

Abstract
Mouse hepatitis virus type 3 (MHV3), a coronavirus, is an excellent animal model for the study of immunological disorders related to acute and chronic hepatitis. In this study, we have verified if the fulminant hepatitis induced by MHV3 could be related to an impairment of innate immunity. Groups of three C57BL/6 mice were infected with the pathogenic L2-MHV3 or attenuated YAC-MHV3 viruses, and the natural killer (NK) cell populations from liver, spleen and bone marrow were analysed. The percentage of intrahepatic NK1·1+T cell receptor (TCR)− cells did not increase while NK1·1+TCRinter cells decreased in both L2-MHV3- and YAC-MHV3-infected mice. Concurrently, splenic and myeloid NK1·1+ cells decreased in L2-MHV3-infected mice. However, the cytotoxic activity of NK cells increased in liver and decreased in bone marrow from pathogenic L2-MHV3-infected mice while no modification was detected in YAC-MHV3-infected mice. Flow cytometric analysis revealed that both normal and larger splenic or myeloid NK cells decreased more in pathogenic L2-MHV3-infected mice than in attenuated YAC-MHV3-infected mice. In vitro viral infections of interleukin (IL)-15-stimulated lymphoid cells from liver and bone marrow revealed that L2-MHV3 induced higher decreases in cell viability of NK1·1+ cells than the YAC-MHV3 variant. The NK cell decreases were due to the viral permissivity leading to cytopathic effects characterized by cell rounding, syncytia formation and apoptosis. Larger NK+ syncytia were observed in L2-MHV3-infected cells than in YAC-MHV3-infected cells. These results suggest that NK cell production is impaired by viral infection favouring fulminant hepatitis.
Keywords: apoptosis, IL-15, mouse hepatitis virus, natural immunity, NK cells
INTRODUCTION
Hepatitis is an inflammatory disease induced by various causes, such as bacteria and viruses. The viral persistence in liver is associated with a chronic hepatitis leading to cirrhosis, hepatic carcinoma and death [1–3]. The role of natural killer (NK) cells is to provide a first line of defence against bacteria, tumour cells and virus-infected cells through the exertion, without priming, of a cytotoxic function as well as a production of interferon (IFN)-γ [4,5]. However, the fulminance of the hepatitis cannot result from a deficiency in antiviral adaptative immune responses following a decrease in T and B lymphoid cells. The acute hepatitis suggests a virus-mediated deficiency in innate immunity mechanisms. However, it was reported recently that the hepatitis C envelope protein can bind to natural killer (NK) cells and impair their cytotoxic properties and IFN-γ production, thus altering the host's natural defences and innate immunity against viral hepatitis [6,7]. The liver contains a population of NK cells and a unique population of T cells with an intermediate level of αβ-T cell receptor (TCR) cells expressing the NK1·1 marker (NK-T cells) [8,9]. The level of NK cell subsets in the liver during the first days of the infection depends on both their recruitment from peripheral lymphoid organs and the efficiency of NK cell production by the bone marrow. NK cells are large granular lymphocytes derived from bone-marrow (BM) progenitor cells which require an intact microenvironment as well as interleukin (IL)-15 production for their complete maturation into lytic cells [10,11]. However, the role of intrahepatic NK cells in the viral hepatitis process is not elucidated.
Mouse hepatitis virus type 3 (MHV3) is an excellent animal model for the study of immunological disorders related to hepatitis. The MHV3 virus induces acute and/or chronic hepatitis associated with viral persistence and immunodeficiency [12,13]. The sensitivity of mice to MHV3 infection varies according to the strain, age and immune status of the animal. The development of a fulminant hepatitis in susceptible C57BL/6 mice which leads to the death of the animal within 3–5 days post-infection (p.i.) is accompanied by a loss of T lymphocytes in spleen and thymus as well as a decrease of splenic and myeloid B lymphocytes [14–16]. We have previously generated an attenuated viral variant, the YAC-MHV3, which induces a subclinical hepatitis in susceptible C57BL/6 mice without inducing immune disorders in the lymphoid organs, including bone marrow, and in T and B lymphocytes [15–18].
We have reported previously that intrahepatic mononuclear cells increased in the liver of L2-MHV3-infected mice within 3 days of infection but not in attenuated YAC-MHV3-infected mice [19,20]. Percentages of intrahepatic CD4+TCRinter and CD4+TCRhigh cell subsets increased in C57BL/6 mice infected with the pathogenic L2-MHV3 but not in liver from the attenuated YAC-MHV3-infected mice. However, percentages of intrahepatic total NK cells did not increase in either L2-MHV3- or YAC-MHV3-infected mice. The in vivo significance of an unresponsiveness of intrahepatic NK cells has not yet been elucidated.
Controversial results on the protective role of NK cells in MHV infections were reported 20 years ago. At that time, it was observed that NK cell activity increased in peritoneal exudates from MHV3-infected C57BL/6 mice suggesting that NK cells did not play an important role in the defence of mice against MHV3 infection [21,22]. Stohlman et al. [22] have observed no IFN-γ production in spite of increase of NK cell cytotoxicity favouring viral replication as it was demonstrated that IFN-γ is involved in the protection against MHV3-induced hepatitis [23]. In addition, the NK depletion increased both inflammatory foci and virus titres in the liver of MHV-infected mice [24]. On the other hand, the natural resistance of A/J mice to the fulminant hepatitis induced by MHV3 has been shown to depend on a bone marrow subpopulation showing features similar to those of NK cells [25]. This observation suggests that the integrity of the bone marrow may play a crucial role in the resistance against the acute phase of hepatitis via the production of NK cells. Taken together, these observations suggest that NK cell disorders may be involved in pathogenic L2-MHV-infected mice. Then, the maturation process of NK cells in bone marrow would be impaired in pathogenic L2-MHV3-infected mice, as demonstrated previously for myeloid pre-B and B lymphocytes [15], leading to a decreased production of cytotoxic NK cells and lower recruitment of NK cells in liver.
In this paper, we report that both NK cell production by the bone marrow and NK cell recruitment by the liver are impaired differently by pathogenic and attenuated viral strains due to virus-induced cell death and apoptosis.
MATERIALS AND METHODS
Mice
C57BL/6 mice were purchased from Charles River Laboratories (St-Constant, Quebec, Canada). Before being used, the animals were tested for the presence of anti-MHV3 antibodies by an enzyme-linked immunosorbent assay (ELISA) test using an MHV3 preparation as antigen. During the experiments, the animals were housed in a sterile atmosphere (Forma Scientific, Marietta, OH, USA). Female mice between 8 and 12 weeks of age were used in all experiments.
Viruses
Pathogenic MHV3 was a cloned substrain produced in L2 cells (L2-MHV3) as described previously [26]. The YAC-MHV3 variant was a cloned virus derived from persistently infected lymphoid YAC cell [17]. Viruses were produced on L2 cells before use and their pathogenic properties were verified regularly.
In vivo viral infections
Groups of three mice were infected intraperitoneally (i.p.) with 1000 TCID50 of pathogenic L2-MHV3 or attenuated YAC-MHV3. Mock-infected mice received a similar volume of RPMI-1640 (Gibco Laboratories, Grand Island, NY, USA). At various times post-infection (p.i.), mice were killed by CO2 anoxia. Liver, spleen and bone marrow were collected and lymphoid cells were isolated.
Cells
L2 cells, a continuous mouse fibroblast cell line, were grown in RPMI-1640 supplemented with glutamine (2 mm), antibiotics (penicillin, 100 U/ml and streptomycin, 100 mg/ml) (Gibco Laboratories) and 5% fetal calf serum (FCS) (Hy-Clone Laboratories, Professional Diagnostics, Alberta, Canada). L2 cells were used for virus propagation and titration.
YAC-1 cells, a continuous mouse lymphocyte cell line, were grown in RPMI-1640 with glutamine (2 mm), antibiotics (penicillin, 100 U/ml and streptomycin, 100 mg/ml) and 10% FCS. These cells were used in the cytotoxicity assays.
Intrahepatic mononuclear cells (MNC) were isolated from the livers of three mice in each experimental group or from uninfected mice according to experiments, as described previously [19]. Then, the livers were pressed through a 70 µm cell strainer, which was then washed with 20 ml of RPMI-1640 (Gibco Laboratories) containing 20% FCS and antibiotics. The cell suspensions were then deposited over a 5 ml cushion of FCS to allow debris sedimentation. The top layer was then centrifuged on a discontinuous Percoll gradient [45%, 67% Percoll in phosphate buffered saline (PBS); Amersham Pharmacia, Uppsala, Sweden] for 30 min at 1000 g. MNC were collected at the interface of the 45% and 67% Percoll layers. The cells were then washed in RPMI-1640 containing 20% FCS and counted electronically (Coulter Counter, Coulter Electronics, Hialeah, FL, USA).
Splenic lymphocytes were obtained from three mice in each experimental group. Spleens were pressed through a 70 µm cell strainer (Falcon, Fisher Scientific Co., Montreal, Canada) and collected in RPMI-1640 supplemented with 20% FCS.
Bone marrow lymphocytes were isolated from the femurs of three mice from each experimental group or from uninfected mice, according to experiments. Femurs were left in 70% ethanol for 2–5 min for disinfection and washed subsequently with RPMI-1640. The femurs were then excised from the surrounding muscle tissue, both ends were cut with scissors and the marrow was flushed using a syringe filled with RPMI-1640 and fitted with a 0·45-mm diameter needle. Clusters within the bone marrow suspension were dissociated by vigorous pipetting. The cell suspensions were then deposited over a 5-ml cushion of FCS to allow debris sedimentation.
Splenic and myeloid cell suspensions were enriched in lymphocytes by passage through a Lymphoprep gradient (Cedarlane, Hornby, Ontario, Canada). The cell suspensions were then washed, resuspended in RPMI-1640 with 20% FCS and counted electronically.
In vitroviral infections in NK cells
Lymphocytes were isolated from the liver or bone marrow of groups of four to six mice, as described above, and pooled together. Cells were seeded in 24-well plates at a concentration of 106 cells/ml in RPMI-1640 media supplemented with antibiotics and 20% FCS. Recombinant murine IL-15 (rmIL-15) (Endogen, Woburn, MA, USA) was added at a final concentration of 20 ng/ml. The plates were incubated at 37°C, under 5% CO2 atmosphere for 7 days, and were then infected with 0·1–1 multiplicity of infection (m.o.i.) of L2-MHV3 or YAC-MHV3 viruses. The plates were then incubated for an additional 24 h. Non-adherent cells were collected, counted electronically and immunolabelled for flow cytometric analysis.
Flow cytometric analysis
Percentages and number of events of myeloid and splenic NK1·1+ and B220+ cells were determined by a double immunolabelling: 106 cells were resuspended in 1 ml of RPMI-1640 containing 20% FCS, and incubated on ice for 30 min with optimal dilutions of antimouse NK1·1-phycoerythrin (PE) (clone PK136, mouse IgG2aκ, Pharmingen, Toronto, Canada) and antimouse CD45R/B220-fluorescein isothiocyanate (FITC) (clone RA3–6B2, rat IgG2aκ, Pharmingen) or FITC-antiαβ-TCR (clone H57-597, Pharmingen) monoclonal antibodies (MoAbs). Cells were then washed in RPMI-1640 containing 20% FCS and fixed overnight at 4°C in PBS, pH 7·2, containing 1% formaldehyde (Fisher Scientific). Flow cytometric analysis was performed on a fluorescence-activated cell sorting flow cytometer (FACScan) with Cell Quest software (Becton-Dickinson, Mountain View, CA, USA). Ten thousand cells were analysed per sample and percentages of various subpopulations were determined by a multiparametric analysis.
Virus titration
Liver, spleen, bone marrow from infected mice and infected rmIL-15-stimulated myeloid cells were kept frozen at − 70°C until titration. The organs were then thawed, triturated in RPMI-1640 medium and centrifuged while the cells were thawed and centrifuged. The supernatants were used as viral suspension. They were then serially diluted in 10-fold steps using RPMI-1640 and tested on L2 cells cultured in 96-well microtitre plates. Cytopathic effects, characterized by syncytia formation and cell lysis, were recorded at 72 h p.i. and virus titres expressed as log10 TCID50. All titrations were performed in triplicate.
TUNEL test
Infected rmIL-15-stimulated myeloid cells were collected, washed, counted electronically and adjusted to a concentration of 106 cells/ml before being cytocentrifuged on coated glass slides (100 µl PBS with 5% BSA for 10 min, 1500 r.p.m) (Cytospin, Shandon Southern Instruments, Sewickly, PA, USA) for 5 min at 1100 r.p.m. The slides were then prepared as indicated on the commercial kit (Boehringer Mannheim, Laval, Canada). Briefly, the cells were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature and washed twice in PBS. Cells were then permeabilized for 2 min at 4°C using a 0,1% sodium citrate buffer solution containing 0,1% Triton X-100 and washed twice in PBS. The cytospots were stained with the TUNEL reagent for 60 min at 37°C in a humidified atmosphere. After washing, the slides were mounted in a medium containing 90% glycerol in PBS and 0·1% p-phenylenediamine (Fisher Scientific). A fluorescence microscope (Leitz Dialux 22, Ernst Leitz ltd., Midland, Ontario, Canada) was used for analysis. The percentage of positive cells was determined by counting 500 cells. A double immunolabelling using TUNEL-TMR (Boeringher Mannheim) and FITC-anti-Pan NK mAb (clone DX5, Pharmingen) was also performed on in vitro infected myeloid lymphocytes. Experiments were conducted in triplicate.
Cytotoxicity test
LDH release was used to measure NK cell cytotoxicity against YAC-1 cells (Roche Molecular Biochemicals, Mannheim, Germany). Briefly, intrahepatic splenic and myeloid NK cells, extracted from three mice in each experimental group, were serially diluted using RPMI 1640 with 1% FCS and seeded on a 96-well tissue culture plate. The target cells, YAC-1 cells, were added to the first series of effector cells at an optimal concentration (effector–target cell ratio). In the second series, assay medium was added to the effector cells to determine their spontaneous lactate dehydrogenase (LDH) release (effector cell control). The absorbance of the treated supernatants was read at a wavelength of 490 nm. The percentage of cytotoxicity was determined as follows:
 |
Statistical analysis
Percentage differences were determined using Student's t-test. Comparison was performed between cells from infected versus control mice for in vivo studies and infected cells versus control cells for in vitro studies. Cytotoxicity test results were analysed by an anova test.
RESULTS
Viral replication of pathogenic L2-MHV3 and attenuated YAC-MHV3 viruses in liver, spleen and bone marrow from C57BL/6 mice
Replication of pathogenic L2-MHV3 and attenuated YAC-MHV3 viruses was verified in liver, spleen and isolated bone marrow lymphoid cells from infected C57BL/6 mice. As shown in Table 1, infectious viruses were produced as soon as 24 h in liver, spleen and bone marrow from L2-MHV3-infected mice. Lower virus titres were detected in liver and bone marrow from YAC-MHV3 infected mice. The viral titres observed at 72 h p.i. in the liver and bone marrow of L2-MHV3-infected mice increased more than in those infected with YAC-MHV3 variant.
Table 1.
Infectious virus titres in liver, spleen and bone marrow from pathogenic L2-MHV3- and attenuated YAC-MHV3-infected C57BL/6 mice
| Virus titres (log10 TCID50/ml) | |||
|---|---|---|---|
| Organ | Virus | 24 h p.i. | 72 h p.i. |
| Liver | Control | <1·6 | <1·6 |
| L2-MHV3 | 3·1 ± 0·5 | 5·2 ± 0·5 | |
| YAC-MHV3 | 2·1 ± 0·3 | 3·4 ± 0·4 | |
| Spleen | Control | <1·6 | <1·6 |
| L2-MHV3 | 2·3 ± 0·3 | 4·1 ± 0·5 | |
| YAC-MHV3 | <1·6 | 2·5 ± 0·3 | |
| Bone marrow | Control | <1·6 | <1·6 |
| L2-MHV3 | 2·8 ± 0·2 | 4·8 ± 0·5 | |
| YAC-MHV3 | 1·8 ± 0·3 | 2·3 ± 0·4 | |
NK1·1+ cell subsets from livers of C57BL/6 mice infected with pathogenic L2-MHV3 or attenuated YAC-MHV3 viruses
To verify if the fulminant hepatitis induced by the pathogenic L2-MHV3 strain in susceptible C57BL/6 mice is related to a deficiency in NK cell subsets, groups of three C57BL/6 mice were infected with 1000 TCID50 of pathogenic L2-MHV3 or attenuated YAC-MHV3 variant. Mice were killed at 72 h p.i. and intrahepatic mononuclear cells were isolated. A macroscopic examination of the liver revealed that hepatic lesions were more extensive in L2-MHV3-infected mice than in YAC-MHV3-infected mice, as reported previously (Lamontagne and Dupuy, 1984) (Table 2). The number of mononuclear cells isolated from the livers was higher in L2-MHV3-infected mice (5·23 ± 1·5 × 106 cells/liver) (P < 0·001) than in YAC-MHV3- or mock-infected mice (1·5 ± 1·2 × 106 and 1·2 ± 0·2 × 106 cells/liver, respectively). The intrahepatic lymphoid cells were then phenotyped by double-immunolabellings with FITC-antiαβ-TCR and PE-anti-NK1·1 MoAbs and the percentages of NK1·1+TCR- (NK cells), NK1·1+TCRinter (NK-T cells), NK1·1-TCRinter and NK1·1-TCRhigh were evaluated by flow cytometric analysis. Percentage of intrahepatic NK1·1+TCR- cells did not increase in both L2-MHV3- and YAC-MHV3-infected mice while NK1·1+TCRinter cells decreased (P < 0·05–0·001) (Table 2). However, the absolute numbers of NK1·1+TCR− and NK1·1+TCRinter increased in L2-MHV3-infected mice (P < 0·01) but not in YAC-MHV3-infected mice. In contrast, the percentages of NK1·1−TCRinter cells increased in the liver of L2-MHV3-infected mice only (P < 0·001), as reported previously [19]. Absolute numbers of NK1·1+TCRinter and NK1·1−TCRhigh cells, however, increased strongly in L2-MHV3-infected mice (nine and three times, respectively) (P < 0·001) while the NK1·1−TCRinter cells increased less in YAC-MHV3-infected mice (P < 0·05).
Table 2.
Percentages and absolute numbers of NK1·1+/αβ-TCR cell subsets in liver from mock-infected, L2-MHV3- and YAC-MHV3-infected C57BL/6 mice
| % NK1·1+ cell subsets (absolute number ×105)2 | |||||
|---|---|---|---|---|---|
| Viral infection | Hepatitis1 | NK1·1+TCR- | NK1·1+TCRinter | NK1·1−TCRinte | NK1·1−TCRhigh |
| Mock-infected | – | 5·3 ± 1·4 | 17·0 ± 1·5 | 9·4 ± 1·2 | 8·1 ± 0·3 |
| (1·00 ± 0·27) | (3·21 ± 0·28) | (1·78 ± 0·23) | (1·53 ± 0·06) | ||
| L2-MHV3 | + + + | 6·5 ± 2·8 | 8·4 ± 0·9** | 20·1 ± 0·4** | 7·0 ± 1·4 |
| (5·43 ± 2·34)** | (7·01 ± 0·75)** | (16· ± 0·33)** | (5·8 ± 1·17)** | ||
| YAC-MHV3 | + | 5·1 ± 1·9 | 9·3 ± 0·5*** | 12·1 ± 1·4 | 5·7 ± 2·3 |
| (1·49 ± 0·56) | (2·72 ± 0·15) | (3·54 ± 0·41)* | (1·67 ± 0·67) | ||
Hepatitis was determined by a macroscopic examination of liver: + no discoloration, small necrotic foci; + ++ discolouration, extensive necrotic foci; and high friability.
Intrahepatic lymphocytes were purified by Percoll gradient, double-labelled with anti-NK1·1−PE and anti-TCR-FITC MoAbs and analysed using a FACScan flow cytometer. Lymphoid cells were gated according to FSC/SSC parameters and cell numbers were evaluated based on a total of 10 000 recorded events. Absolute numbers were calculated by comparing the percentage of cells in each region with the total number of cells obtained from liver.
P < 0·05;
P < 0·01;
P < 0·001.
Splenic NK1·1+ and B cells from C57BL/6 mice infected with pathogenic L2-MHV3 or attenuated YAC-MHV3 viruses
To determine if the lower increase in NK+ cell subsets than other lymphoid cells in the liver from L2-MHV3-infected mice results from a deletion of peripheral NK cells, splenic cells were isolated from groups of three mice infected with L2-MHV3 or YAC-MHV3 viruses at 24 and 72 h p.i. Splenic lymphoid cells were then immunolabelled using anti-NK1·1−PE and anti-B220-FITC MoAbs and analysed by flow cytometry. As shown in Fig. 1, two cell clusters were detected according to FSC/SSC parameters: one showing typical lymphoid FSC/SSC parameters (region R1: normal cells) and a second, characterized by an increased FSC parameter (region R2: large cells) (Fig. 1, section I). In Fig. 1, sections I-A to I-C, the percentage of large cells (R2 region) increased in both infected groups of mice. Phenotypic analysis of splenic lymphoid cells revealed that the percentage of normal NK1·1+ cells increased slightly in YAC-MHV3-infected mice only (Fig. 1, sections I-D to I-F). In addition, the percentages of larger NK+ cells increased in L2-MHV3-infected mice while larger B220+ cells decreased (Fig. 1; sections I-G to I-I).
Fig. 1.

Section I: flow cytometric analysis of normal (R1 region) (d, e, f) and larger (R2 region) (g, h, i) NK+ and B220+ lymphoid cells in spleen from mock-infected (a, d and g), and L2-MHV3- (b, e and h) and YAC-MHV3- (c, f and i) infected C57BL/6 mice at 72 h p.i. Myeloid lymphocytes were double-labelled using anti-NK1·1-PE and anti-B220-FITC MoAbs and were analysed using a FACScan flow cytometer on a total of 10 000 events recorded. Section II: absolute numbers of normal (a and c) and larger (b and d) NK1·1+ cells (a and b) and B220+ lymphocytes (c and d) in spleen from mock-infected (▪), L2-MHV3- (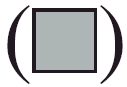 ) and YAC-MHV3- (□) infected C57BL/6 mice at 24 h p.i. Splenic lymphocytes were double-labelled using anti-NK1·1−PE and anti-B220-FITC MoAbs and were analysed using a FACScan flow cytometer on a total of 10 000 events recorded. The results are representative of three experiments. *P < 0·05; **P < 0·001.
) and YAC-MHV3- (□) infected C57BL/6 mice at 24 h p.i. Splenic lymphocytes were double-labelled using anti-NK1·1−PE and anti-B220-FITC MoAbs and were analysed using a FACScan flow cytometer on a total of 10 000 events recorded. The results are representative of three experiments. *P < 0·05; **P < 0·001.
The statistical analysis of splenic NK1·1+ and B220+ cells in the groups of L2-MHV3- and YAC-MHV3-infected mice for 24 and 72 h p.i. indicated that the absolute numbers of both normal and larger NK1·1+ cells decreased strongly in L2-MHV3-infected mice as soon as 24 h p.i. (P < 0·05–0·001) (Fig. 1, sections II-A and II-B). The absolute numbers of large splenic NK1·1+ cells higher decreased in L2-MHV3- than in YACMHV3-infected mice (P < 0·05 and 0·01). However, absolute numbers of both normal and large splenic B220+ lymphocytes decreased only in mice infected with the pathogenic L2-MHV3 strain (P < 0·001).
Production of NK1·1+ and B220+ cells in bone marrow from mock-infected, L2-MHV3- and YAC-MHV3-infected mice
To verify if the loss of NK1·1+ cells in the spleen of L2-MHV3-infected mice reflects a lower production of NK1·1+ cells by the bone marrow, groups of C57BL/6 mice were then infected with either the pathogenic L2-MHV3 or the attenuated YAC-MHV3 and were killed at 24 and 72 h p.i. Myeloid lymphocytes were isolated, double-labelled with anti-NK1·1−PE and anti-B220-FITC MoAbs and analysed by flow cytometry. Two distinct cell populations were observed according to their FSC/SSC parameters, similar to those seen previously in the spleen (Fig. 2). As shown in Fig. 2, sections I-A to I-C, myeloid lymphoid cells are distributed in both the R1 (normal cells) and R2 (large cells) regions. The percentage of normal lymphoid cells (R1 region) from L2-MHV3-infected mice only decreased while larger cells (R2 region) increased (Fig. 2, section I-B). Phenotypic analysis of bone marrow lymphoid cells revealed that the percentages of both normal and larger NK1·1+ cells decreased strongly in L2-MHV3 (Fig. 2, sections I-E and I-H) while the larger NK cells decreased less in YAC-MHV3-infected mice (Fig. 2, sections I-F and I-I). Normal B220+ cells increased in L2-MHV3-infected mice while larger B220+ cells rather decreased (Fig. 2, sections I-E and I-H).
Fig. 2.

Section I: flow cytometric analysis of normal (R1 region) (d, e, f) and larger (R2 region) (g, h, i) NK1·1+ and B220+ lymphoid cells in bone marrow from mock-infected (a, d and g) and L2-MHV3- (b, e and h) and YAC-MHV3- (c, f and i) infected C57BL/6 mice at 72 h p.i. Myeloid lymphocytes were double-labelled using anti-NK1·1−PE and anti-B220−FITC MoAbs and were analysed using a FACScan flow cytometer on a total of 10 000 events recorded. Section II: absolute numbers of normal (a and c) and larger (b and d) NK1·1+ cells (a and b) and B220+ lymphocytes (c and d) in bone marrow from mock-infected (▪), L2-MHV3- (□) and YAC-MHV3- (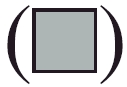 ) infected C57BL/6 mice at 24 h p.i. Myeloid lymphocytes were double labelled using anti-NK1·1−PE and anti-B220−FITC MoAbs and were analysed using a FACScan flow cytometer on a total of 10 000 events recorded. The results are representative of three experiments. *P < 0·05; **P < 0·01; ***P < 0·001.
) infected C57BL/6 mice at 24 h p.i. Myeloid lymphocytes were double labelled using anti-NK1·1−PE and anti-B220−FITC MoAbs and were analysed using a FACScan flow cytometer on a total of 10 000 events recorded. The results are representative of three experiments. *P < 0·05; **P < 0·01; ***P < 0·001.
The analysis of myeloid NK1·1+ and B220+ cells from groups of L2-MHV3- and YAC-MHV3-infected mice at 24 and 72 h p.i. indicated that the number of normal NK1·1+ cells decreased in the bone marrow from L2-MHV3-infected mice as soon as 24 h p.i. (Fig. 2, sections II-A) (P < 0·001). However, normal and larger NK1·1+ cells decreased more at 72 h p.i. in L2-MHV3-infected mice only (P < 0·001) (Fig. 2, sections II-A and II-B). The numbers of larger B220+ cells from YAC-MHV3-infected mice increased at 24 h p.i. (P < 0·05) while normal and larger B220+ cells decreased at 72 h p.i. in L2-MHV3-infected mice only (P < 0·01 and 0·001) (Fig. 2, sections II-C and II-D). Normal B220+ cells from YAC-MHV3-infected mice also decreased at 72 h p.i. (P < 0·05).
Cytotoxic properties of liver, spleen and bone marrow NK1·1+ cells from L2-MHV3- and YAC-MHV3-infected mice
To verify whether the cytotoxicity mediated by NK cells is also impaired in the liver, spleen or bone marrow of L2-MHV3-infected mice, lymphoid cells from these organs were collected from groups of three mice infected with L2-MHV3 or YAC-MHV3 and cytotoxicity against YAC-1 cells was assayed. As shown in Fig. 3, cytotoxic activity increased in the liver (P < 0·001) but disappeared in the bone marrow (P < 0·001) in L2-MHV3-infected mice. In YAC-MHV3-infected mice, a low decrease in NK cell cytotoxicity was also detected but only in the bone marrow (P < 0·05). A non-significant increase of cytotoxic response was detected in liver from YAC-MHV3-infected mice.
Fig. 3.

Determination of the percentage of cytotoxic activity of the splenic (a), myeloid (b) and intrahepatic (c) NK1·1+ cells from mock-infected (▪), L2-MHV3 (⋄) and YAC-MHV3 (▴) -infected C57BL/6 mice at 72 h p.i. Cytotoxicity of lymphoid cells from liver, spleen and bone marrow against YAC-1 target cells was evaluated by release of LDH activity in the supernatant. The optical density was recorded using an ELISA reader (490 nm filter). The experiments were conducted in triplicate. *P < 0·05; **P < 0·001.
In vitroinfections of IL-15-stimulated bone marrow NK1·1+ cells with either the pathogenic L2-MHV3 or the attenuated YAC-MHV3 strain
To verify if the depletion of bone marrow NK1·1+ cells is related with cell permissivity to viral replication, in vitro viral infections were performed on rmIL-15-stimulated bone marrow lymphoid cells. Bone marrow lymphoid cells were incubated with 20 ng/ml of rmIL-15, to increase the number of NK1·1+ cells, and then infected with 0·1–1 m.o.i. of L2-MHV3 or YAC-MHV3 viruses for 24 h. The rmIL-15 treatment increased the number of viable cells by 152 ± 18%. However, L2-MHV3 and YAC-MHV3 infections reduced the number of rmIL-15-treated cells by 55 ± 5% and 33 ± 0·3%, respectively (P < 0·001–0·01).
The cytofluorometric analysis of rmIL-15-stimulated myeloid cells revealed that the percentage of larger cells decreased in L2-MHV3-infected cells only (P < 0·05) (Fig. 4, section I). The phenotypic analysis of these cells, using PE-anti-NK1·1 and anti-FITC-B220 MoAbs, indicated that the larger NK1·1+ cells increased in rmIL-15-treated myeloid cells when compared with the untreated cells (P < 0·001) (Fig. 4, section I). However, the percentages of larger NK1·1+ cells decreased when infected with L2-MHV3 or YAC-MHV3 viruses (P < 0·001) (Fig. 4). Normal B220+ lymphocytes only decreased in L2-MHV3-infected cells (P < 0·05) (results not shown). No significant alterations in the percentages of normal NK1·1+ cells were observed in L2-MHV3- or YAC-MHV3-infected cells.
Fig. 4.

Section I: percentages of total (a) and NK1·1+ (b) normal (▪) and larger (□) cells in control, L2-MHV3- and YAC-MHV3-infected myeloid cells stimulated in vitro with IL-15 from C57BL/6 mice. Myeloid lymphocytes were incubated with 20 ng/ml of rmIL-15 for 7 days, then infected with 0·1–1 m.o.i. of L2-MHV3 or YAC-MHV3 for 24 h. Non-adherent cells were collected and immunolabelled with anti-NK1·1+PE and anti-B220−FITC MoAbs. Lymphoid cells were gated according to FSC/SSC parameters obtained on a FACScan flow cytometer and the percentages of cells were evaluated based on a total of 10 000 events recorded. Results are representative of three experiments. +P < 0·01(compared with negative control); *P < 0·05 and **P < 0·01 (compared with control + IL-15). Section II: syncytia of NK+TUNEL+ cells in L2-MHV3-infected IL-15-stimulated myeloid cells. TUNEL labelling (a) and FITC-DX5 antibody labelling (b).
Cytopathic effects (CPE) and infectious virus titres in the supernatants from both L2-MHV3- or YAC-MHV3-infected rIL-15-treated bone marrow lymphoid cells were then verified. Typical CPE of MHV viruses, characterized by cell rounding and syncytia formation, were detected in bone marrow lymphoid cells infected with L2-MHV3. However, less extensive CPE were detected in YAC-MHV3-infected cells. Viral titration revealed that infectious viruses were produced by bone marrow lymphoid cells infected in vitro with both L2-MHV3 (4·6 ± 0·28 log10TCID50/ml) and YAC-MHV3 (3·95 ± 0·21 log10TCID50/ml) viruses.
To verify whether the viral replication is involved an induction of apoptosis, a TUNEL assay was performed on bone marrow rmIL-15-stimulated lymphoid cells. The results indicated that most of the rmIL-15-stimulated lymphoid cells became apoptotic when infected with L2-MHV3 (86·6 ± 1·7%, P < 0·001). However, the attenuated YAC-MHV3 induced a lower level of apoptosis (49·9 ± 3·1%, P < 0·001).
To confirm that the apoptotic NK cells resulted from viral infection, L2-MHV3- and YAC-MHV3-infected apoptotic NK cells were identified using a double labelling with TUNEL reagents and anti-NK (DX5) MoAbs. Syncytia formed by fused NK+ cells contained apoptotic nuclei, as shown in Fig. 4, section II, were found in both L2-MHV3- and YAC-MHV3-infected cells. However, larger syncytia were observed in L2-MHV3-infected cells than in YAC-MHV3-infected cells. Qualitative evaluation of TUNEL+ NK+ syncytia revealed that a higher number of syncytia occurred in L2-MHV3 (4–5 syncytia by microscopic field)-infected cells than in YAC-MHV3 (1–3 syncytia by microscopic field).
In vitro infections of intrahepatic IL-15-stimulated NK1·1/αβ-TCR cell subsets with the pathogenic L2-MHV3 and the attenuated YAC-MHV3 viruses
To verify whether the recruited intrahepatic NK cells are also permissive to viral infection, intrahepatic lymphoid cells were isolated from uninfected C57BL/6 mice and infected in vitro with 0·1–1 m.o.i. of L2-MHV3 or YAC-MHV3 for 24 h. The cells were then double-labelled with PE-anti-NK1·1 and FITC-antiαβ-TCR MoAbs and the percentages of the NK1·1/αβ-TCR cell subsets were recorded. As shown in the Fig. 5, the percentages of NK1·1+αβ-TCR- or NK1·1+αβ-TCRinter cells decreased in the presence of the L2-MHV3 virus (P < 0·05). However, NK1·1+αβ-TCR- cells increased in the presence of YAC-MHV3 (P < 0·01). However, no significant differences between the percentages of control, L2-MHV3- or YAC-MHV3-infected NK1·1−TCRinter or NK1·1−TCR+ cells were observed.
Fig. 5.

Percentages of NK1·1+TCR−, NK1·1+TCRinter, NK−TCRinter and NK−TCRhigh cells in in vitro L2-MHV3- and YAC-MHV3-infected intrahepatic lymphoid cells stimulated with IL-15. Intrahepatic lymphocytes were incubated with 20 ng/ml of rmIL-15 for 7 days, then infected with 0·1–1 m.o.i. mock-infected (▪), L2-MHV3- (□) and YAC-MHV3- (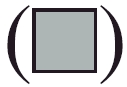 ) infected for 24 h and immunolabelled with anti-NK1·1+PE and antiαβ-TCR MoAbs. Lymphoid cells were gated according to FSC/SSC parameters obtained on a FACScan flow cytometer and the percentages were evaluated based on a total of 10 000 events recorded. Results are representative of three experiments. *P < 0·05; **P < 0·01.
) infected for 24 h and immunolabelled with anti-NK1·1+PE and antiαβ-TCR MoAbs. Lymphoid cells were gated according to FSC/SSC parameters obtained on a FACScan flow cytometer and the percentages were evaluated based on a total of 10 000 events recorded. Results are representative of three experiments. *P < 0·05; **P < 0·01.
DISCUSSION
In this work, we have demonstrated that the pathogenic and attenuated MHV3 viruses both induced NK cell death and apoptosis but lead to different levels of NK cell deletion in the bone marrow and recruitment by the liver.
This is the first report to indicate that NK1·1+ cells can be target cells in a coronavirus infection involving a decrease in innate immunity. Also, we have shown that, in pathogenic L2-MHV3-infected mice, cytotoxic NK cells were less recruited in the liver than other lymphoid cell subsets during the first days of infection. The decrease of splenic NK1·1+ cells observed in L2-MHV3-infected mice indicates that the viral infection may favour a recruitment of NK1·1+ cells by the liver, an in situ depletion of splenic NK1·1+ cells and/or an impairment of the production of NK1·1+ cells by the bone marrow due to virus-induced cell death.
The hypothesis of recruitment of splenic NK1·1+ cells in the livers of L2-MHV3-infected mice is supported by two observations: the increase in intrahepatic mononuclear cell numbers, including NK1·1+ cells, and the simultaneous decrease of splenic NK1·1+ cells. However, the recruitment of NK1·1+ cells in L2-MHV3-infected mice remained lower than that of other lymphoid cell subsets, indicating that the recruitment of NK1·1+ cells in the infected liver could explain partially the loss of splenic NK1·1+ cells in L2-MHV3-infected mice. These results are in agreement with previous studies demonstrating that, in contrast to other murine viral infections, only the first wave of recruited large granular cells, including NK1·1+ and CTL cells, occurs in the liver or peritoneal exudates of MHV-infected mice in the first 3 days p.i. [27,28]. The increase of absolute numbers in intrahepatic NK1·1+TCR− cells in L2-MHV3-infected mice, however, remained very low compared to the decrease in splenic NK1·1+ cells, suggesting either that most NK1·1+ cells are recruited in other target organs or that production of new NK1·1+ cells by the bone marrow is impaired.
The fact that splenic NK1·1+ cells are normally mature and do not undergo mitosis [29] suggests that a depletion of these cells might be the result of a decrease in bone marrow production. This hypothesis was verified through the analysis of the NK1·1+ subpopulation in the bone marrow of L2-MHV3-infected mice. We have showed that both normal and larger lymphoid cells decreased in virus-infected mice. The phenotyping of the cells indicated that normal NK1·1+ decreased as soon as 24 h p.i. while large B cells decreased later, indicating that myeloid NK1·1+ cells are more sensitive to virus-mediated depletion than B lineage cells. We can expect that the NK1·1+ cell decrease in the first 24 h p.i. is due to a recruitment in liver. However, normal and larger NK1·1+ cells decreased strongly at 72 h p.i. in bone marrow from L2-MHV3-infected mice only, suggesting a virus-induced decrease of these cells rather than a low recruitment in liver. The in vitro L2-MHV3 infection of rIL-15 stimulated cells, permitting the increase of the NK1·1+ cells rather than B cells [11,30], revealed that the depletion of NK1·1+ cells resulted from virus-induced cytopathic effects, viral replication and apoptosis occurring in syncytia, as demonstrated by the production of infectious particles and TUNEL assay. The permissivity of NK cells to L2-MHV3 infection involves the expression of the MHV receptor on the cell surface. CEACAM isoform 1 of the biliary glycoprotein (Bgp) molecules expressed on the cell surface are known to act as MHV receptor [31,32]. These molecules are present in many cell types, including hepatocytes, macrophages, endothelial cells, thymic epithelial cells and B lymphocytes [32,33]. There are no reports concerning the expression of CEACAM-1 on mouse NK1·1+ cells, but a similar protein was detected recently on human T cells and activated NK1·1+ cells [34], suggesting that viral permissivity of NK1·1+ cells to MHV3 virus may be related to the presence of a specific MHV-receptor. Preliminary results from in vitro L2-MHV3 infections of NK1·1+ cells cultured in the presence of myeloid dendritic cells revealed that the NK1·1+ cells expressed intracellular viral proteins, thus reinforcing the hypothesis of viral replication in NK cells. In the present study, B220+ cells decreased slightly in L2-MHV3-infected myeloid cells, just as we had observed previously in infected myeloid B lineage cells [15].
We can thus establish a relationship between the NK1·1+ cells and pathogenic properties of the MHV3 virus, as the use of the attenuated YAC-MHV3 variant demonstrated that splenic NK1·1+ cells were less depleted in contrast to that observed in L2-MHV3-infected mice. In vitro YAC-MHV3 infection of IL-15-stimulated myeloid cells also revealed higher cell viability and lower percentages of apoptotic cells than those observed in L2-MHV3-infected cells. However, infectious particle production was similar to that seen in L2-MHV3-infected cells. In spite of a similar production of infectious viruses in the first 24 h p.i., the apparent discrepancy in the sensitivity of NK1·1+ cells to the attenuated YAC-MHV3 can be explained by the fact that a lower number of IL-15-stimulated myeloid cells can be efficiently infected by the YAC-MHV3 variant. We had already observed higher CPE and increased cell death in L2-MHV3-infected macrophages when compared to those occurring in YAC-MHV3-infected cells in spite of comparable infectious virus titres in the first 24 h p.i. [35].
The fact that the death of virus-infected NK1·1+ cells involves syncytia formation and apoptosis suggests that MHV3 replication induces an apoptotic process. It was demonstrated recently that MHV3 can induce apoptosis in macrophages from 8 h to 12 h p.i. [36,37]. Interestingly, viral pathogenicity seems directly correlated with syncytia formation rather than apoptosis, as lower numbers of NK cell syncytia were observed in YAC-MHV3-infected cells even though in vitro apoptosis levels were similar to those induced by the pathogenic L2-MHV3 strain. The new E protein of MHV viruses has been related to the apoptotic process in virus-infected cells [38].
Many previous studies have demonstrated that MHV3 is the most virulent serotype of the MHV viruses because it induces a fulminant hepatitis leading to the death of the animal within 3 days p.i. [12,14]. We have observed previously that splenic T and B cell subsets decreased in L2-MHV3-infected mice [15,16,39]. However, the fulminant hepatitis observed in C57BL/6 cannot be explained by T and B cell depletions alone. Budowski et al. [24] have shown a higher replication of MHV and a more extensive hepatitis along with higher numbers of inflammatory foci in the livers of asialo-GM1-depleted mice, hinting at the important role of NK1·1+ cells in limiting the spread of the viral replication during the first days of the infection. Rather, Stohlman et al. [22] have demonstrated that, in C57BL/6 mice, MHV-JHM induced an increase of natural cytotoxicity against YAC-1 target cells without detectable IFN-γ production, such as seen in lymphocytic choriomeningitis virus-infected mice. In addition, the NK1·1+ cell response was variable among mice, raising the possibility of MHV-mediated disorders in NK1·1+ functions. Our results are in agreement with these observations, but also suggest that higher cytotoxicity of intrahepatic NK cells from L2-MHV3-infected mice reflects extensive histopathological lesions leading to a fulminant hepatitis and a rapid death rather than an efficient antiviral natural response.
The permissivity of NK1·1+ cells to viral infection and apoptosis is a new viral evasion mechanism which might favour viral replication in hepatocytes and other resident liver cells during the first days p.i. In addition, it is also probable that an impaired production of bone marrow NK1·1+ cells may not result solely from the viral permissivity of IL-15-stimulated cells but may also be related to a maturation defect resulting from viral replication in myeloid dendritic cells, as already observed in the thymus [18].
The MHV3-infection is a powerful animal model for the study of the relationship between viral tropism for NK1·1+, T and B cells and the induction of the hepatitis process. Further work is in progress to identify bone marrow target cells involved in the NK1·1+ maturational defects as well as the implications of NK1·1+ permissivity in the hepatitis process.
Acknowledgments
This work was supported by a grant from NSERC–Canada. The authors thank Mr Christian Pagé and Christophe Dugré for their technical assistance as well as Dr Roland Savard and Mr Jean-Philippe Laverdure for revising this manuscript.
REFERENCES
- 1.Brinton MA, Nathanson N. Genetic determinants of virus susceptibility: epidemiologic implication of murine models. Epidemiol Rev. 1981;3:115–39. doi: 10.1093/oxfordjournals.epirev.a036230. [DOI] [PubMed] [Google Scholar]
- 2.Byrne JA, Oldstone MBA. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus: clearance of virus in vivo. J Virol. 1985;51:682–6. doi: 10.1128/jvi.51.3.682-686.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sissons JPG, Borysiewicz LK. Viral immunopathology. Br Med Bull. 1985;41:34–40. doi: 10.1093/oxfordjournals.bmb.a072021. [DOI] [PubMed] [Google Scholar]
- 4.Herberman RB. NK11+ cells and other natural effector cells. New York: Academic Press; 1982. [Google Scholar]
- 5.Gosselin J, Tomoiu A, Gallo RC, Flamand L. Interleukin-15 as an activator of natural killer cell-mediated antiviral response. Blood. 1999;94:4210–19. [PubMed] [Google Scholar]
- 6.Crotta S, Stilla A, Wack A, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195:35–41. doi: 10.1084/jem.20011124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tseng CTK, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell function. J Exp Med. 2002;195:43–9. doi: 10.1084/jem.20011145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe H, Watanabe H, Miyaji C, et al. Relationship between intermediate TCR cells and NK1.1+T cells in various immune organs. NK1.1+ T cells are present within a population of intermediate TCR cells. J Immunol. 1995;155:2972–83. [PubMed] [Google Scholar]
- 9.Tsukahara A, Seki S, Iai T, et al. Mouse liver T cells: their change with aging and in comparison with peripheral T cells. Hepatology. 1997;26:301–9. doi: 10.1002/hep.510260208. [DOI] [PubMed] [Google Scholar]
- 10.Moore TA, Bennett M, Kumar V. Murine natural killer cell differentiation: past, present and future. Immunol Res. 1996;15:151–62. doi: 10.1007/BF02918504. [DOI] [PubMed] [Google Scholar]
- 11.Puzanov IJ, Williams NS, Schatzel J, Sivakumar PV, Bennett M, Kumar V. Ontogeny of NK1.1+ cells and the bone marrow microenvironment: where does IL-15 fit in? Res Immunol. 1997;148:195–201. doi: 10.1016/s0923-2494(97)84225-3. [DOI] [PubMed] [Google Scholar]
- 12.Le Prevost C, Levy-Leblond E, Virelizier JL, Dupuy JM. Immunopathology of mouse hepatitis virus type 3 infection. I. Role of humoral and cell-mediated immunity in resistance mechanism. J Immunol. 1975;114:221–5. [PubMed] [Google Scholar]
- 13.Levy-Leblond E, Oth O, Dupuy JM. Genetic study of mouse sensivity to MHV3 infection: influence of the H-2 complex. J Immunol. 1979;112:1359–62. [PubMed] [Google Scholar]
- 14.Dupuy JM, Levy-Leblond E, Le Prevost C. Immunopathology of mouse hepatitis virus type 3 infection. II. Effect of immunosuppression in resistant mice. J Immunol. 1975;114:221–5. [PubMed] [Google Scholar]
- 15.Jolicoeur P, Lamontagne L. Mouse hepatitis virus 3 pathogenicity expressed by a lytic viral infection in bone marrow 14,8+µ+ B lymphocytes subpopulations. J Immunol. 1989. pp. 3722–30. [PubMed]
- 16.Lamontagne L, Descoteaux JP, Jolicoeur P. Mouse hepatitis virus 3 replication in T and B lymphocytes correlate with viral pathogenicity. J Immunol. 1989;142:4458–65. [PubMed] [Google Scholar]
- 17.Lamontagne L, Dupuy JM. Persistent infection with mouse hepatitis virus 3 in mouse lymphoid cell lines. Infect Immun. 1984;44:716–23. doi: 10.1128/iai.44.3.716-723.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamontagne L, Jolicoeur P. Mouse hepatitis virus 3 thymic cell interactions correlating with viral pathogenicity. J Immunol. 1991;146:3152–9. [PubMed] [Google Scholar]
- 19.Lamontagne L, Massicotte E, Page C. Mouse hepatitis viral infection induces and extrathymic differenciation of the specific intrahepatic αβ-TCRinterLFA-1high T-cell population. Immunology. 1997;160:376–84. doi: 10.1111/j.1365-2567.1997.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamontagne L, Lusignan S, Page C. Recovery from mouse hepatitis virus infection depends on recruitment of CD8+ cells rather than activation of intrahepatic CD4+αβ−TCRinter or NK-T cells. Clin Immunol. 2001;101:345–56. doi: 10.1006/clim.2001.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schindler L, Engler H, Kirchner H. Activation of natural killer cells and induction of interferon after injection of mouse hepatitis virus type 3 in mice. Infect Immun. 1982;35:869–73. doi: 10.1128/iai.35.3.869-873.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stohlman SA, Brayton PR, Harmon RC, Stevenson D, Ganges RG, Matsushima GK. Natural killer cell activity during mouse hepatitis virus infection. response in the absence of interferon. Int J Cancer. 1983;31:309–14. doi: 10.1002/ijc.2910310310. [DOI] [PubMed] [Google Scholar]
- 23.Mello IG, Vassao RC, Pereira CA. Virus specificity of the antiviral state induced by IFN gamma correlates with the resistance to MHV3 infection. Arch Virol. 1993;132:281–9. doi: 10.1007/BF01309539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bukowski JF, Woda BA, Habo S, Okumura K, Welsh RM. NK1.1+ cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J Immunol. 1983;131:1531–8. [PubMed] [Google Scholar]
- 25.Tardieu M, Hery C, Dupuy JM. Neonatal susceptibility to MHV3 infection in mice. II. Role of natural effector marrow cells in transfer of resistance. J Immunol. 1980;124:418–23. [PubMed] [Google Scholar]
- 26.Dupuy JM, Rodrigue D. Heterogeneity in evolutive patterns of inbred mice infected with a cloned substrain of mouse hepatitis virus type 3. Intervirology. 1981;16:114–17. doi: 10.1159/000149255. [DOI] [PubMed] [Google Scholar]
- 27.Natuk RJ, Welsh RM. Accumulation and chemostaxis of natural killer/large granular lymphocytes at sites of virus replication. J Immunol. 1987;138:877–83. [PubMed] [Google Scholar]
- 28.McIntyre KW, Welsh RM. Accumulation of natural killer and cytotoxic T large granular lymphocytes in the liver during virus infection. J Exp Med. 1986;164:1667–81. doi: 10.1084/jem.164.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams NS, Moore TA, Schatzle JD, et al. Generation of lytic natural killer 1.1+ly49- cells from multipotential murine bone marrow progenitor in a stroma free culture: definition of cytokine requirements and developmental intermediates. J Exp Med. 1997;186:1609–14. doi: 10.1084/jem.186.9.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bykovskaia SN, Buffo M, Zhang H, et al. The generation of human dendritic and NK1.1+ cells from hemopoietic progenitors induced by interleukin-15. J Leuk Biol. 1999;66:659–66. doi: 10.1002/jlb.66.4.659. [DOI] [PubMed] [Google Scholar]
- 31.Tan K, Zelus BD, Meijers R, et al. Crystal structure of murine sCEACAM1a[1,4]: a coronavirus receptor in the CEA family. EMBO J. 2002;21:2076–86. doi: 10.1093/emboj/21.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Godfraind C, Kilgreth SG, Cardellichio CB, et al. Tissue and cellular distribution of an adhesion molecule in the carcinoembryonic antigen family that serves as receptor for mouse hepatitis virus. Lab Invest. 1995;73:615–27. [PubMed] [Google Scholar]
- 33.Godfraind C, Coutelier JP. Morphological analysis of mouse hepatitis virus A59-induced pathology with regard to viral receptor expression. Histol Histopathol. 1998;13:181–99. doi: 10.14670/HH-13.181. [DOI] [PubMed] [Google Scholar]
- 34.Moller MJ, Kammerer R, Grunert F, von Kleist S. Biliary glycoprotein (BGP) expression on T cells and on a natural-killler-cell subpopulation. Int J Cancer. 1996;65:740–5. doi: 10.1002/(SICI)1097-0215(19960315)65:6<740::AID-IJC5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 35.Lamontagne L, Décarie D, Dupuy JM. Host cell resistance to mouse hepatitis virus type 3 is expressed in vitro in macrophages and lymphocytes. Viral Immunol. 1989;2:37–45. doi: 10.1089/vim.1989.2.37. [DOI] [PubMed] [Google Scholar]
- 36.Belyavsky M, Belyavskaya E, Levy GA, Leibowitz JL. Coronavirus MHV3-induced apoptsis in macrophages. Virology. 1998;250:41–9. doi: 10.1006/viro.1998.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belyavsky M, Levy GA, Leibowitz JL. The pattern of induction of apoptosis during infection with MHV3 correlates with strain variation in resistance and susceptibility to lethal hepatitis. Adv Exp Med Biol. 1998;440:619–25. doi: 10.1007/978-1-4615-5331-1_80. [DOI] [PubMed] [Google Scholar]
- 38.An S, Chen C, Yu X, Leibowitz JL, Makino S. Induction of apoptosis in murine coronavirus-infected cultured cells and demonstration of E protein as an apoptosis inducer. J Virol. 1999;73:7853–9. doi: 10.1128/jvi.73.9.7853-7859.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jolicoeur P, Lamontagne L. Impaired T and B cells subpopulations involved in a chronic disease induced by mouse hepatitis virus type 3. J Immunol. 1994;153:1318–27. [PubMed] [Google Scholar]