

Abstract
Complement activation with formation of biologically potent mediators like C5a and the terminal C5b-9 complex (TCC) contributes essentially to development of inflammation and tissue damage in a number of autoimmune and inflammatory conditions. A particular role for complement in the ischaemia/reperfusion injury of the heart, skeletal muscle, central nervous system, intestine and kidney has been suggested from animal studies. Previous experiments in C3 and C4 knockout mice suggested an important role of the classical or lectin pathway in initiation of complement activation during intestinal ischaemia/reperfusion injury while later use of factor D knockout mice showed the alternative pathway to be critically involved. We hypothesized that alternative pathway amplification might play a more critical role in classical pathway-induced C5 activation than previously recognized and used pathway-selective inhibitory mAbs to further elucidate the role of the alternative pathway. Here we demonstrate that selective blockade of the alternative pathway by neutralizing factor D in human serum diluted 1 : 2 with mAb 166–32 inhibited more than 80% of C5a and TCC formation induced by solid phase IgM and solid- and fluid-phase human aggregated IgG via the classical pathway. The findings emphasize the influence of alternative pathway amplification on the effect of initial classical pathway activation and the therapeutic potential of inhibiting the alternative pathway in clinical conditions with excessive and uncontrolled complement activation.
Keywords: complement, inflammation, autoimmunity, factor D, alternative pathway activation
INTRODUCTION
The complement system with its central position in innate and adaptive immunity mediates a variety of effector functions. During the past decades it has become evident that it is also an important mediator of tissue damage in disease [1,2], in particular in ischaemia/reperfusion (I/R) injury [3,4]. Clinical evidence points to complement and immune complexes as critical players in mediating reperfusion injury [5]. Intestinal I/R injury can also be initiated by clonally specific natural IgM antibodies via the classical pathway [6].
Results from earlier studies using C3 and C4 knockout (–/–) mice suggested an important role of the classical or lectin pathway in inducing complement activation during intestinal I/R injury. However, in a separate study in the same model using factor D –/– mice the alternative pathway was shown to be critically involved in the I/R injury [7]. We hypothesized that absence of amplification via the alternative pathway in factor D –/– mice could reduce the inflammatory response and tissue injury triggered by initial activation of the classical pathway and used pathway-selective inhibitory monoclonal antibodies (mAbs) to further elucidate the role of the alternative pathway in the activation of the complement system. Selective blockade of the alternative pathway was induced by neutralizing factor D with mAb 166–32 [8,9] and the classical pathway was blocked by anti-C2 mAb 175–62 after initial activation of the classical pathway induced by solid phase IgM and solid- and fluid phase human aggregated IgG.
MATERIALS AND METHODS
Monoclonal antibodies
Mouse mAbs against human factor D (clone 166–32, IgG1), C2 (clone 175–62, IgG1), and an isotype-matched control mAb (clone G3-519, anti-HIV gp120, IgG1) were generated and purified under identical conditions as described previously [8,9].
Normal human serum
Complement-sufficient normal human serum (NHS) was collected from five healthy volunteers and stored as aliquots at −70°C. To approach physiological conditions NHS was tested at a final dilution as low as 1 : 2 and in further two-fold dilutions. In activation and inhibition experiments, the amounts of activating substance or purified antibody in the well are recorded as transformed to amount/ml of undiluted serum.
Complement reagents
C1q depleted (C1qD) human serum and purified human C1q were obtained from Quidel (San Diego, CA, USA), C2-depleted (C2D) human serum and factor B-depleted (fBD) human serum from Advanced Research Technologies (San Diego, CA, USA).
Assay of classical pathway complement activation by human IgM on solid phase
The assay was performed according to Roos et al. [10] with slight modifications. Purified polyclonal human IgM (Sigma-Aldrich, St. Louis, MO, USA) in phosphate-buffered saline (PBS) pH 7·4, at 40 µg/ml NHS and in further two-fold dilutions, were coated on Costar 3590 flat-bottomed polystyrene 96-well plates (Corning Inc, Corning, NY, USA) overnight at 20°C, washed thoroughly in PBS with 0·1% Tween 20 (Sigma-Aldrich). The remaining binding sites in the wells were then saturated with a blocking buffer (PBS containing 1% bovine serum albumin (BSA) and 0·1% Tween 20) for 1 h at 37°C. Following washing, NHS (diluted 1 : 2) in veronal buffer pH 7·5 (100 µl) containing 0·5 m m MgCl2, 2 m m CaCl2, 0·05% Tween 20 and 0·1% gelatin was added to each well for complement activation for 60 min at 37°C. To stop further activation the microtitre plate was incubated on ice and ethylenediaminetetraacetic acid (EDTA) was immediately added to each well to a final concentration of 20 m m. Terminal complement complex (TCC) in fluid phase was measured by an ELISA based on reaction with mAb aE11 specific for a neoepitope exposed when C9 is incorporated in the complex, as described previously [11]. An aliquot of the supernatant fluid was pipetted off from each well for quantification based on comparison with a standard curve prepared by dilutions of Zymosan (Sigma-Aldrich) activated human serum defined to contain 1000 AU TCC/ml. The supernatant fluid was tested in different dilutions to ensure that the O.D. values fell on the steep part of the standard curve, the deduced TCC value being multiplied with the dilution to calculate the TCC value in each well transformed as AU/ml of undiluted NHS. To measure the amount of TCC deposited on the surface of the washed wells, mAb aE11 diluted 1 : 5000 was added to the wells. The bound antibody was detected with HRP conjugated sheep antimouse immunoglobulin (Amersham Biosciences, Little Chalfont, UK) using 2,2′-azino-di-[3-ethylbenzthiazoline-sulphonate] (ABTS) (Sigma-Aldrich) as substrate. OD values were used to represent the relative amount of deposited TCC in the wells.
Assay of classical pathway activation by aggregated human IgG on solid phase
Soluble human aggregated IgG (HAIGG) was prepared by heating human IgG (Gammaglobulin Pharmacia & Upjohn, Stockholm, Sweden) 10 mg/ml in PBS pH 7·4 for 10 min at 63°C, immediately followed by cooling on ice. The protein was then stored in aliquots at 4°C. Activation of NHS 1 : 2 for 30 min at 37°C and assay for generation of TCC in fluid phase and deposition of TCC on solid phase were identical to the procedures for human IgM on solid phase described above.
Assay of classical pathway activation by aggregated human IgG in fluid phase
Serially diluted HAIGG (from 200 µg/ml) was added to 100 µl NHS 1 : 2 for complement activation for 30 min at 37°C. TCC was measured in fluid phase as for the solid phase IgM experiment described above.
Assay of classical pathway activation by aggregated human IgG in whole blood
Whole blood from a complement-sufficient healthy individual was collected using the thrombin-specific hirudin analogue lepirudin (Refludan, batch 18261611C, Hoechst, Frankfurt am Main, Germany, 50 µg/ml blood) as anticoagulant [12]. In contrast to heparin, lepirudin does not interfere with complement activation. The procedure for complement activation was identical to that for NHS. Plasma TCC levels were then measured.
Assay of other activation products
C3bc, C5a and C3bBbP complex were measured by ELISA as described by Garred et al. [13], Bergh et al. [14] and Mollnes et al. [12].
Haemolytic assay of classical pathway complement activation
Serially diluted NHS (from 1 : 2) in veronal buffer pH 7·2 containing 0·5 m m MgCl2 and 0·2 m m CaCl2 was mixed with sheep red blood cells (RBCs) sensitized with rabbit antisheep RBC antibody (Institut Virion\Serion GmbH, Würzburg, Germany) in a V-bottomed Sero-Wel96-well microtitre plate (Bibby Sterilin Ltd, Stone, Staffordshire, UK) for incubation at 37°C for 1 h. Then the microtitre plate was centrifuged at 300 × g and supernatant from each well was collected to a flat-bottomed 96-well Costar microtitre plate (Corning) for reading of OD at 405 nm. Percent haemolysis was calculated according to Fung et al. [8].
Haemolytic assay of alternative pathway complement activation
Serially diluted NHS (from 1 : 2) in veronal buffer pH 7·2 containing 7 m m EGTA and 3·5 m m MgCl2 and 3 mg BSA/ml were mixed with washed unsensitized rabbit RBCs in a V-bottomed microtitre plate (Bibby Sterilin) for incubation at 37°C for 1 h followed by centrifugation and quantification of haemolysis as in the classical pathway assay.
Inhibition assays
mAbs were added to serum followed by incubation for 5 min at room temperature prior to activation and assay. Percentage remaining activity after inhibition was calculated as follows:
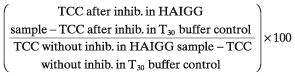 |
RESULTS
Inhibition of alternative pathway activation by antifactor D mAb 166–32
Factor D is a rate-limiting serine protease in the amplification loop of the alternative pathway [15]. Therefore it is a target of choice for effective inhibition of the alternative pathway. Anti-factor D mAb 166–32 was generated by immunizing mice with human factor D and was shown to be effective in inhibiting complement activation [8,9]. Component C2 is specific for the classical and lectin pathways in the complement cascade. Anti-C2 mAb 175–62 was generated by immunizing mice with purified human C2a obtained by digesting human C2 with human C1s. The antibody binds both C2 and C2a (Fung et al. unpublished observations). To characterize the specificity of the antifactor D mAb 166–32 and anti-C2 mAb 175–62 used in this study, haemolytic assays were conducted using normal human serum (NHS), C2-depleted (C2D) and factor B-depleted (fBD) human sera.
Anti-factor D, anti-C2 or isotype-matched control mAbs were added to NHS at 100 µg/ml and the serum was serially diluted for haemolytic alternative pathway and classical pathway assays. The antifactor D mAb inhibited alternative pathway in NHS very effectively while the anti-C2 mAb had no effect on alternative pathway haemolytic activity (Fig. 1a). The antifactor D mAb had no effect on classical pathway haemolytic activity while the anti-C2 mAb inhibited classical pathway efficiently from serum dilution 1 : 8 (Fig. 1b). The marginal effect of anti-C2 in serum dilutions 1 : 2 and 1 : 4 is consistent with the high sensitivity of RBCs to complement lysis by the classical pathway. Normal alternative pathway haemolytic activity was lost at serum dilution 1 : 16 while classical pathway haemolysis in NHS was lost at serum dilution 1 : 1024, consistent with the well-known difference in the sensitivity of haemolysis in the two assay systems.
Fig. 1.

Specificity of inhibition by antifactor D and anti-C2 mAbs in haemolytic assays. Anti-factor D mAb 166–32 (•) completely inhibited alternative pathway haemolytic activity in normal human serum (NHS) while anti-C2 mAb 175–62 (▴) and the control mAb (○) had no effect (a). Anti-factor D showed no inhibition of classical pathway haemolysis in NHS in contrast to anti-C2 (b). Normal alternative pathway haemolysis (□) and no classical pathway haemolysis (▪) were found in C2-depleted (C2D) serum (c), while the opposite pattern was obtained in factor B depleted serum (d). Anti-factor D inhibited alternative pathway haemolysis in C2D serum completely while anti-C2 had no effect (e). The opposite pattern was obtained in factor B depleted serum (f).
To further characterize the specificity of the antifactor D and anti-C2 mAbs, haemolytic assays were conducted with C2D and fBD human sera. First, the pathway specificity of the sera was tested. The C2D serum showed normal alternative pathway haemolytic activity but no classical pathway haemolytic activity (Fig. 1c). Conversely, the fBD serum had no alternative pathway haemolytic activity but normal classical pathway haemolytic activity (Fig. 1d). In inhibition experiments, the antifactor D, the anti-C2, or the isotype-matched control mAb at 100 µg/ml was added to the C2D and fBD sera which were then two fold serially diluted from 1 : 2 for haemolytic assays. The antifactor D mAb completely inhibited alternative pathway haemolytic activity in C2D serum while the anti-C2 mAb had no effect on the alternative pathway (Fig. 1e). The anti-C2 mAb completely inhibited classical pathway haemolytic activity in fBD serum while the antifactor D mAb had no effect on the classical pathway (Fig. 1f).
Alternative pathway haemolysis was used to determine the concentration of antifactor D mAb required to completely neutralize factor D in NHS at 1 : 2 dilution. Complete inhibition of haemolysis was achieved at 5 µg/ml of the antibody. Thus, 10 µg/ml was chosen to ensure complete inhibition of the alternative pathway in NHS at 1 : 2 dilution in the subsequent experiments by ELISAs. Based on similar experiments to ensure complete inhibition of the classical pathway, the anti-C2 mAb was used at 50 µg/ml serum.
Localization of the mAb 166–32 reactive epitope in the factor D molecule
Inasmuch as the antifactor D mAb 166–32 binds human factor D but not pig factor D, we used pig/human hybrid constructs of factor D to map the binding epitope of the antibody which is located in the ‘methionine loop’ of factor D (-CNRRTHHDGAITER LMC-) between Cys154 and Cys170. Arg156, His159 and Leu168 are critical for the antibody binding. This antibody binding epitope is distant from the catalytic site of the trypsin-like domain of factor D in which there is extensive homology with other serine proteases of the complement system (C1r, C1s, MASP1, MASP2) (Fung et al. unpublished observations). This antibody binding site may be responsible for the interaction between factor D and factor B. Extensive search in the gene bank revealed no significant homology of this epitope sequence with other known proteins. This is consistent with the observations in the haemolytic assays that mAb 166–32 reacts specifically with factor D and inhibits the alternative pathway, but does not interfere with classical pathway components.
Inhibition of solid-phase classical pathway activation of normal human serum
Classical pathway activation was induced by purified human IgM adsorbed on the solid phase of microtitre plates in the presence of NHS at 1 : 2 dilution. Deposition of TCC, expressed as OD, increased with increasing amounts of immobilized IgM (Fig. 2a). Similar results were observed for TCC in fluid phase (Fig. 2b). TCC formation was abolished by the antifactor D mAb as indicated in both assays. Classical pathway activation was also induced with human aggregated IgG (HAIGG) on the solid phase. Similar results were obtained as with IgM. The amount of TCC on solid phase and in fluid phase was proportional to the amount of immobilized HAIGG. A reduction of 87% of TCC formation in the fluid phase by the antifactor D mAb was attained at 10 µg HAIGG/ml NHS.
Fig. 2.

Anti-factor D inhibition of classical pathway activation in NHS. Anti-factor D mAb 166–32 (•) abolished solid phase (a) and fluid phase (b) formation of TCC induced by human IgM on the solid phase (Δ; no antibody added). Similarly, antifactor D (•) profoundly inhibited fluid phase formation of TCC induced by human aggregated IgG (HAIGG) in the fluid phase (Δ; no antibody added) (c). The points and error bars indicate mean values and range from two independent experiments.
Inhibition of fluid-phase classical pathway activation of normal human serum
Fluid phase classical pathway complement activation was induced by adding HAIGG directly into NHS. TCC formed in the fluid phase increased with increasing amounts of HAIGG. Inhibition by the antifactor D mAb was obtained with a reduction of 80% of TCC formation at 20 µg HAIGG/ml NHS and >80% at 10 µg HAIGG/ml NHS (Fig. 2c). Inhibition of HAIGG-induced activation in the fluid phase is illustrated in Fig. 3. To ensure optimal conditions for these inhibition experiments, an increased amount of HAIGG was used to induce stronger classical pathway activation in the control without any antibody added. In this control 200 µg HAIGG/ml NHS induced formation of 66·0 AU (arbitrary units) TCC/ml NHS (Fig. 3a). No inhibition was induced by the isotype-matched control mAb with 66·4 AU TCC/ml being generated. The anti-C2 mAb blocked activation with generation of only 1·2 AU TCC/ml NHS. Over 90% of TCC generation was inhibited by antifactor D with formation of 5·2 AU TCC/ml, while inhibition by a combination of the anti-C2 and antifactor D mAbs was even more profound, with a reduction in TCC to 0·8 AU/ml which is identical to the baseline level. Similar findings were obtained for the intermediary activation products C3bc and C5a (Fig. 3b,c).
Fig. 3.

Effect of antifactor D, anti-C2 and control antibody on HAIGG-induced activation in NHS. Anti-factor D mAb 166–32 inhibited more than 90% of fluid phase TCC formation induced by human HAIGG whereas anti-C2 and a combination of anti-C2 and antifactor D completely blocked TCC formation (a). The control antibody had no effect on TCC formation. Similar results were obtained for the activation products C3bc (b), C5a (c) and C3bBbP (d). The data and error bars as for Fig. 2.
The results of assay for the fluid phase alternative pathway convertase C3bBbP were of central importance for evaluating the effect of HAIGG on the alternative pathway. From a baseline value of 28 AU C3bBbP/ml NHS a marked increase to 954 AU/ml was observed after addition of 200 µg HAIGG/ml NHS (Fig. 3d). After addition of the anti-C2 mAb to block the classical pathway, formation of C3bBbP was markedly reduced to 108 AU/ml NHS. Anti-factor D reduced the C3bBbP value to 235 AU/ml NHS. The combination of the anti-C2 and antifactor D mAbs completely blocked C3bBbP formation giving a value similar to when the antifactor D mAb was added to serum without any HAIGG (21 and 26 AU/ml NHS, respectively), indicating that the antifactor D mAb also blocks the spontaneous self-activation of the alternative pathway during the 30 min incubation at 37°C.
To confirm the general notion that HAIGG induces selective activation through the classical pathway in the present set-up and to exclude any direct alternative pathway activation by HAIGG, experiments were performed with C1q-depleted (C1qD) serum. Assay of the alternative pathway specific C3bBbP convertase is shown in Fig. 4. The baseline value in NHS was 28 AU/ml. The increased baseline value in C1qD serum (176 AU/ml) is consistent with in vitro complement activation during immunoadsorption when the C1qD serum was prepared. Addition of HAIGG to NHS induced activation and marked C3bBbP formation in a dose–response manner. By contrast, addition of 2 and 20 µg HAIGG/ml to C1qD serum induced no C3bBbP formation, showing that HAIGG had no direct effect on the alternative pathway. After reconstituting C1qD serum with 200 µg purified C1q/ml, C3bBbP was again generated in a dose–response manner with increasing amounts of HAIGG added, consistent with a restored classical pathway activity leading to alternative pathway amplification. The results of TCC assay corroborated with those on C3bBbP (data not shown).
Fig. 4.

Fluid phase C3bBbP formation after HAIGG-induced activation in NHS, C1q depleted serum (C1qD) and C1qD reconstituted withpurified C1q. Addition of HAIGG to NHS induced activation and marked C3bBbP formation in a dose–response manner. By contrast, addition of HAIGG to C1qD serum induced no C3bBbP formation, showing that HAIGG had no direct effect on the alternative pathway. After reconstituting C1qD serum with purified C1q, C3bBbP was again generated in a dose–response manner with increasing amounts of HAIGG, consistent with a restored classical pathway activity leading to alternative pathway amplification. T0,baseline value in NHS (left), C1qD (middle) and C1qD + C1q (right). The data and error bars as for Fig. 2.
Inhibition of fluid-phase classical pathway activation in whole blood
To study whether similar activation was obtained under more physiological conditions, experiments were repeated with whole blood from a complement-sufficient healthy individual in the model using the thrombin-specific hirudin analogue lepirudin as anticoagulant. In contrast to heparin, lepirudin does not interfere with complement activation (12). Addition of 100 µg HAIGG/ml blood induced a substantial TCC formation in plasma which was inhibited by the anti-C2 and antifactor D mAbs similar to what was found in the serum experiments (data not shown).
DISCUSSION
Interaction between the classical and alternative pathways may have different effects depending on the conditions. Antibodies may, e.g. mask cell-surface controls to facilitate C3b deposition and alternative pathway activation [16]. Furthermore during classical pathway activation, C4b deposition may recruit C3b directly forming a C2 independent classical pathway convertase [17]. Conversely, alternative pathway activation may increase the effect of initial classical pathway activation, consistent with the well-known amplification loop of the alternative pathway [18]. However, surprisingly few observations have been reported on the quantitative effect of the alternative pathway on classical pathway-induced activation [19]. Taking into account the important biological role of C5 activation, our data support a more crucial role for the alternative pathway in C5a and TCC formation than previously recognized. The main reason probably influencing the earlier observations is that classical pathway haemolytic assays were unsuitable to demonstrate the involvement of alternative pathway amplification when carried out at serum dilutions with undetectable alternative pathway activity (>1 : 16,Fig. 1a,c,e). We used high serum concentration (dilution 1 : 2 in the final reaction mixture) to approach physiological conditions and to ensure a fully active alternative pathway. In addition, a substantial amount of the earlier work used complement-depleted serum with a potential loss of bystander complement components during immunoadsorption. The reported effects of alternative pathway amplification on the effect of classical pathway induced activation vary with different methods, and the results are mostly qualitative or semiquantitative.
Our observations on the influence of alternative pathway amplification on the effect of initial classical pathway complement activation can be explained based on the known mechanisms of C3 and C5 convertase formation. The classical pathway C3 convertase C4b2a splits C3 into C3a and C3b resulting in formation of the classical pathway C5 convertase C4b2a3b which splits C5 into C5a and C5b. C5a induces reactions with potent pathophysiologic effects [20], and formation of C5b is the first step in assembly of the later components into the inflammatory and lytic TCC. In the alternative pathway low amounts of spontaneously formed C3(H2O) binds factor B which is cleaved into Ba and Bb by factor D. C3(H2O)Bb splits more C3, resulting in self-amplification with formation of the alternative pathway C5 convertase C3bBb3b. Incorporation of additional C3b molecules in this complex results in markedly increased C5 convertase activity [21]. C3b initially formed from C3 cleavage by the classical pathway C3 convertase augments the formation of alternative pathway C5 convertase. Thereby C3b deposition becomes independent of the initial trigger by the classical and lectin pathways and is a main component of the alternative pathway convertase. Studies on the formation of classical pathway C5 convertase on antibody-coated sheep RBCs indicate that one in every four C4b molecule is bound with a catalytic C2a molecule [22]. On the other hand, every molecule of C3b deposited on unsensitized rabbit RBCs binds a catalytic Bb molecule [21]. Therefore every molecule of C3b has the potential to form an alternative pathway C5 convertase complex. In general, fewer classical pathway C5 convertase sites are generated than alternative pathway C5 convertase sites once the complement system is activated, explaining the amplification of the classical pathway by the alternative pathway.
The complement system is strictly regulated both at the surface and in the fluid phase [23]. C3b is rapidly inactivated to iC3b by factors I and H in the fluid phase. Sine C5 binds only to C3b and not to iC3b, the classical pathway C5 convertase tends to generate only small amounts of TCC when regulation is kept under control. Extensive consumption of the terminal components with formation of substantial amounts of TCC requires C3b to be bound on a ‘protected surface’, facilitating factor B instead of factor H binding to C3b, thereby generating an alternative pathway C5 convertase. Evidence for this effect was provided by Reiter and Fishelson [24] showing that when C3b is coupled to IgG it becomes highly resistant to inactivation by Factors I and H, i.e. that IgG provides a protected surface.
The physiological benefit of alternative pathway amplification is particularly important when the classical and lectin pathways are not functioning at their full capacity. For example in newborns, maternal IgG decreases while their immune system is still not fully developed. A broad array of antibodies in limited amounts may still be essential in defense against bacterial infection when assisted by local alternative pathway amplification. On the other hand, alternative pathway activation can cause serious inflammation and tissue damage in various clinical conditions if complement is inappropriately activated or if activation occurs systemically with breakdown of the control mechanisms. These ‘dual roles’ of the complement system represent a double-edge sword of innate immunity.
Therapeutic intervention of the complement system is currently of great interest. The key issue is to select optimal steps or pathways for safe and effective inhibition. Our findings indicate a crucial role of factor D in the activation of the complement cascade since antifactor D mAb 166–32 in addition to inhibiting the alternative pathway per se, inhibited >80% of classical pathway induced C5a and TCC formation. The therapeutic benefits of targeting the alternative pathway should be validated in experimental models of various human diseases, including I/R injury, immune-complex diseases and sepsis. HAIGG was used in our studies as a prototype model similar to previous studies of immune complex deposition [25] and lupus nephritis [26]. MRL/lpr mice develop a spontaneous lupus-like disease characterized by immune complex glomerulonephritis. Decreased tissue damage has been demonstrated in factor B-deficient [27] as well as factor d-deficient [28] MRL/lpr mice. In a mouse model of arthritis triggered by immune complex deposition, manifestation of clinical disease was shown to be dependent of alternative pathway activation [29]. In the antiphospholipid syndrome [APS] antiphospholipid autoantibodies react with lipid-binding proteins on endothelial cells to trigger complement activation which causes cell damage, a procoagulant state, thrombosis, and fetal death secondary to placental insufficiency [30]. In an experimental model of APS it was recently shown that both the classical and alternative pathways are required to generate sufficient C5 cleavage to cause fetal loss [31]. In all these in vivo models the striking common feature is an increased effect of initial classical pathway activation induced by the alternative pathway.
Terminal complement activation is closely associated with the development of experimental lethal sepsis [20]. Excessive alternative pathway activation has also been documented in patients with Neisseria infection with poor outcome [32]. It seems appropriate to suggest that uncontrolled alternative pathway activation during sepsis is a main cause of the homeostatic breakdown and lethal outcome in the most severe form of the disease. Our combined data indicate that uncontrolled alternative pathway activation is responsible for systemic ‘explosive’ complement activation. Therefore, selective inhibition of the alternative pathway with preserved function of the initial classical and lectin pathway activation could attenuate severe inflammation and tissue injury and be an attractive therapeutic approach for these clinical indications.
Together, our studies provide for the first time quantitative evidence to explain the previous observations and highlight the predominant role of the alternative pathway in amplification of effector functions in the complement system.
ACKNOWLEDGEMENTS
We thank Grethe Bergseth and Dorte Christiansen for performing complement activation assays, Kåre Bergh for providing the antibodies for the C5a assay, Matthew Moyle for useful discussion of experiments, and John P. Atkinson for critical reading and valuable suggestions to the manuscript.
Financial supports were kindly provided to T. E. Mollnes by Tanox Inc., Research Council of Rikshospitalet, Norwegian Council on Cardiovascular Disease, Norwegian Foundation for Health and Rehabilitation, Sonneborn Chairtable Trust, Odd Fellow Foundation, Norsk Revmatikerforbund and Gythfeldt's legacy.
REFERENCES
- 1.Walport JM. Advances in immunology: Complement – first of two parts. New Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Mollnes TE, Song W-C, Lambris JD. Complement in inflammatory tissue damage and disease. Trends Immunol. 2002;23:61–4. doi: 10.1016/s1471-4906(01)02129-9. [DOI] [PubMed] [Google Scholar]
- 3.Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med. 1971;133:885–900. doi: 10.1084/jem.133.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisman HF, Bartow T, Leppo MK, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–51. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 5.Chan RK, Ibrahim SI, Verna N, Carroll M, Moore FD, Jr, Hechtman HB. Ischaemia-reperfusion is an event triggered by immune complexes and complement. Br J Surg. 2003;90:1470–8. doi: 10.1002/bjs.4408. [DOI] [PubMed] [Google Scholar]
- 6.Zhang M, Austen WG, Jr, Chiu I, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2004;101:3886–91. doi: 10.1073/pnas.0400347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stahl GL, Xu Y, Hao L, Miller M, Buras JA, Fung M, Zhao H. Role for the alternative complement pathway in ischemia/reperfusion injury. Am J Pathol. 2003;162:449–55. doi: 10.1016/S0002-9440(10)63839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung M, Loubser PG, Ündar A, Mueller M, Sun C, Sun WN, Vaughn WK, Fraser CD., Jr Inhibition of complement, neutrophil, and platelet activation by an anti-factor D monoclonal antibody in simulated cardiopulmonary bypass circuits. J Thorac Cardiovasc Surg. 2001;122:113–22. doi: 10.1067/mtc.2001.114777. [DOI] [PubMed] [Google Scholar]
- 9.Ündar A, Eichstaedt HC, Clubb FJ, Jr, Fung M, Lu M, Bigley JE, Vaughn WK, Fraser CD., Jr Novel anti-factor D monoclonal antibody inhibits complement and leukocyte activation in a baboon model of cardiopulmonary bypass. Ann Thorac Surg. 2002;74:355–62. doi: 10.1016/s0003-4975(02)03656-1. [DOI] [PubMed] [Google Scholar]
- 10.Roos A, Bouwman LH, Munoz J, et al. Functional characterization of the lectin pathway of complement in human serum. Mol Immunol. 2003;39:655–68. doi: 10.1016/s0161-5890(02)00254-7. [DOI] [PubMed] [Google Scholar]
- 11.Mollnes TE, Lea T, Frøland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoepitope of the complex. Scand J Immunol. 1985;22:197–202. doi: 10.1111/j.1365-3083.1985.tb01871.x. [DOI] [PubMed] [Google Scholar]
- 12.Mollnes TE, Brekke O-L, Fung M, et al. Essential role of the C5a receptor in E. coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002;100:1869–77. [PubMed] [Google Scholar]
- 13.Garred P, Mollnes TE, Lea T. Quantification in enzyme-linked immunosorbent assay of a C3 neoepitope expressed on activated human complement factor C3. Scand J Immunol. 1998;27:329–35. doi: 10.1111/j.1365-3083.1988.tb02354.x. [DOI] [PubMed] [Google Scholar]
- 14.Bergh K, Iversen OJ. Production of monoclonal antibodies against the human anaphylatoxin C5a des Arg and their application in the neoepitope-specific sandwich-ELISA for the quantification of C5a des Arg in plasma. J Immunol Meth. 1992;152:79–87. doi: 10.1016/0022-1759(92)90091-7. [DOI] [PubMed] [Google Scholar]
- 15.Volanakis JE, Narayana SVL. Complement factor D, a novel serine protease. Protein Sci. 1996;5:553–64. doi: 10.1002/pro.5560050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ratnoff WD, Fearon DT, Austen KF. The role of antibody in the activation of the alternative complement pathway. Springer Semin Immunopath. 1983;6:361–71. doi: 10.1007/BF02116280. [DOI] [PubMed] [Google Scholar]
- 17.Farries TC, Steuer KL, Atkinson JP. The mechanism of activation of the alternative pathway of complement by cell-bound C4b. Mol Immunol. 1990;27:1155–61. doi: 10.1016/0161-5890(90)90104-8. [DOI] [PubMed] [Google Scholar]
- 18.Fearon DT, Austen KF. Current concepts in immunology: the alternative pathway of complement – a system for host resistance to microbial infection. New Engl J Med. 1980;303:259–63. doi: 10.1056/NEJM198007313030505. [DOI] [PubMed] [Google Scholar]
- 19.Gupta-Bansal R, Parent JB, Brunden KR. Inhibition of complement alternative pathway function with anti-properdin monoclonal antibodies. Mol Immunol. 2000;37:191–201. doi: 10.1016/s0161-5890(00)00047-x. [DOI] [PubMed] [Google Scholar]
- 20.Ward PA. The dark side of C5a in sepsis. Nature Rev Immunol. 2004;4:133–42. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 21.Rawal N, Pangburn MK. Formation of high-affinity C5 convertases of the alternative pathway of complement. J Immunol. 2001;166:2635–42. doi: 10.4049/jimmunol.166.4.2635. [DOI] [PubMed] [Google Scholar]
- 22.Rawal N, Pangburn MK. Formation of high affinity C5 convertase of the classical pathway of complement. J Biol Chem. 2003;278:38476–83. doi: 10.1074/jbc.M307017200. [DOI] [PubMed] [Google Scholar]
- 23.Mollnes TE, Lachmann PJ. Regulation of complement. Scand J Immunol. 1988;27:127–42. doi: 10.1111/j.1365-3083.1988.tb02331.x. [DOI] [PubMed] [Google Scholar]
- 24.Reiter Y, Fishelson Z. Targeting of complement to tumor cells by heteroconjugates composed of antibodies and of the complement component C3b. J Immunol. 1989;142:2771–7. [PubMed] [Google Scholar]
- 25.Rostagno AA, Gallo G, Gold LI. Binding of polymeric IgG to fibronectin in extracellular matrices: an in vitro paradigm for immune-complex deposition. Mol Immunol. 2002;38:1101–11. doi: 10.1016/s0161-5890(02)00040-8. [DOI] [PubMed] [Google Scholar]
- 26.Kaneko N, Masuyama J, Nara H, Hirata D, Iwamoto M, Okazaki H, Minota S, Yoshio T. Production of thromboxane A2 and prostaglandin I2 affected by interaction of heat aggregated IgG, endothelial cells, and platelets in lupus nephritis. J Rheumatol. 2002;29:2106–13. [PubMed] [Google Scholar]
- 27.Watanabe H, Garnier G, Circolo A, et al. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164:786–94. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 28.Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS. Effects of complement factor D deficiency on the renal disease of MRL/lpr mice. Kidney Int. 2004;65:129–38. doi: 10.1111/j.1523-1755.2004.00371.x. [DOI] [PubMed] [Google Scholar]
- 29.Ji H, Ohmura K, Mahmood U, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–68. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 30.Atkinson JP. Complement system on the attack in autoimmunity. J Clin Invest. 2003;112:1639–41. doi: 10.1172/JCI20309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girardi G, Berman J, Redecha P, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112:1644–54. doi: 10.1172/JCI18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandtzaeg P, Høgåsen K, Kierulf P, Mollnes TE. The excessive complement activation in fulminant meningococcal septicemia is predominantly caused by alternative pathway activation. J Infect Dis. 1996;173:647–55. doi: 10.1093/infdis/173.3.647. [DOI] [PubMed] [Google Scholar]