

Abstract
Reactive oxygen species generated during various metabolic and biochemical reactions have multifarious effects that include oxidative damage to DNA leading to various human degenerative and autoimmune diseases. The highly reactive hydroxy radical (·OH) can interact with chromatin and result in a wide range of sugar and base-derived products, DNA–protein cross-links and strand breaks. Studies from our laboratory have demonstrated that after modification the DNA becomes highly immunogenic and the induced antibodies exhibit variable antigen-binding characteristics. Systemic lupus erythematosus, a prototype autoimmune disease, is characterized by the presence of autoantibodies to multiple nuclear antigens. The detection of 8-hydroxyguanosine in the immune complex derived DNA of systemic lupus erythematosus patients reinforces the evidence that reactive oxygen species may be involved in its pathogenesis. Increased apoptosis and decreased clearance of apoptotic cells as observed in systemic lupus erythematosus (SLE) might well be a contributory factor in systemic autoimmunity. Clinically, titres of autoantibodies are closely related to the degree of renal inflammation. Anti-DNA antibodies may combine with circulating antigen and contribute to the deposition of immune complexes in renal glomeruli.
Keywords: apoptosis, anti-DNA antibodies, autoantibodies, ROS, SLE, systemic autoimmunity
Reactive oxygen species
A free radical is any species capable of independent existence containing one or more unpaired electrons [1]. The unpaired electron alters the chemical reactivity of the molecule/atom, making it more reactive than the corresponding non-radical form. The oxygen free radicals include superoxide anion radical (O2·–), singlet oxygen (1O2), hydroxyl radical (·OH) and perhydroxyl radical (HO2·) and are termed collectively the ‘reactive oxygen species’ (ROS). The usual route of O2 metabolism is through its complete reduction to H2O by accepting four electrons. However, with a single electron reduction several free radicals and hydrogen peroxide (H2O2) are formed. In vivo, ROS are generated by oxidant enzymes, phagocytic cells, ionizing radiation, etc. Superoxide anion is believed to be the first radical formed, mainly by the electron transport chain when O2 picks up a single electron. Radicals such as ·OH, HO2· and H2O2 are formed from O2·– [2,3]. O2·– undergoes a dismutation reaction catalysed by the enzyme superoxide dismutase (SOD) to form H2O2, which by itself is not reactive enough to cause damage to macromolecules. It is, however, a very important oxidant since it can cross biological membranes and form the highly reactive ·OH by interaction with transition metal ions such as Fe2+ or Cu+.
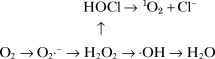
H2O2 is reduced by three general mechanisms. First, it is a substrate for two enzymes, catalase and glutathione peroxidase, that catalyse its conversion to H2O and O2 [4], a detoxification mechanism. Secondly, H2O2 is converted by myeloperoxidase (MPO) in neutrophils to hypochlorous acid (HOCl), a strong oxidant that acts as a bactericidal agent in phagocytic cells. Reaction of HOCl with H2O2 yields 1O2. Thirdly, H2O2 is converted in a spontaneous reaction catalysed by transition metal ions to the highly reactive ·OH.
 |
Among the ROS, ·OH is the most potent damaging radical which can react with all biological macromolecules (lipids, proteins, nucleic acids and carbohydrates). It is extremely reactive and can lead to formation of DNA-protein cross-links, single- and double-strand breaks, base damage, lipid peroxidation and protein fragmentation [5,6]. It may also be generated by ionizing radiation [7]:
The cellular generation of ·OH may occur in two steps [8]:
- Reduction of H2O2 by the Fenton reaction:
- Interaction of O2·– with H2O2 by the Haber–Weiss reaction:
Among the more susceptible targets of ·OH are polyunsaturated fatty acids. Abstraction of a hydrogen atom from a molecule of polyunsaturated fatty acid initiates the process of lipid peroxidation. A hydrogen atom is abstracted from a second molecule, leading to a new free radical. Protein structure and functions are also modified by ROS. Metal ion catalysed protein oxidation results in addition of carbonyl groups, cross-linking and fragmentation. Aldehydes of lipid peroxidation can react with sulphydryl (cysteine) or basic amino acids (histidine, lysine) affecting their biological characteristics. Similarly, modification of individual nucleotide bases, single strand breaks and cross-linking are the typical effects of ROS on nucleic acids [9].
The damage to DNA by ·OH includes single-strand breaks, base modifications and conformational changes. Nitrogenous bases react preferentially with ·OH rather than sugar moiety by 4–6-fold. Thymine and guanine are most susceptible to modifications followed by cytosine and adenine. Thymine glycol is the major oxidation product, its presence in urine serves as an indicator of endogenous DNA damage. Cytosine glycols are also formed which can undergo deamination to form uracil derivatives that base pair preferentially with adenine, instead of guanine. Reduction of guanine leads to ring opening forming formamidopyrimidine (FAPy) derivative of guanine (FAPyG). Oxidation leads to the formation of 8-oxo-deoxyguanine (8-oxodG), a major product. Its measurement in urine is used as a biomarker of endogenous oxidative DNA damage [10].
ROS generation through normal cellular metabolism and by exogenous stimulus is a constant problem for which cells have developed multiple defense mechanisms to survive [11,12]. An imbalance between free radical generation and sequestration leads to oxidative stress. ROS are generated by mitochondria through the electron transport chain as toxic by products of oxidative phosphorylation [13]. In addition, free radical production and disturbances in redox status can modulate the expression of a variety of immune and inflammatory molecules [14–16] leading to inflammatory processes, exacerbating inflammation and affecting tissue damage [17]. It has been suggested that abnormal immunity is related to oxidative imbalance [18,19] and antioxidant functions are linked to anti-inflammatory and/or immunosuppressive properties [20–22]. Neutrophils, which constitute about 60% of the circulating leucocytes and are the most abundant cellular components of the immune system, produce ROS resulting in oxidative damage and inflammation. The phagocytosis of bacteria, secretion of proteolytic enzymes and immunomodulatory agents are accompanied by ‘respiratory burst’, involving a sudden increase in oxidative metabolism that results in the production of ROS [23].
Systemic autoimmune disorders
In some autoimmune diseases, such as Goodpasture's syndrome, antibodies are directly related to pathogenesis of the disease, but in many instances the relationship between the disease and prevalent autoantibodies is vague. The diseases may be organ-specific, such as diabetes, autoimmune thyroiditis, Goodpasture's syndrome and primary biliary cirrhosis or systemic, such as progressive systemic sclerosis (PSS, scleroderma) and systemic lupus erythematosus (SLE). Damage due to inflammatory processes is seen more often in systemic diseases than organ-specific disease. With systemic diseases, inflammation is associated with vasculitis, skin rash, swelling of joints, cutaneous ulceration, peripheral gangrene, neuropathy and visceral abnormalities. These clinicopathological features define a group of diseases that over the years have come to be known as collagen-vascular diseases, immunological diseases of the connective tissue or rheumatic diseases [24]. Mixed connective tissue disease (MCTD) was first presented as a distinct rheumatic disease syndrome characterized by high titres of antibodies specific to a ribonucleoprotein (anti-RNA antibodies) and the clinical features were similar to that of other defined connective tissue diseases. The most common clinical feature were Raynaud's phenomenon, puffy hands, arthritis and myositis, also characteristic of other connective diseases. The MCTD patients also have positive rheumatoid factor and high serum levels of immunoglobulins, suggesting an abnormal humoral immune response [25,26].
In systemic autoimmune diseases, individuals develop autoantibodies directed against a variety of cellular components. It is remarkable that a particular set of autoantibody specificities is associated with each disease. Hence, these autoantibody profiles constitute a useful and valuable tool in their diagnosis. Some of these autoantibody specificities show a unique disease-restriction such as the anti-Sm autoantibody found exclusively in the sera of SLE patients. Other specificities show a broader association, such as anti-U1 RNP antibody found in high titres in patients with MCTD and in low titres in SLE [27]. This clinical and serological heterogeneity has been identified and characterized in detail that is associated with specific autoantibodies. Thus, antibodies to DNA have been linked closely to nephritis. Antibodies to U1 RNP are found in SLE without nephritis and the overlapping syndrome of scleroderma, myositis, SLE or MCTD. Antibodies to histone are associated with drug-induced lupus erythematosus and antibodies to Ro (SSA) are related to cutaneous lupus erythematosus, neonatal lupus erythematosus, SLE–Sjogren's syndrome overlap, antinuclear antibody (ANA) negative SLE and Sjogren's syndrome (Table 1) [28].
Table 1.
Antibodies associated with systemic autoimmune diseases
| Antibody | Antigen | Disease |
|---|---|---|
| Antinuclear antibody | Nuclear antigens(dsDNA, ssDNA, histone, Sm,RNP, SS-A/Ro, SS-B/La) | SLE |
| Antinuclear antibody | Nuclear antigens(RNP, 70 kDa protein) | MCTD |
| Antinuclear antibody | Nuclear antigens(DNA, PM1, Jo-1) | Polymyositis,Dermatomyositis |
| Antinuclear antibody | Nuclear antigens(SS-A/Ro, SS-B/La) | Sjogren's syndrome |
| Antinuclear antibody | Nuclear antigens(nucleolar DNA, RNP, Scl70) | PSS |
| Rheumatoid factor(RF, anti-Fc) | Fc portion of IgG | RA |
| Antibodies to platelets | Platelet membrane antigens | SLE |
| Antibodies to RBC | Red blood cell antigens | SLE |
SLE: systemic lupus erythematosus, PSS: progressive systemic sclerosis, Sm: Smith antigen. MCTD: mixed connective tissue disease, RA: rheumatoid arthritis, RNP: ribonucleoprotein, SS-A/Ro: Sjogren's syndrome antigen A/Robert antigen, SS-B/La: Sjogren's syndrome antigen B.
Manifestations of autoimmunity are often complex and heterogeneous. It has been postulated that immune response against host antigens could result from genetic predisposition, exaggerated random B cell activity, cross-reactivity between foreign and host antigens. The foreign antigens arise as a consequence of infection, inflammation, drug administration, environmental factors, free radicals [29, 30, 31, 32, 33, 34, 35] and scores of other modifying agents. ROS have gained considerable interest in recent years as plausible causative agents in the pathogenesis of several human degenerative diseases [36]. The ·OH is generated in substantial amounts in chronic inflammatory conditions due to increased oxidative stress. Lymphocytes isolated from patients suffering from rheumatoid arthritis (RA) and SLE contain increased levels of 8-oxodG [37,38]. A study on blood monocytes isolated from SLE patients indicated an impairment in the removal of 8-oxodG from cellular DNA due to a deficient repair system, that may result in cell death and release of oxidized DNA. ROS have been known to cause damage to DNA and their relevance in the induction and development of cancer is well documented [39–42]. These radicals have also been implicated as causative agents of ageing [43] and of several human diseases including multiple sclerosis, Parkinson's disease and autoimmune disorders [44–46]. It has been proposed that in chronic inflammatory diseases such as RA and SLE, DNA–anti-DNA antibody complex(es) deposit in tissues and induce inflammation [47]. The phagocytic cells may then release ROS at the site of injury [48]. These oxygen species being highly reactive may penetrate cellular membranes and react with nuclear DNA [49,50].
Humoral autoimmunity
Two diseases that have been considered prototypes for systemic autoimmunity are SLE and RA. SLE is a multi-systemic disorder characterized by a variety of autoantibodies and abnormal lymphocyte function that may be responsible for many of the clinical manifestations that are important in diagnosis. A hallmark of this disease is the presence of antinuclear antibodies. ANA are prototype autoantibodies that mark the course of rheumatic diseases. These antibodies target a diverse range of macromolecules including DNA, RNA, proteins and protein–nucleic acid complexes. Because of the close association between ANA and clinical diagnosis, these antibodies have become a key component in the evaluation of patients [51]. While antibodies to single-stranded DNA are formed in several inflammatory conditions including RA, antibodies to double-stranded DNA serve as an immunochemical marker in the diagnosis of SLE [52]. Antibodies to DNA have been associated particularly with SLE, which is considered to be a prototype autoimmune disease [53,54]. Serum obtained from SLE individuals have been shown to possess anti-DNA antibodies of diverse antigenic specificity [55–58]. These anti-DNA autoantibodies have been used to evaluate therapeutic effects and clinical features of SLE patients. Native DNA is no longer regarded as the antigen initiating the disease mainly because immunization with nDNA does not produce SLE-like symptoms. A few of the possible candidates could be polynucleotides, denatured DNA, RNA or modified DNA [59] (Table 2).
Table 2.
Antigenic specificity of anti-ROS-DNA monoclonal antibody
| Inhibitor | Max % inhibition at 20 µg/ml | % relative affinity |
|---|---|---|
| 400 bp ROS-DNA | 76·5 | 100 |
| ROS-DNA | 84 | 146·6 |
| 200 bp ROS-DNA | 59·7 | 44 |
| Native DNA | 65·1 | 40 |
| UV treated 400 bp DNA | 35·9 | – |
| Poly(dT) | 34·8 | – |
| ROS-poly(dT) | 68·2 | 122·2 |
| Poly(G) | 27 | – |
| ROS-poly(G) | 47·2 | – |
Adapted from Ashok and Ali [39].
It is reported that anti-DNA antibodies found typically in SLE have a greater capacity to bind to ROS-modified DNA [60] (Table 3). Recent studies from our laboratory have demonstrated that after modification with ROS, DNA becomes highly immunogenic and the induced antibodies exhibit variable binding to native DNA [61–63]. Blount et al. have postulated ROS-modified DNA as a more discriminating antigen for the diagnosis of SLE [64,65]. Monoclonal anti-DNA antibodies react more strongly with denatured DNA than nDNA [66–68]. It has been proposed that ROS generated in vivo can cause DNA damage, thus altering its structure and immunogenicity, resulting in the antibodies cross reactive to nDNA [60]. The detection of 8-oxodG, a marker of oxidative DNA damage in the immune complex derived DNA of SLE reinforces the evidence that ROS may be involved in its aetiology [37].
Table 3.
Inhibition of SLE antibody binding to native DNA and ROS-DNA fragments of varying size
| % Inhibition | ||
|---|---|---|
| Size of DNA | Native DNA | ROS-DNA |
| Native DNA | 60 | 75 |
| 800 base pair | 58 | 76 |
| 600 bp | 55 | 78 |
| 300 bp | 65 | 72 |
| 200 bp | 50 | 70 |
| 120 bp | 60 | 72 |
| 70 bp | 30 | 55 |
| 50 bp | 30 | 50 |
| Mean | 51 | 68·5 |
| SD | ±12·9 | ±9·6 |
Adapted from: Ara J, Ali R. Reactive oxygen species modified DNA fragments of varying size are preferred antigen for human anti-DNA autoantibodies. Immunol Lett 1992; 34:195–200.
Cellular autoimmunity
The production of autoantibodies in certain autoimmune diseases has been attributed to the selective stimulation of autoreactive B lymphocytes by self-antigens or cross-reaction of antigens with self [69]. In autoimmune prone individuals, B lymphocytes are hyperresponsive to various polyclonal activators (such as viruses, drugs) that bypass the T cell regulatory mechanism. These and several other factors cause immune dysfunction, leading to polyclonal B cell activation [70]. B cell proliferation in SLE has been suggested to be T cell-dependent and the persistence of autoreactive B and T lymphocytes is thought to be responsible for hypergammaglobulinaemia and autoantibody production [71].
A major mechanism by which undigested, intact nuclear antigens are generated and released in vivo is by the process of ‘programmed cell death’ (PCD), or apoptosis. It is characterized by the ordered digestion of nuclear chromatin yielding intact oligonucleosomes that are released into the extracellular matrix.
The immune system represents a prototype for complex multi-cellular organs where during infection antigen-specific lymphocytes need to rapidly proliferate. After clearance of the infectious microbes, lymphocytes need to die in order to prevent unregulated proliferation. It is therefore evident that the control of apoptosis is critical for the homeostasis of the immune system.
Many signals that may originate either endogenously or exogenously have been shown to influence life processes. These include hormones, immune killing, ROS, genetic and physical trauma, oncogene expression, etc. The execution of PCD is often associated with characteristic morphological and biochemical changes. Apoptotic hallmarks include membrane blebbing, cell shrinkage, chromatin condensation, DNA cleavage and fragmentation, etc. [72]. During apoptotic breakdown many nuclear constituents are post-translationally modified altering antigenicity. It is therefore speculated that failure to achieve PCD and clear apoptotic cell fragments may be a key pathological factor leading to autoimmune disorders. Autoimmunity could result from a failure to kill an autoreactive cell or by inducing autoimmunity against apoptotically modified cellular constituents. Therefore, the process of apoptosis may provide a source of nuclear antigens to drive the autoantibody response and provide antigens in SLE [73]. The serological hallmark of SLE, the appearance of antinucleic acid autoantibodies suggests a polyclonal B cell activation. The aetiopathogenesis of lupus, which is still not fully understood, is a multi-factorial one involving environmental factors; drugs, infectious agents, chemicals, free radicals may lead to a profound alteration of the immune system. Changes in the immune system include the appearance of different autoantibodies with differ-ent specificity, altered T cell function, defective phagocytosis, etc. Reports have suggested that apoptosis is abnormal in autoimmune diseases and may play a role in the induction of autoimmunity.
Studies on apoptosis and clearance of apoptotic cells in lupus have shed light on the development and course of the disease. During maturation of the immune system, apoptosis of autoreactive lymphocytes in the central lymphoid organs underies the development of tolerance. Whenever apoptotic cells accumulate by an increased rate of apoptosis, decreased elimination or both, tolerance can be broken. Disturbances in any one of the many factors that regulate the apoptotic process might change the balance in the immune system and may predispose for the development of autoimmune phenomenon [74]. Levels of apoptotic lymphocytes are higher in SLE patients than normal healthy individuals [75]. It has been observed that even during inactive disease, increased amount of activated T and B cells are found in the peripheral blood of these patients [76] and during exacerbations, lymphocyte activation is further increased [77]. The increased presence of apoptotic cells as demonstrated in the peripheral blood of SLE patients can be accounted for by an increased level of activation-induced cell death [78]. Adequate removal of apoptotic cells therefore also seems important for the prevention of excessive autoantigen exposure. Evidence that abnormal phagocytosis of apoptotic cells might be a relevant factor in the development of autoimmune diseases has been shown [79].
One of the mechanisms by which elimination of autoreactive lymphocytes takes place is PCD, and a defect in apoptosis may thus contribute to the development of autoimmune diseases [80]. An increased rate of apoptosis has been demonstrated in lymphocytes from SLE patients in vitro [81]. During the process of apoptosis, the release of excessive quantities of intact nucleosomes may be a source of nuclear antigens that drives an immune response, inducing anti-DNA and antihistone antibody production. These observations imply that a failure of mechanism regulating the physiological clearance of apoptotic cells may contribute to the generation and maintenance of chronic autoimmune diseases [82–84]. There is convincing evidence that in the sera of SLE patients, circulating DNA does not exist in free form but as multimeric complexes of different sizes bound to histones [85]. Such nucleosome-like particles are a major immunogen for pathogenic autoantibody response [86,87] and their complexes with autoantibodies can induce renal lesions by binding to glomerular capillary walls [88].
Collagen and fibronectin deposition and the composition of inflammatory infiltrate in the skin as well as quantitative and functional T cell deficiency in peripheral blood lymphocytes in PSS have been demonstrated [89]. PSS is a systemic rheumatic disease that has been shown to contain autoantibodies to nucleolar and intranuclear components [90]. Garg and Ali have demonstrated the presence of autoantibodies against native and ROS-poly(G) in the sera of SLE and PSS patients [91]. Anti-cardiolipin antibodies may be involved in the pathogenesis of PSS by causing vascular damage through the inhibition of prostacyclin production in vascular endothelium [92].
The origin of autoantibody remains an enigma and the production of anti-DNA antibodies is even more complicated. Even though nucleic acid antigens are by themselves poorly immunogenic, their antigenicity can be enhanced by modification with agents such as ROS. Autoantibodies produced against such modified conformations is the hallmark of the systemic human disease, SLE. B cell hyperactivity and the production of pathogenic autoantibodies are the main immunological events in the pathogenesis of SLE. One approach to study the pathogenesis of this disease and how the autoantibody response is initiated and sustained is to analyse variable genes expressed by antibodies that is the trademark of the disease causing pathogenic autoantibodies. Quantification of this repertoire has revealed the presence of a specific expansion of IgG clonotypes that impart reactivity with disease related autoantigens [93]. The nucleotide sequence of autoantibodies derived from human lupus present in immune complex(es) and renal eluates of subjects with active disease show features of diversification with a high rate of replacement or silent mutations and the clustering of mutations in the hypervariable regions. These characteristics imply that a pure polyclonal activation cannot be the only mechanism responsible for autoantibody production [94]. An antigen-driven process is more likely to play a role in their generation. It has been suggested that the antibody may be stimulated by nucleic acid antigens [95,96] or pathogens [97,98]. B cells whose paratopes have complementary determining regions which are formed by amino acids that can promote DNA binding may be selectively stimulated by nucleic acid related structures.
Conclusions
The mechanism of autoantibody production in diseases such as SLE has not yet been identified. If antigen selection is an important aspect of differentiation, the nature of the stimulating antigen also remains to be determined. The origin of antibodies remains obscure, although modified DNA appears to be a causative factor in RA and SLE. It is possible that the consequent production of autoantibodies may be the result of ROS attack on DNA, causing changes in structure at the macromolecular level. It is therefore postulated that in chronic inflammatory diseases, ROS generated by phagocytic cells may cause damage to DNA and autoantibodies to a self-antigen are produced. Alternatively, a defect in the control of apoptosis and delayed clearance of apoptotic cells provide sustained interaction between oxygen free radicals and apoptotic cell macromolecules including DNA, generating neoepitopes which subsequently results in autoimmunity and generation of polyspecific autoantibodies (Fig. 1). It has been shown in our laboratory that damage of double-stranded DNA or DNA fragments by ROS results in an increased binding of human anti-DNA autoantibodies.
Fig 1.

Possible role of ROS in systemic autoimmunity.
It has now been established clearly that not only oxygen but also nitrogen free radicals play an important role in the pathogenesis of several human diseases. Reactive nitrogen species is produced by the reaction of nitric oxide with O2–. or peroxide. Nitric oxide radical participates in some pathological conditions such as arthritis, autoimmune diseases, vasculitis, asthma, hypertension, etc. It is also an unstable molecule, like ROS but less reactive, and can react with proteins O2 and O2·–.
Acknowledgments
The authors wish to thank University Grants Commission, the Indian Council of Medical Research and the Council of Scientific and Industrial Research for funding through various research projects.
References
- 1.Halliwell B, Gutteridge JMC. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grisham MB. Reactive metabolites of oxygen and nitrogen in biology and medicine. Austin: RG Landes Co.; 1992. [Google Scholar]
- 3.Nappi AJ, Vass E. Hydroxyl radical formation via iron-mediated Fenton Chemistry is inhibited by methylated catechols. Biochim Biophys Acta. 1998;1425:159–67. doi: 10.1016/s0304-4165(98)00062-2. [DOI] [PubMed] [Google Scholar]
- 4.Maddipati KR, Marnett LJ. Characterization of the major hydroperoxide-reducing activity of human plasma. Purification and properties of a selenium-dependent glutathione peroxidase. J Biol Chem. 1987;262:17398–403. [PubMed] [Google Scholar]
- 5.Lloyd RV, Hanna PM, Mason RP. The origin of the hydroxyl radical oxygen in the Fenton reaction. Free Rad Biol Med. 1997;22:885–8. doi: 10.1016/s0891-5849(96)00432-7. [DOI] [PubMed] [Google Scholar]
- 6.Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Rad Biol Med. 1995;18:321–36. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 7.Ward JF, Evans JW, Limoli CL, Calabro-Jones PM. Radiation and hydrogen peroxide induced free radical damage to DNA. Br J Cancer. 1987;55:105–12. [PMC free article] [PubMed] [Google Scholar]
- 8.Mates JM, Gomez CP, Blanca M. Chemical and biological activity of free radical scavengers in allergic diseases. Clin Chim Acta. 2000;296:1–15. doi: 10.1016/s0009-8981(00)00215-1. [DOI] [PubMed] [Google Scholar]
- 9.Arouma OI. Free radicals and food. Chem Br. 1993;29:210–4. [Google Scholar]
- 10.Linn S. DNA damage by iron and hydrogen peroxide in vitro and in vivo. Drug Metab Rev. 1998;30:313–26. doi: 10.3109/03602539808996315. [DOI] [PubMed] [Google Scholar]
- 11.Ha HC, Sirisoma NS, Kuppusamy P, Zweier JL, Woster PM, Casero RA. The natural polyamine spermine functions directly as a free radical scavenger. Proc Natl Acad Sci USA. 1998;95:11140–5. doi: 10.1073/pnas.95.19.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halliwell B, Vitamin C. poison, prophylactic or panacea? Trends Biochem Sci. 1999;24:255–9. doi: 10.1016/s0968-0004(99)01418-8. [DOI] [PubMed] [Google Scholar]
- 13.Melov S, Schneider JA, Day BJ, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–63. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 14.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–9. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 15.Kaouass M, Deloyer P, Gouders I, Peulen O, Dandrifosse G. Role of interleukin-1 beta, interleukin-6 and TNF-alpha in intestinal maturation induced by dietary spermine in rats. Endocrine. 1997;6:187–94. doi: 10.1007/BF02738963. [DOI] [PubMed] [Google Scholar]
- 16.Kagaya K, Miyakawa Y, Watanabe K, Fukazawa Y. Antigenic role of stress-induced catalase of Salmonella typhimurium in cell mediated immunity. Infect Immun. 1992;60:1820–5. doi: 10.1128/iai.60.5.1820-1825.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai KJ, Hung IJ, Chow CK, Stern A, Chao SS, Chin DTY. Impared production of nitric oxide, superoxide and hydrogen peroxide in glucose 6-phosphate dehydrogenase-deficient granulocytes. FEBS Lett. 1998;436:411–4. doi: 10.1016/s0014-5793(98)01174-0. [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Zhou J, Xu H, Jiang Y, Zhu G. Effect of selenium supplementation on mice infected with LP-BM5 MuLV, a murine AIDS model. Biol Trace Elem Res. 1997;59:187–93. doi: 10.1007/BF02783244. [DOI] [PubMed] [Google Scholar]
- 19.Galan P, Preziosi P, Monget AL. Effects of trace elements and/or vitamin supplementation on vitamin and mineral status, free radical metabolism and immunological markers in elderly long hospitalized subjects. Int J Vit Nutr Res. 1997;67:450–60. [PubMed] [Google Scholar]
- 20.DeWaart FG, Portengen L, Doekes G, Verwaal CJ, Kok FJ. Effect of 3 months vitamin E supplementation on indices of the cellular and humoral immune response in elderly subjects. Br J Nutr. 1997;78:761–4. doi: 10.1079/bjn19970193. [DOI] [PubMed] [Google Scholar]
- 21.Chen F, Lu Y, Demers LM, et al. Role of hydroxyl radical in silica-induced NF-kappa B activation in macrophages. Ann Clin Lab Sci. 1998;28:1–13. [PubMed] [Google Scholar]
- 22.Shankar AH, Prasad AS. Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr. 1998;68:447–63. doi: 10.1093/ajcn/68.2.447S. [DOI] [PubMed] [Google Scholar]
- 23.PithonCuri TC, DeMelo MP, Palanch AC, Miyasaka CK, Curi R. Percentage of phagocytosis, production of O2·–, H2O2 and NO and antioxidant enzyme activities of rat neutrophils in culture. Cell Biochem Funct. 1998;16:43–9. doi: 10.1002/(SICI)1099-0844(199803)16:1<43::AID-CBF761>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Levinson SS. Humoral mechanisms in autoimmune disease. J Clin Immunoassay. 1994;17:72–84. [Google Scholar]
- 25.Lundberg I, Hedfors E. Clinical course of patients with anti-RNP antibodies. A prospective study of 32 patients. J Rheumatol. 1991;18:1511–9. [PubMed] [Google Scholar]
- 26.Fritzler MJ, Ali R, Tan EM. Antibodies from patients with mixed connective tissue disease react with heterogeneous nuclear ribonucleoprotein or ribonucleic acid (hnRNP/RNA) of the nuclear matrix. J Immunol. 1984;132:1216–22. [PubMed] [Google Scholar]
- 27.Rokeach L, Hoch SO. B-cell epitopes of Sm autoantigens. Mol Biol Rep. 1992;16:165–74. doi: 10.1007/BF00464704. [DOI] [PubMed] [Google Scholar]
- 28.Reichlin M, Harley JB. Antibodies to Ro (SSA) and the heterogeneity of systemic lupus erythematosus. J Rheumatol. 1987;14:112–7. [PubMed] [Google Scholar]
- 29.Dixit K, Ali R. Antigen binding characteristics of antibodies induced against nitric oxide modified plasmid DNA. Biochim Biophys Acta. 2001;1528:1–8. doi: 10.1016/s0304-4165(01)00162-3. [DOI] [PubMed] [Google Scholar]
- 30.Islam N, Ali R. Immunological studies on DNA-lysine photoadduct. Biochem Mol Biol Int. 1998;45:453–64. doi: 10.1080/15216549800202842. [DOI] [PubMed] [Google Scholar]
- 31.Tasneem S, Ali R. Binding of SLE autoantibodies to native poly(I), ROS-poly(I) and native DNA. A comparative study. J Autoimmunity. 2001;17:199–205. doi: 10.1006/jaut.2001.0546. [DOI] [PubMed] [Google Scholar]
- 32.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 33.Alam K, Ali R. Human anti-DNA autoantibodies and induced antibodies against ROS-modified DNA show similar antigen binding characteristics. Biochem Mol Biol Int. 1999;47:881–90. doi: 10.1080/15216549900201983. [DOI] [PubMed] [Google Scholar]
- 34.Ara J, Ali R. Polynucleotide specificity of anti-ROS DNA antibodies. Clin Exp Immunol. 1993;94:134–9. doi: 10.1111/j.1365-2249.1993.tb05990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naparstek Y. Annu Rev Immunol. Vol. 11. 1993. The role of autoantibodies in autoimmune disease; pp. 79–104. [DOI] [PubMed] [Google Scholar]
- 36.Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–9. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 37.Lunec J, Herbert K, Blount S, Griffiths HR, Emery P. 8-hydroxydeoxyguanosine. A marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett. 1994;348:131–8. doi: 10.1016/0014-5793(94)00583-4. [DOI] [PubMed] [Google Scholar]
- 38.Bashir S, Harris G, Denman M, Blake DR, Winyard PG. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann Rheum Dis. 1993;52:659–66. doi: 10.1136/ard.52.9.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashok BT, Ali R. Binding of human anti-DNA autoantibodies to reactive oxygen species modified DNA and probing oxidative DNA damage in cancer using monoclonal antibody. Int J Can. 1998;78:404–9. doi: 10.1002/(sici)1097-0215(19981109)78:4<404::aid-ijc2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 40.Joenje H. Genetic toxicology of oxygen. Mutat Res. 1989;219:193–208. doi: 10.1016/0921-8734(89)90001-5. [DOI] [PubMed] [Google Scholar]
- 41.Stich HF, Anders F. The involvement of reactive oxygen species in oral cancers of betel quid/tobacco chewers. Mutat Res. 1989;214:47–61. doi: 10.1016/0027-5107(89)90197-8. [DOI] [PubMed] [Google Scholar]
- 42.Meneghini R. Genotoxicity of active oxygen species in mammalian cells. Mutat Res. 1988;195:215–30. [PubMed] [Google Scholar]
- 43.Ashok BT, Ali R. The aging paradox: free radical theory of aging. Exp Gerontol. 1999;34:293–303. doi: 10.1016/s0531-5565(99)00005-4. [DOI] [PubMed] [Google Scholar]
- 44.Ames BN. Endogenous oxidative DNA damage, aging and cancer. Free Rad Res Commun. 1989;7:121–8. doi: 10.3109/10715768909087933. [DOI] [PubMed] [Google Scholar]
- 45.Marx JL. Oxygen free radicals linked to many diseases. Science. 1987;235:529–31. doi: 10.1126/science.3810154. [DOI] [PubMed] [Google Scholar]
- 46.Halliwell B, Grootveld M. The measurement of free radical reactions in humans. Some thoughts for future experimentation. FEBS Lett. 1987;213:9–14. doi: 10.1016/0014-5793(87)81455-2. [DOI] [PubMed] [Google Scholar]
- 47.Naparstek Y, Madaio MP. Are DNA antibodies actually pathogenic? Lupus. 1997;6:307–9. doi: 10.1177/096120339700600318. [DOI] [PubMed] [Google Scholar]
- 48.Allan IM, Vaughan ATM, Milner AE, Lunec J, Bacon PA. Structural damage to the lymphocyte nuclei by H2O2 or gamma irradiation is dependent on the mechanism of ·OH radical production. Br J Cancer. 1988;58:34–7. doi: 10.1038/bjc.1988.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lunec J, Griffiths HR, Blake DR. Oxygen free radicals in inflammation. ISI Atlas Sci. 1987;1:45–58. [Google Scholar]
- 50.Stollar BD, Anti DNA. antibodies. Clin Immunol Allergy. 1981;1:243–59. [Google Scholar]
- 51.Pisetsky DS. Antinuclear antibodies. Diagn Lab Immunol. 1994;14:371–85. [Google Scholar]
- 52.Swaak T, Smeenk R. Detection of anti-dsDNA as a diagnostic tool: a prospective study in 441 non-systemic lupus erythematosus patients with anti-dsDNA antibody. Ann Rheum Dis. 1985;44:245–51. doi: 10.1136/ard.44.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Isenberg D, Shoenfeld Y. The origin and significance of anti-DNA antibodies. Immunol Today. 1987;8:279–81. doi: 10.1016/0167-5699(87)90189-7. [DOI] [PubMed] [Google Scholar]
- 54.Tan EM. Autoantibody to nuclear antigens (ANA): their immunobiology and medicine. Adv Immunol. 1982;33:167–240. doi: 10.1016/s0065-2776(08)60836-6. [DOI] [PubMed] [Google Scholar]
- 55.Pollard KM, Jones JE, Tan EM, Theofilopoulos AN, Dixon FJ, Rubin RL. Polynucleotide specificity of murine monoclonal anti-DNA antibodies. Clin Immunol Immunopathol. 1986;40:197–208. doi: 10.1016/0090-1229(86)90022-x. [DOI] [PubMed] [Google Scholar]
- 56.Tasneem S, Ali R. Antigenicity of poly(I) and ROS-poly(I) and their recognition of human anti-DNA autoantibodies. Immunol Invest. 2001;30:335–45. doi: 10.1081/imm-100108167. [DOI] [PubMed] [Google Scholar]
- 57.Arif Z, Ali R. Antigenicity of poly(dA-dT) poly(dA-dT) photo-crosslinked with 8-methoxypsoralen. Arch Biochem Biophys. 1996;329:191–8. doi: 10.1006/abbi.1996.0208. [DOI] [PubMed] [Google Scholar]
- 58.Ahmad J, Ashok BT, Ali R. Reactive oxygen species modified thymine and poly(dT) represent unique epitope for human anti-DNA autoantibodies. Immunol Lett. 1997;58:69–74. doi: 10.1016/s0165-2478(97)02711-9. [DOI] [PubMed] [Google Scholar]
- 59.Stollar BD. Immunochemistry of DNA. Int Rev Immunol. 1989;5:1–22. doi: 10.3109/08830188909086987. [DOI] [PubMed] [Google Scholar]
- 60.Blount S, Griffiths HR, Lunec J. Reactive oxygen species induce antigenic changes in DNA. FEBS Lett. 1989;245:100–4. doi: 10.1016/0014-5793(89)80200-5. [DOI] [PubMed] [Google Scholar]
- 61.Ahmad R, Alam K, Ali R. Antigen binding characteristics of antibodies against hydroxyl radical modified thymidine monophosphate. Immunol Lett. 2000;71:111–5. doi: 10.1016/s0165-2478(99)00177-7. [DOI] [PubMed] [Google Scholar]
- 62.Ashok BT, Ali R. Antigen binding characteristics of experimentally-induced antibodies against hydroxyl radical modified native DNA. Autoimmunity. 1999;29:11–9. doi: 10.3109/08916939908995968. [DOI] [PubMed] [Google Scholar]
- 63.Ashok BT, Ahmad J, Qadri A, Ali R. Anti-ROS-DNA monoclonal antibody as molecular probe for DNA damage. Biochem Mol Biol Int. 1997;43:1219–29. doi: 10.1080/15216549700205051. [DOI] [PubMed] [Google Scholar]
- 64.Blount S, Griffiths HR, Lunec J. Reactive oxygen species damage to DNA and its role in systemic lupus erythematosus. Mol Asp Med. 1991;12:93–105. doi: 10.1016/0098-2997(91)90005-7. [DOI] [PubMed] [Google Scholar]
- 65.Blount S, Griffiths HR, Lunec J. Reactive oxygen species modified human DNA eliciting a more discriminating antigen for the diagnosis of systemic lupus erythematosus. Clin Exp Immunol. 1990;81:384–9. doi: 10.1111/j.1365-2249.1990.tb05343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahmad J, Ashok BT, Ali R. Detection of oxidative DNA damage by a monoclonal antibody: role of lysyl residues in antigen binding. Immunol Lett. 1998;62:87–92. doi: 10.1016/s0165-2478(98)00024-8. [DOI] [PubMed] [Google Scholar]
- 67.Ashok BT, Ali R. Binding of circulating antibodies to reactive oxygen species modified-DNA and detecting DNA damage by a monoclonal antibody probe. Mech Ageing Dev. 1998;103:69–80. doi: 10.1016/s0047-6374(98)00025-6. [DOI] [PubMed] [Google Scholar]
- 68.Wu DP, Gilkeson GS, Armitage J, Reich CE, Pisetsky DS. Selective recognition of DNA antigenic determinants by murine monoclonal anti-DNA antibodies. Clin Exp Immunol. 1990;82:33–7. [PMC free article] [PubMed] [Google Scholar]
- 69.Hardin JA. The lupus autoantigens and the pathogenesis of SE. Arth Rheum. 1986;29:457–61. doi: 10.1002/art.1780290401. [DOI] [PubMed] [Google Scholar]
- 70.Klinman DM, Steinberg AD. Inquiry into murine and human lupus. Immunol Rev. 1995;144:157–93. doi: 10.1111/j.1600-065x.1995.tb00069.x. [DOI] [PubMed] [Google Scholar]
- 71.Linker-Israeli M, Quismorio FP, Horwitz DA. CD8+ lymphocytes from patients with systemic lupus erythematosus sustain, rather than suppress, spontaneous polyclonal IgG production and synergize with CD4+ cells to support autoantibody synthesis. Arth Rheum. 1990;33:1216–25. doi: 10.1002/art.1780330823. [DOI] [PubMed] [Google Scholar]
- 72.Mignotte B, Vayssiere J-L. Mitochondria and apoptosis. Eur J Biochem. 1998;252:1–15. doi: 10.1046/j.1432-1327.1998.2520001.x. [DOI] [PubMed] [Google Scholar]
- 73.Lorenz H-M, Herrmann M, Kalden JM. The pathogenesis of autoimmune diseases. Scand J Clin Laboratory Invest. 2001;61(Suppl. 235):16–26. doi: 10.1080/003655101753352004. [DOI] [PubMed] [Google Scholar]
- 74.Bijl M, Limburg PC, Kallenberg CGM. New insights into the pathogenesis of systemic lupus erythematosus (SLE): the role of apoptosis. Neth J Med. 2001;59:66–75. doi: 10.1016/s0300-2977(01)00131-0. [DOI] [PubMed] [Google Scholar]
- 75.Courtney PA, Crockard AD, Williamson K, et al. Increased peripheral blood neutrophils in systemic lupus erythematosus. relations with disease activity, antibodies to double stranded DNA and neutropenia. Ann Rheum Dis. 1999;58:1241–50. doi: 10.1136/ard.58.5.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bijl M, vanLopik T, Limburg PC. Do elevated levels of serum soluble fas contribute to the persistance of activated lymphocytes in systemic lupus erythematosus? J Autoimmunity. 1998;11:457–63. doi: 10.1006/jaut.1998.0233. [DOI] [PubMed] [Google Scholar]
- 77.Gordon C, Salmon M. Update on systemic lupus erythematosus: autoantibodies and apoptosis. Clin Med. 2001;1:10–4. doi: 10.7861/clinmedicine.1-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Funauchi M, Sugiyama M, Sukyoo B, Ikoma S, Ohno M, Kinoshita K, Kanamaru A. A possible role of apoptosis for regulating autoreactive responses in systemic lupus erythematosus. Lupus. 2001;10:284–8. doi: 10.1191/096120301680416977. [DOI] [PubMed] [Google Scholar]
- 79.Hermann M, Voll RE, Zopller OM, et al. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophage from patients with systemic lupus erythematosus. Arth Rheum. 1998;41:1241–50. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 80.Mountz JD, Wu J, Cheng J, Zhou T. Autoimmune disease. A problem of defective apoptosis. Arth Rheum. 1994;37:1415–20. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- 81.Emlen W, Neibur J, Kadera R. Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J Immunol. 1994;152:3685–92. [PubMed] [Google Scholar]
- 82.Casiano CA, Tan EM. Recent developments in the understanding of antinuclear antibodies. Int Arch Allergy Immunol. 1996;114:308–13. doi: 10.1159/000237385. [DOI] [PubMed] [Google Scholar]
- 83.Cabral AR, Alarcon-Segovia D. Autoantibodies in systemic lupus erythematosus. Curr Opin Rheumatol. 1997;9:387–92. doi: 10.1097/00002281-199709000-00003. [DOI] [PubMed] [Google Scholar]
- 84.Vaishnaw AK, McNally JD, Elkon KB. Apoptosis in rheumatic diseases. Arth Rheum. 1997;40:1917–27. doi: 10.1002/art.1780401102. [DOI] [PubMed] [Google Scholar]
- 85.Rumore PM, Steinman CR. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J Clin Invest. 1990;86:69–74. doi: 10.1172/JCI114716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: a major immunogen for pathogenic autoantibody-inducing T cells of lupus. J Exp Med. 1993;177:1367–81. doi: 10.1084/jem.177.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gilbert D, Brard F, Jovelin F, Tron F. Do naturally occurring autoantibodies participate in the constitution of the pathological B-cell repertoire in systemic lupus erythematosus? J Autoimmunity. 1996;9:247–57. doi: 10.1006/jaut.1996.0031. [DOI] [PubMed] [Google Scholar]
- 88.Morioka T, Fujigaki Y, Batsford SR, et al. Anti-DNA antibody derived from a systemic lupus erythematosus patient forms histone-DNA-anti-DNA complexes that bind to rat glomeruli in vivo. Clin Exp Immunol. 1996;104:92–6. doi: 10.1046/j.1365-2249.1996.d01-658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gruschwitz MS, Shoenfeld Y, Krupp M, et al. Antinuclear antibody profile in UCD line 200 chickens: a model for progressive systemic sclerosis. Int Arch Allergy Immunol. 1993;100:307–13. doi: 10.1159/000236430. [DOI] [PubMed] [Google Scholar]
- 90.Reimer G, Scheer U, Peters JM, Tan EM. Immunolocalization and partial characterization of a nucleolar antigen (PM-Scl) associated with polymyositis/scleroderma overlap syndromes. J Immunol. 1986;137:3802–8. [PubMed] [Google Scholar]
- 91.Garg DK, Ali R. Reactive oxygen species modified polyguanylic acid: immunogenicity and implications for systemic autoimmunity. J Autoimmunity. 1998;11:371–8. doi: 10.1006/jaut.1998.0208. [DOI] [PubMed] [Google Scholar]
- 92.Schorer AE, Wickham NW, Watson KV. Lupus anticoagulant induces a selective defect in thrombin-mediated endothelial pro-stacyclin release and platelet aggregation. Br J Haematol. 1989;71:399–407. doi: 10.1111/j.1365-2141.1989.tb04298.x. [DOI] [PubMed] [Google Scholar]
- 93.Zouali M. The structure of human lupus anti-DNA antibodies. Methods. 1997;11:27–35. doi: 10.1006/meth.1996.0384. [DOI] [PubMed] [Google Scholar]
- 94.Klinman DM, Steinberg AD. Systemic autoimmune disease arises from polyclonal B cell activation. J Exp Med. 1987;165:1755–60. doi: 10.1084/jem.165.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bloom DD, Davignon JL, Cohen PL, Eisenberg RA, Clarke SH. Overlap of the anti-Sm and anti-DNA responses of MRL/MP-lpr/lpr mice. J Immunol. 1993;150:1579–90. [PubMed] [Google Scholar]
- 96.Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol. 1991;146:176–82. [PubMed] [Google Scholar]
- 97.Zouali M, Druilhe P, Gentilini M, Eyquem A. High titres of anti-T antibodies and other haemagglutins in human malaria. Clin Exp Immunol. 1982;50:83–91. [PMC free article] [PubMed] [Google Scholar]
- 98.Fredriksen K, Osei A, Sundsfjord A, Traavik T, Rekvig OP. On the origin of anti-double stranded (ds) DNA antibodies: systemic lupus erythematosus related anti-dsDNA antibodies are induced by polyomavirus BK in lupus-prone (NZW × NZW) F1 hybrids, but not in normal mice. Eur J Immunol. 1994;24:66–70. doi: 10.1002/eji.1830240111. [DOI] [PubMed] [Google Scholar]