. A new complement regulatory protein of human melanoma cells. J Immunol. 1990;144:3862–7. [PubMed] [Google Scholar]
- 14.Lauf PK. Immunological and physiological characteristics of the rapid immune hemolysis of neuraminidase-treated sheep red cells produced by fresh guinea pig serum. J Exp Med. 1975;142:974–88. doi: 10.1084/jem.142.4.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobsen F. Increase of the in vitro complement-dependent cytotoxicity against autologous invasive human bladder tumor cells by neuraminidase treatment. Acta Pathol Microbiol Immunol Scand. 1982;90:187–92. doi: 10.1111/j.1699-0463.1982.tb01437.x. [DOI] [PubMed] [Google Scholar]
- 16.Fishelson Z, Kopf E, Paas Y, et al. Protein phosphorylation as a mechanism of resistance against complement damage. Prog Immunol. 1989;7:205–8. [Google Scholar]
- 17.Kraus S, Fishelson Z. Cell desensitization by sublytic C5b-9 complexes and calcium ionophores depends on activation of protein kinase C. Eur J Immunol. 2000;30:1272–80. doi: 10.1002/(SICI)1521-4141(200005)30:5<1272::AID-IMMU1272>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol. 2001;123:366–74. doi: 10.1046/j.1365-2249.2001.01477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohanian SH, Schlager SI. Humoral immune killing of nucleated cells: mechanisms of complement-mediated attack and target cell defense. Crit Rev Immunol. 1981;1:165–209. [PubMed] [Google Scholar]
- 20.Carney DF, Koski CL, Shin ML. Elimination of terminal complement intermediates from the plasma membrane of nucleated cells: the rate of disappearance differs for cells carrying C5b-7 or C5b-8 or a mixture of C5b-8 with a limited number of C5b-9. J Immunol. 1985;134:1804–9. [PubMed] [Google Scholar]
- 21.Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J Immunol. 1987;138:246–53. [PubMed] [Google Scholar]
- 22.Reiter Y, Ciobotariu A, Jones J, et al. Complement membrane attack complex, perforin, and bacterial exotoxins induce in K562 cells calcium-dependent cross-protection from lysis. J Immunol. 1995;155:2203–10. [PubMed] [Google Scholar]
- 23.Reiter Y, Ciobotariu A, Fishelson Z. Sublytic complement attack protects tumor cells from lytic doses of antibody and complement. Eur J Immunol. 1992;22:1207–13. doi: 10.1002/eji.1830220515. [DOI] [PubMed] [Google Scholar]
- 24.Reiter Y, Fishelson Z. Complement membrane attack complexes induce in human leukemic cells rapid expression of large proteins (L-CIP) Mol Immunol. 1992;29:771–81. doi: 10.1016/0161-5890(92)90187-3. [DOI] [PubMed] [Google Scholar]
- 25.Brodbeck WG, Mold C, Atkinson JP, et al. Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol. 2000;165:3999–4006. doi: 10.4049/jimmunol.165.7.3999. [DOI] [PubMed] [Google Scholar]
- 26.Chen S, Caragine T, Cheung NK, et al. CD59 expressed on a tumor cell surface modulates decay-accelerating factor expression and enhances tumor growth in a rat model of human neuroblastoma. Cancer Res. 2000;60:3013–8. [PubMed] [Google Scholar]
- 27.Jurianz K, Ziegler S, Donin N, et al. K562 erythroleukemic cells are equipped with multiple mechanisms of resistance to lysis by complement. Int J Cancer. 2001;93:848–54. doi: 10.1002/ijc.1406. [DOI] [PubMed] [Google Scholar]
- 28.Gotze O, Muller-Eberhard HJ. The C3-activator system: an alternate pathway of complement activation. J Exp Med. 1971;134:90s–108s. [PubMed] [Google Scholar]
- 29.Blomberg K, Hautala R, Lovgren J, et al. Time-resolved fluorometric assay for natural killer activity using target cells labelled with a fluorescence enhancing ligand. J Immunol Meth. 1996;193:199–206. doi: 10.1016/0022-1759(96)00063-4. [DOI] [PubMed] [Google Scholar]
- 30.Jurianz K, Maslak S, Garcia-Schuler H, et al. Neutralization of complement regulatory proteins augments lysis of breast carcinoma cells targeted with rhumAb anti-HER2. Immunopharmacology. 1999;42:209–18. doi: 10.1016/s0162-3109(99)00006-5. [DOI] [PubMed] [Google Scholar]
- 31.Toullec D, Pianetti P, Coste H, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–81. [PubMed] [Google Scholar]
- 32.Combest WL, Bloom TJ, Gilbert LI. Polyamines differentially inhibit cyclic AMP-dependent protein kinase-mediated phosphorylation in the brain of the tobacco hornworm, Manduca sexta. J Neurochem. 1988;51:1581–91. doi: 10.1111/j.1471-4159.1988.tb01128.x. [DOI] [PubMed] [Google Scholar]
- 33.Chijiwa T, Mishima A, Hagiwara M, et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–72. [PubMed] [Google Scholar]
- 34.Collard CD, Vakeva A, Morrissey MA, et al. Complement activation after oxidative stress. Role of the lectin complement pathway. Am J Pathol. 2000;156:1549–56. doi: 10.1016/S0002-9440(10)65026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi E, Nakano H, Morimoto M, et al. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;159:548–53. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- 36.Seya T, Kojima A, Hara T, et al. Enhancement of lymphocyte-mediated K562 cytotoxicity by antibodies against complement membrane cofactor protein (CD46) and decay-accelerating factor (CD55) Immunobiology. 1991;183:115–24. doi: 10.1016/S0171-2985(11)80191-9. [DOI] [PubMed] [Google Scholar]
- 37.Marchbank KJ, Morgan BP, van den Berg CW. Regulation of CD59 expression on K562 cells: effects of phorbol myristate acetate, cross-linking antibody and non-lethal complement attack. Immunology. 1995;85:146–52. [PMC free article] [PubMed] [Google Scholar]
- 38.Goslings WR, Blom DJ, de Waard-Siebinga I, et al. Membrane-bound regulators of complement activation in uveal melanomas. CD46, CD55, and CD59 in uveal melanomas. Invest Ophthalmol Vis Sci. 1996;37:1884–91. [PubMed] [Google Scholar]
- 39.Bjorge L, Hakulinen J, Wahlstrom T, et al. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. 1997;70:14–25. doi: 10.1002/(sici)1097-0215(19970106)70:1<14::aid-ijc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Koretz K, Bruderlein S, Henne C, et al. Expression of CD59, a complement regulator protein and a second ligand of the CD2 molecule, and CD46 in normal and neoplastic colorectal epithelium. Br J Cancer. 1993;68:926–31. doi: 10.1038/bjc.1993.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hakulinen J, Meri S. Expression and function of the complement membrane attack complex inhibitor protectin (CD59) on human breast cancer cells. Laboratory Invest. 1994;71:820–7. [PubMed] [Google Scholar]
- 42.Jarvis GA, Li J, Hakulinen J, et al. Expression and function of the complement membrane attack complex inhibitor protectin (CD59) in human prostate cancer. Int J Cancer. 1997;71:1049–55. doi: 10.1002/(sici)1097-0215(19970611)71:6<1049::aid-ijc22>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 43.Frey A, Giannasca KT, Weltzin R, et al. Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med. 1996;184:1045–59. doi: 10.1084/jem.184.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brasoveanu LI, Fonsatti E, Visintin A, et al. Melanoma cells constitutively release an anchor-positive soluble form of protectin (sCD59) that retains functional activities in homologous complement-mediated cytotoxicity. J Clin Invest. 1997;100:1248–55. doi: 10.1172/JCI119638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blok VT, Daha MR, Tijsma OM, et al. A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Laboratory Invest. 2000;80:335–44. doi: 10.1038/labinvest.3780038. [DOI] [PubMed] [Google Scholar]
- 46.Pangburn MK, Muller-Eberhard HJ. Complement C3 convertase. cell surface restriction of beta1H control and generation of restriction on neuraminidase-treated cells. Proc Natl Acad Sci USA. 1978;75:2416–20. doi: 10.1073/pnas.75.5.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ray PK, Sundaram K. Cytolysis of neuraminidase-treated autochthonous lymphoid cells by autologous serum. Clin Exp Immunol. 1975;19:529–32. [PMC free article] [PubMed] [Google Scholar]
- 48.Turianskyj FH, Gyenes L. The effect of neuraminidase on the sensitivity of tumor cells toward lysis by antibody and complement or by sensitized lymphocytes. Transplantation. 1976;22:24–30. doi: 10.1097/00007890-197607000-00004. [DOI] [PubMed] [Google Scholar]
- 49.Kipnis TL, David JR, Alper CA, et al. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc Natl Acad Sci USA. 1981;78:602–5. doi: 10.1073/pnas.78.1.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarvis GA, Vedros NA. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect Immun. 1987;55:174–80. doi: 10.1128/iai.55.1.174-180.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ram S, Sharma AK, Simpson SD, et al. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med. 1998;187:743–52. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kazatchkine MD, Fearon DT, Austen KF. Human alternative complement pathway. membrane-associated sialic acid regulates the competition between B and beta1 H for cell-bound C3b. J Immunol. 1979;122:75–81. [PubMed] [Google Scholar]
- 53.Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA. 1990;87:3982–6. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiter Y, Fishelson Z. Killing of human tumor cells by antibody C3b conjugates and human complement. Targeted Diagn Ther. 1989;2:119–35. [PubMed] [Google Scholar]
- 55.Morgan BP, Campbell AK. The recovery of human polymorphonuclear leucocytes from sublytic complement attack is mediated by changes in intracellular free calcium. Biochem J. 1985;231:205–8. doi: 10.1042/bj2310205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bjorge L, Jensen TS, Matre R. Characterisation of the complement-regulatory proteins decay-accelerating factor (DAF, CD55) and membrane cofactor protein (MCP, CD46) on a human colonic adenocarcinoma cell line. Cancer Immunol Immunother. 1996;42:185–92. doi: 10.1007/s002620050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hindmarsh EJ, Marks RM. Decay-accelerating factor is a component of subendothelial extracellular matrix in vitro, and is augmented by activation of endothelial protein kinase C. Eur J Immunol. 1998;28:1052–62. doi: 10.1002/(SICI)1521-4141(199803)28:03<1052::AID-IMMU1052>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 58.Marchbank KJ, van den Berg CW, Morgan BP. Mechanisms of complement resistance induced by non-lethal complement attack and by growth arrest. Immunology. 1997;90:647–53. doi: 10.1046/j.1365-2567.1997.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fishelson Z, Hochman I, Greene LE, et al. Contribution of heat shock proteins to cell protection from complement-mediated lysis. Int Immunol. 2001;13:983–91. doi: 10.1093/intimm/13.8.983. [DOI] [PubMed] [Google Scholar]
- 60.Frade R. Structure and functions of proteases which cleave human C3 and are expressed on normal or tumor human cells: some are involved in tumorigenic and metastatic properties of human melanoma cells. Immunopharmacology. 1999;42:39–45. doi: 10.1016/s0162-3109(99)00028-4. [DOI] [PubMed] [Google Scholar]
- 61.Paas Y, Bohana-Kashtan O, Fishelson Z. Phosphorylation of the complement component, C9, by an ecto-protein kinase of human leukemic cells. Immunopharmacology. 1999;42:175–85. doi: 10.1016/s0162-3109(99)00027-2. [DOI] [PubMed] [Google Scholar]