

Abstract
Interleukin (IL)-18 is a member of the IL-1 cytokine family. Its expression is increased in rheumatoid arthritis synovium, and its proinflammatory effects have been demonstrated in experimental models of murine arthritis. Here, we investigate the actions of varying doses of recombinant rat IL-18 (rIL-18) on the course of type II collagen-induced arthritis (CIA) in BB rats, including clinical and immune events, plus splenic cytokine production. Small doses of rIL-18 (10 and 50 µg/rat) administered intraperitoneally (i.p.) increased arthritis incidence and severity (P < 0·01) when a low-potency CII preparation was used for immunization. IgG1 and IgG2a anti-CII antibody levels were significantly greater in rats given 10 and 50 µg rIL-18 doses than controls. rIL-18 significantly increased levels of proinflammatory cytokines [interferon (IFN)-γ, IL-2, tumour necrosis factor (TNF)-α and IL-6] produced by splenocyte cultures. Larger doses of rIL-18 (300 µg/rat) suppressed arthritis and immunity. To ascertain whether the pro-arthritic effects of IL-18 could be attenuated, rats were treated with neutralizing rabbit anti-rIL-18 IgG before immunization with a high-potency CII preparation. When given serially for 3 weeks, the incidence and severity of CIA, in addition to anti-CII IgG2a and splenic IL-6 and IFN-γ production, were all significantly reduced. Similar results were noted when antibody was given twice, just before arthritis onset. These results demonstrate that IL-18 plays an important proinflammatory role in the pathogenesis of CIA which is achieved, in part, by an immunostimulatory action. Neutralizing endogenous IL-18 with antibodies attenuated CIA, CII immunity and cytokine responses. These studies support the use of IL-18 antagonists as treatments for inflammatory arthritis.
Keywords: anti-IL-18 antibody, autoimmunity, collagen-induced arthritis, cytokine, IL-18, type II collagen
INTRODUCTION
Collagen-induced arthritis (CIA) is an experimental model of autoimmune inflammatory arthritis, which shares features with human rheumatoid arthritis (RA). Both humoral and cellular immune responses are essential for the induction and perpetuation of CIA. The pathogenic mechanisms of autoimmune arthritis indicate that both MHC and non-MHC genes play critical roles in the clinical manifestation of arthritis [1]. The importance of cytokine production, e.g. interleukin (IL-1), tumour necrosis factor (TNF)-α, IL-12 and IL-6 in autoimmune diseases has been studied intensively in animal models and human diseases, thus providing insight into the events leading to joint destruction [2–6].
IL-18 is a member of the IL-1 cytokine family [7,8] which, like IL-1, is released as a propeptide and cleaved by an IL-1β-converting enzyme (ICE) to yield a biologically active product [8]. It is expressed by a variety of cell types including: monocytes/macrophages, dendrocytes, Kupffer cells, keratinocytes, intestinal epithelial cells, skin cells, synovial fibroblasts, articular chondrocytes and osteoblasts [9–12]. The initial function of IL-18 was thought to be an interferon (IFN)-γ-inducing factor [13,14]; however, it has also been shown to possess a broad range of activities in regulating innate and acquired immune responses. IL-18 acts synergistically with IL-12 to induce naive CD4+ T cells to differentiate into Th1 cells [9,15] and to stimulate IFN-γ production by Th1 and natural killer (NK) cells [7,13,16,17]. IL-18 promotes inflammation by enhancing TNF-α, IL-1β, IL-2, IL-6 and granulocyte-macrophage colony stimulating factor (GM-CSF) gene expression, in addition to chemokines IL-8 and macrophage inflammatory protein (MIP)-α[17–19]. Moreover, IL-18 up-regulates the expression of adhesion molecules such as intercellular adhesion molecule (ICAM)-1 [20,21] and enhances prostaglandin E2, inducible nitric oxide synthetase (iNOS) and leukotriene B4 production by mononuclear and mesenchymal cells [9,10,15,22]. Lastly, IL-18 has been shown to promote inflammation by the recruitment and activation of neutrophils [22,23].
With regard to joint injury, Gracie et al. [24] have shown IL-18 mRNA and protein to be significantly more abundant in the joints of rheumatoid patients than patients with osteoarthritis. It is believed that IL-18 may play an important role in inducing and sustaining articular Th1-type cell activity and promote TNF-α production. The capacity of IL-18 to activate synovial fluid (SF) neutrophils to release cytokines, chemokines and leukotriene B4 is also significantly enhanced in RA patients [22,23]. Preliminary studies of experimental CIA in mice show that treatment with IL-12 plus IL-18, or with IL-18 alone, enhance the incidence and severity of joint injury [24,25]. In contrast, IL-18 knock-out mice are resistant to CIA [26], as are mice treated with neutralizing anti-IL-18 antibodies [23]. A naturally occurring IL-18 protein antagonist is also able to attenuate disease in CIA [27].
In the present study we investigate the role of IL-18 in CIA in the BB rat, because this model has certain advantages over the murine model, one being that rats require only the use of incomplete Freund's adjuvant (IFA) to induce arthritis. Thus, by eliminating Mycobacterium tuberculosis (MT), which is needed to assure a high incidence of CIA in mice, its potential confounding effects on cytokine production are eliminated. Additional advantages include a short latent period before the onset of arthritis and a greater amount of synovium to study. Rats are also susceptible to adjuvant arthritis, streptococcal cell wall-induced arthritis and type XI collagen-induced arthritis, thus providing alternative models for comparative studies.
Our data presented here demonstrate that IL-18 is capable of significantly modulating the incidence and severity of arthritis and IgG2a anticollagen antibody production in rats, depending on the quantity of rIL-18 administered. IL-18 is also shown to promote the synthesis of Th1-type cytokines, as well as proinflammatory cytokines, which are central to joint injury in CIA. Furthermore, by treating rats with a neutralizing anti-IL-18 antibody the incidence and severity of CIA could be significantly reduced, as were IgG2a antibodies to CII and the splenic production of IL-6 and IFN-γ. These findings suggest that IL-18 works through an array of pathways and that agents which block its actions may have therapeutic potential in treating human inflammatory arthritis.
MATERIALS AND METHODS
Preparation of collagen
Type II collagen was isolated from fetal bovine cartilage by limited-pepsin digestion and the purity determined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing and non-reducing conditions [28]. Denatured CII was prepared by heating native CII at 80°C for 15 min. Two different preparations of CII were used, as described below.
Animals, immunization and scoring protocols
Male and female, diabetic-resistant BB (RT1.Du) rats were purchased from the University of Massachusetts (Worchester, MA, USA), bred in our animal facility, housed and studied in accordance with AALAC guidelines. The rats were fed standard rodent chow and water ad libitum. Native bovine CII was dissolved in 0·05 m acetic acid (2 or 4 mg/ml) and emulsified in IFA (Difco, Detroit, MI, USA) [28]. Arthritis was induced in rats 8–10 weeks old. No difference in the incidence or severity of CIA has been observed in male and female rats, so equal or comparable numbers of both sexes were used in all studies. Mild arthritis at a low incidence was induced in some groups by injection with 100 µg of a bovine CII preparation purified 8 years earlier. Although this preparation was initially highly arthritogenic, its potency waned with time. Severe arthritis at a high incidence was induced using 300 µg of a fresh preparation of bovine CII. All injections were made intradermally at the base of the tail. Arthritis severity was scored visually using a 0–4 scale with a maximal possible score of 16 per rat [28]. In one study, ankle thickness (medial–lateral) was measured with a Vernier caliper.
Preparation of recombinant rat IL-18
Total RNA was extracted from the draining lymphoid nodes of BB rats with TRIzol Reagent (Life Technologies, Grand Island, NY, USA) using the supplier's protocol. First-strand cDNA synthesis was performed with a cDNA cycle kit (Invitrogen, San Diego, CA, USA). Primer set pairs designed from rat IL-18 sequence data were used to clone IL-18 (sense, ATGGCTG CAATACCAGAAGAAG; antisense, ACTTTGATGTAAGT TAGTAAG). The polymerase chain reaction (PCR) product was confirmed by sequencing, cloned into the pET-19b and expressed in Escherichia coli. IL-18 was extracted under native conditions and purified as a histidine-tagged fusion protein. Its purity was tested by SDS-PAGE and Pierce gel code blue staining, which showed a single protein band at 28 kDa, and by limulus assay (Sigma, St. Louis, MO, USA), which disclosed an endotoxin level below 0·3 U/mg protein (lowest level of detection). Biological activity of rIL-18 was determined by IFN-γ production from non-immune BB rat spleen cell cultures.
Expression of IL-18 in synovium
Isolation of synovial cells, extraction of total RNA and cDNA synthesis were performed as described previously [29]. PCR was performed in 50 µl reaction mixtures containing 5 µl of 10× PCR buffer, 4 µl of 2·5 mm dNTPs, 2·5 µl of primer, 2 µl of 25 mm MgCl2, 10 µl of Q-Solution (Qiagen, Chatsworth, CA, USA), 0·3 µl of HotStar Taq DNA polymerase (Qiagen) and 2 µl of cDNA product. The reaction utilized a DNA thermal cycler (Perkin-Elmer, Foster City, CA, USA) for 35 cycles at 94°C for 45 s, 50°C for 45 s and 72°C for 2 min, with an initial activation step at 95°C for 15 min; the oligonucleotide primer sequences used were:
IL-18: 5′-TGCAATACCAGAAGAAGGC-3′ forward;
IL-18: 5′-CCCCATTTTCATCCTTCC-3′ reverse;
β-actin: 5′-TTGTAACCAACTGGGACGATATGG-3′ forward;
β-actin: 5′-GATCTTGATCTTCATGGTGCTAGG-3′ reverse.
After amplification, the PCR products were resolved by electrophoresis on a 1·5% agarose gel. The gel photographs were analysed on a computer by using NIH Image software. Data are presented as the ratio of IL-18 to the housekeeping gene β-actin.
Administration of rIL-18
Rats immunized with 100 µg of bovine CII in IFA received intraperitoneal (i.p.) injection of rIL-18 diluted in phosphate buffered saline (PBS) at days 0–2 and 7–9 after immunization; control rats received PBS only.
Anti-collagen antibody assay
IgG and IgG subclass anticollagen antibodies were measured by enzyme-linked immunosorbent assay (ELISA) as described previously [30]. A standard curve and antibody values were generated by computer analysis (softmax). All antibody values are expressed as µg/ml of IgG. For antibody subclass analysis, biotin-conjugated mouse antirat IgG1, IgG2a and IgG2b (PharMingen, San Diego, CA, USA) were used as second antibodies followed by the addition of ExtrAvidin peroxidase (Sigma), and substrate. These antibody results are expressed as optical density (OD) at 490 nm.
Cell culture and cytokine measurement
Single cell suspensions (5 × 106/ml) were prepared from individual spleens. The cells were washed in Hanks's balanced salt solution (HBSS) and resuspended in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS), 5 × 10−5m 2-ME, 10 mm HEPES buffer, 4 mm l-glutamine, penicillin/streptomycin and non-essential amino acids. When IL-18 was tested for its ability to stimulate INF-γ and TNF-α production, a submitogenic amount of concanavalin-A (100 ng/ml) was added to cultures to activate T cells. This step was omitted when immune spleen cells were cultured with soluble CII. However, after 24 and 48 h culture with or without bovine CII (25 µg/ml) or IL-18, supernatants were collected from 24-well microtitre plates maintained at 37°C in 5% humidified CO2 and stored at −70°C until assay. Cytokines were measured using commercial ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. The sensitivity limits for each cytokine were: IL-2, 15 pg/ml; IL-6, 10 pg/ml; IFN-γ, 10 pg/ml; TNF-α, 5 pg/ml; and IL-10, 10 pg/ml. Later cytokine measurements were performed by a Bio-Plex assay system (Bio-Rad Laboratories Inc., Hercules, CA, USA) using diluents designed specifically for rat samples.
Generation and administration of anti-IL-18 polyclonal antibody
New Zealand white rabbits (Myrtle's Rabbitry Inc., TN, USA) weighing 2 kg or greater were immunized subcutaneously with 500 µg of rIL-18 emulsified in complete Freund's adjuvant (CFA) (Difco) and boosted monthly with 300 µg of rIL-18 in IFA twice. Serum was collected before immunization and at 2-week intervals after the last boost. Antibody specificity was tested by ELISA using rIL-18 coated plates; a Western blot assay using E. coli lysate revealed a single band. Total IgG was purified from the antiserum by protein-A chromatography. The biological activity of the antibody was tested in vitro for its ability to prevent rIL-18-induced IFN-γ production by rat spleen cells. Rats were immunized with CII in IFA followed by i.p. injection of 5 mg of purified rabbit antirat rIL-18 IgG or normal rabbit IgG; subsequent injections were made thrice-weekly for 3 weeks (days 0, 2, 4, 7, 9, 11, 14, 16 and 18) or twice at days 9 and 11 (prior to arthritis onset).
Statistical analysis
The statistical significance of the data was calculated by Student's t-test for parametric data; arthritis incidence was tested by χ2 testing with Yates's correction. Non-parametric data were analysed by Wilcoxon/Kruskal–Wallis testing. All data are expressed as mean ± s.e.m.; P-values < 0·05 were considered significant.
RESULTS
IL-18 expressed by synovial cells
The expression of mRNA for IL-18 by the synovia of normal and arthritic rats was measured semiquantitatively by reverse transcription-polymerase chain reaction (RT-PCR). As shown in Fig. 1, IL-18 expression peaked abruptly shortly after the onset of arthritis at day 3 of inflammation, which corresponded with maximal paw swelling (i.e. a 4 + score), then dropped rapidly, although remaining above the prearthritic baseline.
Fig. 1.
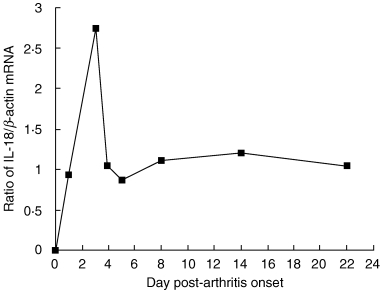
Expression of mRNA for IL-18 in synovium as a function of time as measured by RT-PCR. The data shown are the ratios of mRNA for IL-18 to β-actin. Arthritis scores for each rat constituting one time-point given in the figure are: 0 (normal rat, day 0), 2+, 4+, 4+, 4+, 3+, 3+ and 3+, respectively. Synovium was obtained from the arthritic hind-paws of one rat and pooled for each time point, except for the normal rat where four hind-paws were pooled from two animals.
Recombinant IL-18 stimulates cytokine production by spleen cells
To confirm that rIL-18 induces cytokine production in vitro, splenocytes from normal BB rats were cultured with increasing concentrations of rIL-18 for 48 h along with a submitogenic amount (100 ng/ml) of concanavalin-A (Con-A), and the supernatants assayed for IFN-γ and TNF-α by ELISA. As shown in Fig. 2a, IFN-γ (Th1 cytokine) and TNF-α (monocytes/macrophage cytokine) both increased in a dose-dependent manner in response to rIL-18, indicating that rIL-18 can induce proinflammatory cytokines.
Fig. 2.
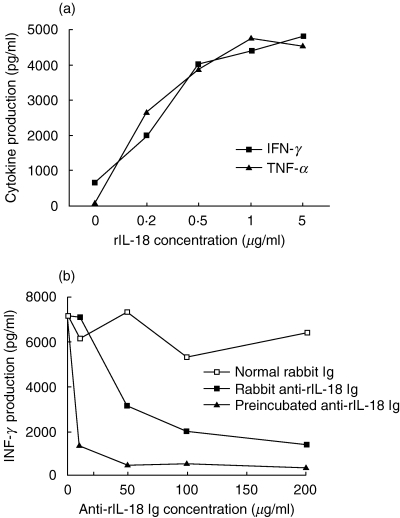
(a) Ability of increasing doses of rIL-18 to stimulate splenocytes to produce IFN-γ and TNF-αin vitro. (b) Production of IFN-γ could also be inhibited in a dose-dependent manner by adding rabbit anti-rIL-18 IgG to the culture system, either 1 h before or with rIL-18. The supernatants were obtained after 48 h of culture and assayed for cytokine by ELISA. Average values are shown; variation between duplicate samples was less than 5% of the average.
To determine whether the in vitro actions of rIL-18 could be neutralized with antibody, anti-rIL-18 IgG was produced in rabbits. As above, spleen cells from normal rats were cultured for 24 h with 200 ng/ml of rIL-18 and increasing concentrations of purified anti-rIL-18 IgG or non-immune rabbit IgG. The supernatants were collected for INF-γ assay. Data in Fig. 2b show that IFN-γ production was decreased by anti-rIL-18 antibody in a dose-dependent manner, and that IFN-γ production was affected even more profoundly when rIL-18 was preincubated with anti-IL-18 antibody before being added to cell culture.
Pretreatment with rIL-18 modulates the development of CIA, immunity to CII and cytokine production
To ascertain whether rIL-18 has a proinflammatory effect on in vivo, as suggested by the preceding study, rats were immunized with bovine CII using a protocol, which induces mild disease (see Materials and methods), and injected i.p. with rIL-18 at increasing doses. Data in Fig. 3a and b show that 50 µg of rIL-18/rat significantly increased the incidence (P < 0·0001) and severity (P < 0·002) of arthritis versus controls given PBS. Recombinant IL-18 administration also worsened arthritis severity at 10 µg/rat, although not significantly. In contrast, pretreatment with 300 µg/rat abrogated arthritis.
Fig. 3.
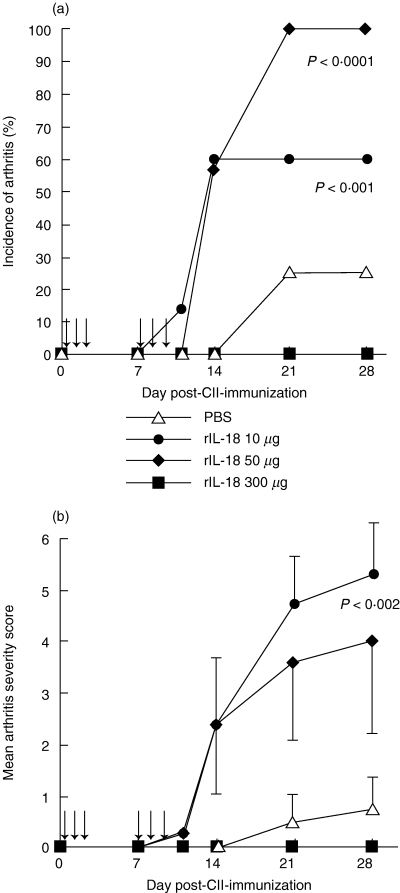
Actions of rIL-18 on the incidence (a) and severity (b) of CIA in the BB rat. Rats (n = 7) given rIL-18 at 10 and 50 µg doses on the day shown (arrows) developed arthritis at a significantly higher incidence of arthritis than controls (n = 8) given PBS (P < 0·001 and <0·0001, respectively); both doses worsened arthritis severity similarly (P < 0·002). In contrast, 300 µg of rIL-18 abrogated arthritis (n = 6). Arrows represent the days of rIL-18 administration; values are expressed as mean ± s.e.m. per group.
Individual sera from both groups were collected at day 28 and assayed for anti-CII antibodies. Data in Fig. 4a show that 50 µg of rIL-18 significantly enhanced total anti-CII-antibody production versus controls and 300 µg of IL-18 (P = 0·009). The analysis of IgG subclasses (Fig. 4b) demonstrated that rats injected with 50 µg of rIL-18 produced higher levels of IgG1 and IgG2a compared to controls (P < 0·02, and P < 0·002, respectively). IgG2b levels tended to be higher than controls but did not reach statistical significance.
Fig. 4.
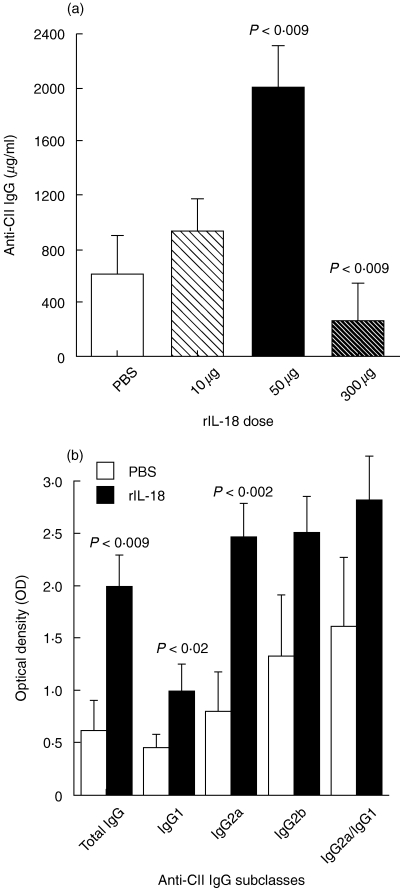
Actions of rIL-18 on the production of CII-specific antibodies. (a) Levels of anti-CII IgG measured in the sera of rats treated with different doses of rIL-18 as shown in Fig. 3a and b. Increased levels were significantly greater at the 50 µg dose, whereas changes with 10 µg were minor and 300 µg was suppressive (P < 0·009). (b) IgG subclass distribution of anti-CII antibodies in rats treated with 50 µg doses of rIL-18 or controls given PBS. The greatest increase was found in the IgG2a subclass (P < 0·002), but increases were also noted in IgG1 (P < 0·02) and IgG2b (P = n.s.). Values are expressed as mean ± s.e.m. per group.
Splenocytes harvested at day 28 from rIL-18 (50 µg dose) and PBS-treated rats were cultured with CII and cytokine release as measured by ELISA. Data shown in Fig. 5 demonstrate that rIL-18-treated rats produced higher levels of Th1-type cytokines IFN-γ and IL-2 in addition to monokines TNF-α and IL-6 (P < 0·04 to P = 0·02) than controls. In comparison the Th2-type cytokine, IL-10, was not affected.
Fig. 5.
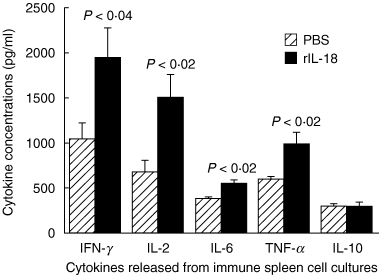
Actions of rIL-18 pretreatment on the production of cytokines by splenocytes obtained from CII-immune rats. The spleens were harvested 4 weeks after immunization as described in Fig. 3 and splenocytes cultured with CII for 48 h. Cytokine proteins were quantified in the supernatants by ELISA. IL-18-treated rats produced significantly greater amounts of proinflammatory cytokines (TNF-α and IL-6), Th1 cell cytokines (IL-2 and INF-γ) than controls (P < 0·04 to < 0·02), did but not produce the Th2 cell cytokine IL-10. Values are expressed as mean ± s.e.m. per group.
Treatment with anti-rIL-18 antibody suppresses arthritis incidence and severity, anti-CII antibody levels and cytokine responses
Two groups of rats were treated with neutralizing anti-rIL-18 IgG or normal rabbit IgG beginning at the time of immunization with a potent CII preparation; the antibody injections were continued thrice-weekly for 3 weeks (Fig. 6). Whereas anti-rIL18 antibodies did not affect the onset time of arthritis, they significantly reduced the incidence (P < 0·05) and severity (P < 0·002) of disease versus controls.
Fig. 6.
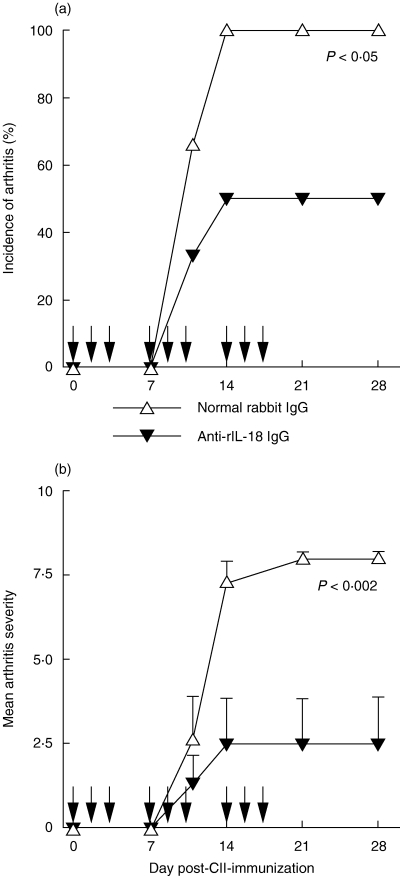
Neutralizing antibodies to rIL-18 attenuate arthritis. Both the incidence (a) and severity (b) of arthritis were significantly reduced in BB rats treated with rabbit anti-rIL-18 IgG versus controls given normal rabbit IgG at the time-points shown above by the arrows. Both groups contained six rats. Values are expressed as mean ± s.e.m. per group.
Data showing serum levels of anti-CII antibodies in these rats are summarized in Table 1. The levels of total anti-CII IgG antibody and subclasses IgG1 and IgG2a were significantly lower in anti-rIL-18-treated rats than controls (P < 0·04, P < 0·03 and P < 0·05, respectively). IgG2b levels were lower in the treatment group, but did not reach statistical significance.
Table 1.
The effects of anti-rIL-18 antibody on anti-CII IgG production*
| IgG subclasses (OD) | |||||
|---|---|---|---|---|---|
| Group | Anti-CII† | IgG1 | IgG2a | IgG2b | IgG2a/IgG1 |
| Normal IgG | 1·577 ± 105 | 0·879 ± 0·111 | 1·579 ± 0·114 | 1·895 ± 0·194 | 1·906 |
| Anti-IL18 | 0·886 ± 272 | 0·554 ± 0·060 | 0·965 ± 0·237 | 1·485 ± 0·520 | 1·987 |
| P-value | <0·04 | <0·03 | <0·05 | n.s. | |
Sera were collected 28 days after immunization and assayed by ELISA; each group contains six rats.
Antibodies to CII are expressed as µg/ml; IgG subclass values are expressed as OD and are not quantitative. n.s., not significant. All values are shown as mean ± s.e.m.
Splenocytes from rats receiving anti-rIL-18 IgG or normal IgG were also cultured with CII and cytokine levels were measured. Figure 7 shows that levels of IL-6 and IFN-γ were again lower in anti-rIL-18 IgG-treated rats (P < 0·03 and < 0·05 versus controls). The levels of IL-2 and TNF-α were also decreased and IL-10 levels were slightly higher in anti-rIL-18-treated rats, although not significantly.
Fig. 7.
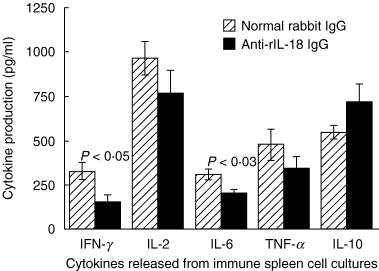
Neutralizing antibodies to rIL-18 decrease splenic cytokine production in CII-immunized rats. Splenocytes from CII-immune rats shown in Fig. 6 were harvested 4 weeks post-immunization and cultured with CII for 48 h. Cytokine proteins were quantified by ELISA. Anti-rIL-18 IgG-treated rats produced less IFN-γ and IL-6 than controls (P < 0·05 to < 0·03). Values are expressed as mean ± s.e.m. per group.
In an effort to dissociate immunosuppressive effects from the anti-inflammatory actions of anti-IL-18 treatment, a second group of rats was given anti-rIL-18 IgG at days 9 and 11 post-CII-immunization, a time when the immune response should have developed, yet prior to arthritis onset. As observed with the multiple-dose protocol, the onset, severity and incidence of arthritis were again significantly reduced compared to controls. Arthritis occurred in 50% of treated rats, which reached a mean severity score of 2·5 versus 100% incidence in controls (P < 0·06, n = 8 per group), with a mean severity score of 7·6 (P < 0·002, days 10–24). Ankle thickness measurements were also performed and were consistently lower throughout the study (P < 0·05–0·007 versus controls; days 10–24), thus confirming our visual scoring (data not shown).
Somewhat unexpectedly, IgG antibody levels to CII were again reduced at day 24 (0·548 ± 0·080 and 1·302 ± 0·218 study versus control mean ± s.e.m., respectively; P = 0·006), indicating that anti-IL-18 treatment need not be initiated before CII/IFA challenge. Immunoglobulin G subclass antibody studies were not performed on these animals.
Cytokine production by splenocytes harvested at 24 days post-immunization were measured. A BioPlex assay system was used for these supernatants under identical culture conditions. The results of this study were similar to those presented in Fig. 7, demonstrating that even a brief, relatively late course of anti-rIL-18 IgG also diminished splenic INF-γ (103 ± 18 versus 258 ± 60, P = 0·026) and GM-CSF (0·45 ± 0·15 versus 1·61 ± 0·35, P = 0·008). The amounts of IL-1α (25 ± 6 versus 65 ± 23) and IL-1β (93 ± 12 versus 147 ± 31) were lower than controls but not statistically different.
DISCUSSION
In addition to TNF-α and IL-1 α/β, IL-18 may be a fundamental driving force in CII-induced autoimmune joint injury. Our studies performed in rats show that IL-18 acts both as a potent immunostimulatory and a proinflammatory agent. The former role is critical in the pathogenesis of CIA, as CII is a T-dependent antigen, thus requiring Th1 lymphocyte participation. These cells are driven by INF-γ, produced by T cells and NK cells, which are stimulated by IL-18 and IL-12 produced by macrophages/monocytes and dendritic cells. Our studies, showing that normal splenocytes cultured with rIL-18 and submitogenic amounts of Con-A produce INF-γ, are consistent with this concept. Relevant to the model of CIA, activated Th1-type cells promote the production of IgG2a and IgG2b anti-CII antibodies, which bind to cartilage, fix and activate complement, thus triggering synovitis [31,32]. As anticipated in our studies, serum levels of IgG2a and IgG2b anti-CII were higher in rIL-18-treated rats than controls. Interestingly, anti-CII antibodies IgG1, which are regulated by Th2-type cytokines, were also found to be elevated. Because Th2 cytokines IL-4 and IL-10 were not affected by rIL-18 treatment, these data suggest that other mechanisms may be involved in B cell function. Overall, our data regarding IgG quantity and subclass, arthritis incidence and arthritis severity confirm the findings of Plater-Zyberk et al., who conducted similar studies with IL-18 in DBA/1 mice [33]. Interestingly, treating rats with a 300 µg dose of IL-18 beginning at the time of immunization suppressed immunity to CII and arthritis, suggesting a bimodal dose–response effect. The mechanism of high-dose IL-18 suppression is unknown, however, and warrants further study. Studies of INF-γ by others show that its effect on murine CIA vary depending on the dose and timing of administration [34–36]. These findings underscore the importance of dose–response studies in ascertaining the role(s) of cytokine in animal models.
Other possible actions of IL-18 include those on synovial T cells. Previous studies have identified T cells isolated from the joints of arthritic BB rats which preferentially express TCR Vβ 8·2 and proliferate on culture with CII [29]. Although these findings are less definitive that those demonstrating the induction of arthritis with monoclonal anti-CII antibodies [32], they do indicate that T cells contribute to joint destruction [29]. One action of IL-18 might be the stimulation of IL-2 production, as demonstrated in spleen cell cultures. IL-2 is increased in the synovium of affected joints in murine CIA, but not unaffected joints [37]. IL-2 is also capable of promoting T cell proliferation and NK cell growth. These cells produce IFN-γ, which has been found in the synovial fluid of patients with early-onset RA [38] and the joints of mice with CIA [39]. Attempts to attenuate CIA in mice by depleting NK cells with a monoclonal antibody, however, were not successful, even though these cells are more abundant in the synovium of inflamed joints [39]. The exact role of NK cells in CIA remains unclear. Additional roles that IL-18 might play in the pathogenesis of synovitis include the promotion of angiogenesis via ICAM-1 and VCAM-1 expression [21].
The ability of IL-18 to act as a proinflammatory molecule was also demonstrated by our studies. Spleen cells harvested from rIL-18-treated CII-immune rats were found to produce significantly greater amounts of TNF-α and IL-6, whereas the Th2 cytokine IL-10 production was unaffected. The possibility that these cytokines can be produced in the joint is supported by our observation that mRNA encoding IL-18 was expressed strongly in arthritic synovium, and peaked at day 3 after arthritis onset. This timing corresponds with the detection of peak mRNA message expression for IL-6, IL-1β, TNF-α and IFN-γ peaks in the synovium of rats affected by CIA, as well as their protein products (our unpublished data). The production of these cytokines in addition to the direct action of IL-18 on neutrophils and chondrocytes, which stimulate the production of chondrocyte cytokines and the release of metalloproteinase enzymes from both cell types, most probably facilitate cartilage destruction [10,22,23].
Given the multiple potentially adverse effects of IL-18, it was not unexpected that neutralizing anti-IL-18 antibodies administered at immunization or shortly before arthritis onset reduced the incidence and severity of CIA. The clinical results observed with these protocols correlated with decreased serum levels of total anti-CII antibody and IgG1 and IgG2a, which also corresponded with the reduction of in vitro and in vivo IFN-γ production. The effects of anti-rIL18 antibody on proinflammatory cytokine production was demonstrated by the significant reduction of splenocyte IL-6 production by rats treated thrice-weekly with anti-rIL-18 antibody. TNF-α levels also tended to be lower, although not significantly. Only a small reduction of IL-6 was seen in rats given two doses of neutralizing antibody, suggesting that earlier or longer exposure to neutralizing antibody was needed to suppress IL-6. As expected, Th2-type cytokine IL-10 levels were increased in some anti-IL-18-treated rats, although not significantly. Others have demonstrated recently that a naturally occurring IL-18 non-receptor binding protein can ameliorate CIA in mice when administered systemically as a fusion protein using the Fc portion of mouse IgG1 [27], or expressed locally following the intra-articular injection of an IL-18 binding protein-transfected adenovirus [40]. Both treatments were found to reduce anti-CII IgG2a antibody levels, the former approach being more effective.
In conclusion, our findings confirm those obtained in mice by other investigators and demonstrate that the rat model of CIA can be used effectively to study the role of IL-18 in arthritis. Our studies also show that IL-18 modulates CIA in a bimodal manner, i.e. 10 and 50 µg doses induce Th1-type cytokines (INF-γ and IL-2) which stimulate anti-CII IgG2a, whereas 300 µg of IL-18 suppresses immunity and arthritis. Moreover, 50 µg doses of IL-18 enhanced the production of proinflammatory cytokines IL-1β and IL-6, which effect bone and cartilage adversely. These results, and those by others showing that IL-18 activates NK cells, neutrophils, chondrocytes and other cell types indicate that IL-18 may play a pivotal role in the pathogenesis of inflammatory arthritis. From a therapeutic standpoint, it is important to note that by neutralizing endogenous IL-18, arthritis and immunity to CII can be attenuated. These properties support the potential use of IL-18 antagonists as biological treatments of inflammatory diseases in man.
Acknowledgments
These studies were funded largely by a Merit Review Grant provided by the Department of Veterans Affairs and with additional help from USPHS grants AR39166 and AR43589.
References
- 1.Griffiths MM, Cremer MA, Harper DS, McCall S, Cannon GW. Immunogenetics of collagen-induced arthritis in rats. Both MHC and non-MHC gene products determine the epitope specificity of immune response to bovine and chick type II collagens. J Immunol. 1992;149:309–16. [PubMed] [Google Scholar]
- 2.Alonzi T, Fattori E, Lazzaro D, et al. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–8. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohshima S, Saeki Y, Mima T, et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA. 1998;95:8222–6. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joosten LA, Helsen MM, van de Loo FA, van den Berg WB. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNF alpha, anti-IL-1 alpha/beta, and IL-1Ra. Arthritis Rheum. 1996;39:797–809. doi: 10.1002/art.1780390513. [DOI] [PubMed] [Google Scholar]
- 5.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler DM, Malfait AM, Mason LJ, et al. DBA/1 mice expressing the human TNF-alpha transgene develop a severe, erosive arthritis: characterization of the cytokine cascade and cellular composition. J Immunol. 1997;159:2867–76. [PubMed] [Google Scholar]
- 7.Kohno K, Kataoka J, Ohtsuki T, et al. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol. 1997;158:1541–50. [PubMed] [Google Scholar]
- 8.Ghayur T, Banerjee S, Hugunin M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–23. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 9.Gillespie MT, Horwood NJ. Interleukin-18: perspectives on the newest interleukin. Cytokine Growth Factor Rev. 1998;9:109–16. doi: 10.1016/s1359-6101(98)00004-5. [DOI] [PubMed] [Google Scholar]
- 10.Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol. 1999;162:1096–100. [PubMed] [Google Scholar]
- 11.Stoll S, Jonuleit H, Schmitt E, et al. Production of functional IL-18 by different subtypes of murine and human dendritic cells (DC): DC-derived IL-18 enhances IL-12-dependent Th1 development. Eur J Immunol. 1998;28:3231–9. doi: 10.1002/(SICI)1521-4141(199810)28:10<3231::AID-IMMU3231>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 12.Udagawa N, Horwood NJ, Elliott J, et al. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. J Exp Med. 1997;185:1005–12. doi: 10.1084/jem.185.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamura H, Tsutsi H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura K, Okamura H, Wada M, Nagata K, Tamura T. Endotoxin-induced serum factor that stimulates gamma interferon production. Infect Immun. 1989;57:590–5. doi: 10.1128/iai.57.2.590-595.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okamura H, Tsutsui H, Kashiwamura S, Yoshimoto T, Nakanishi K. Interleukin-18: a novel cytokine that augments both innate and acquired immunity. Adv Immunol. 1998;70:281–312. doi: 10.1016/s0065-2776(08)60389-2. [DOI] [PubMed] [Google Scholar]
- 16.Hunter CA, Timans J, Pisacane P, et al. Comparison of the effects of interleukin-1 alpha, interleukin-1 beta and interferon-gamma-inducing factor on the production of interferon-gamma by natural killer. Eur J Immunol. 1997;27:2787–92. doi: 10.1002/eji.1830271107. [DOI] [PubMed] [Google Scholar]
- 17.Micallef MJ, Ohtsuki T, Kohno K, et al. Interferon-gamma-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-gamma production. Eur J Immunol. 1996;26:1647–51. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- 18.Netea MG, Kullberg BJ, Verschueren I, Van Der Meer JW. Interleukin-18 induces production of proinflammatory cytokines in mice: no intermediate role for the cytokines of the tumor necrosis factor family and interleukin-1beta. Eur J Immunol. 2000;30:3057–60. doi: 10.1002/1521-4141(200010)30:10<3057::AID-IMMU3057>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 19.Puren AJ, Razeghi P, Fantuzzi G, Dinarello CA. Interleukin-18 enhances lipopolysaccharide-induced interferon-gamma production in human whole blood cultures. J Infect Dis. 1998;178:1830–4. doi: 10.1086/314481. [DOI] [PubMed] [Google Scholar]
- 20.Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA. 2000;97:734–9. doi: 10.1073/pnas.97.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohka H, Yoshino T, Iwagaki H, et al. Interleukin-18/interferon-gamma-inducing factor, a novel cytokine, up-regulates ICAM-1 (CD54) expression in KG-1 cells. J Leukoc Biol. 1998;64:519–27. doi: 10.1002/jlb.64.4.519. [DOI] [PubMed] [Google Scholar]
- 22.Cannetti CA, Leung BP, Culshaw S, McInnes IB, Cunha F, Liew FY. IL18 enhances collagen-induced arthritis by recruiting neutrophils via TNF-alpha and leukotriene B4. J Immunol. 2003;171:1009–15. doi: 10.4049/jimmunol.171.2.1009. [DOI] [PubMed] [Google Scholar]
- 23.Leung BP, Culshaw S, Gracie JA, et al. A role for IL-18 in neutrophil activation. J Immunol. 2001;167:2879–86. doi: 10.4049/jimmunol.167.5.2879. [DOI] [PubMed] [Google Scholar]
- 24.Gracie JA, Forsey RJ, Chan WL, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999;104:1393–401. doi: 10.1172/JCI7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung BP, McInnes IB, Esfandiari E, Wei XQ, Liew FY. Combined effects of IL-12 and IL-18 on the induction of collagen-induced arthritis. J Immunol. 2000;164:6495–502. doi: 10.4049/jimmunol.164.12.6495. [DOI] [PubMed] [Google Scholar]
- 26.Jiang HR, Wei X, Niedbala W, Lumsden L, Liew FY, Forrester JV. IL-18 not required for IRBP peptide-induced EAU: studies in gene-deficient mice. Invest Ophthalmol Vis Sci. 2001;42:177–82. [PubMed] [Google Scholar]
- 27.Banda NK, Vondracek A, Kraus D, et al. Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J Immunol. 2003;170:2100–5. doi: 10.4049/jimmunol.170.4.2100. [DOI] [PubMed] [Google Scholar]
- 28.Rosloniec EF, Kang AH, Myers LK, Cremer MA. Collagen-induced arthritis. In: Coico R, Shevach E, editors. Current protocols in immunology. New York, NY: Wiley and Sons; 1996. pp. 15.5.1–15.5.24. [Google Scholar]
- 29.Ye XJ, Aelion J, Endres RO, Kang AH, Cremer MA. Evidence for preferential T cell receptor V beta gene usage and T cell clonal expansion in the synovium of BB rats with early-onset collagen- induced arthritis. Cell Immunol. 1998;183:81–9. doi: 10.1006/cimm.1998.1249. [DOI] [PubMed] [Google Scholar]
- 30.Cremer MA, Terato K, Seyer JM, et al. Immunity to type XI collagen in mice. Evidence that the alpha 3 (XI) chain of type XI collagen and the alpha 1(II) chain of type II collagen share arthritogenic determinants and induce arthritis in DBA/1 mice. J Immunol. 1991;146:4130–7. [PubMed] [Google Scholar]
- 31.Watson WC, Townes AS. Genetic susceptibility to murine collagen II autoimmune arthritis. Proposed relationship to the IgG2 autoantibody subclass response, complement C5, major histocompatibility complex (MHC) and non-MHC loci. J Exp Med. 1985;162:1878–91. doi: 10.1084/jem.162.6.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terato K, Hasty KA, Reife RA, Cremer MA, Kang AH, Stuart JM. Induction of arthritis with monoclonal antibodies to collagen. J Immunol. 1992;148:2103–8. [PubMed] [Google Scholar]
- 33.Plater-Zyberk C, Joosten LA, Helsen MM, et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J Clin Invest. 2001;108:1825–32. doi: 10.1172/JCI12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakajima H, Takamori H, Hiyama Y, Tsukada W. The effect of treatment with interferon-gamma on type II collagen-induced arthritis. Clin Exp Immunol. 1990;81:441–5. doi: 10.1111/j.1365-2249.1990.tb05353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boissier MC, Chiocchia G, Bessis N, et al. Biphasic effect of interferon-gamma in murine collagen-induced arthritis. Eur J Immunol. 1995;25:1184–90. doi: 10.1002/eji.1830250508. [DOI] [PubMed] [Google Scholar]
- 36.Billiau A, Heremans H, Vermeire K, Matthys P. Immunomodulatory properties of interferon-gamma. An update. Ann NY Acad Sci. 1998;856:22–32. doi: 10.1111/j.1749-6632.1998.tb08309.x. [DOI] [PubMed] [Google Scholar]
- 37.Thornton S, Duwel LE, Boivin GP, Ma Y, Hirsch R. Association of the course of collagen-induced arthritis with distinct patterns of cytokine and chemokine messenger RNA expression. Arthritis Rheum. 1999;42:1109–18. doi: 10.1002/1529-0131(199906)42:6<1109::AID-ANR7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 38.Steiner G, Tohidast-Akrad M, Witzmann G, et al. Cytokine production by synovial T cells in rheumatoid arthritis. Rheumatology (Oxford) 1999;38:202–13. doi: 10.1093/rheumatology/38.3.202. [DOI] [PubMed] [Google Scholar]
- 39.Wang CR. Involvement of natural killer T cells in C57BL/6 mouse model of collagen-induced arthritis. J Microbiol Immunol Infect. 2003;36:15–20. [PubMed] [Google Scholar]
- 40.Smeets RL, van de Loo FA, Arntz OJ, Bennink MB, Joosten LA, van den Berg WB. Adenoviral delivery of IL-18 binding protein C ameliorates collagen-induced arthritis in mice. Gene Ther. 2003;10:1004–11. doi: 10.1038/sj.gt.3301986. [DOI] [PubMed] [Google Scholar]