

Abstract
Site-specific DNA recombination is important for basic cellular functions including viral integration, control of gene expression, production of genetic diversity and segregation of newly replicated chromosomes, and is used by bacteriophage λ to integrate or excise its genome into and out of the host chromosome. λ recombination is carried out by the bacteriophage-encoded integrase protein (λ -int) together with accessory DNA sites and associated bending proteins that allow regulation in response to cell physiology. Here we report the crystal structures of λ -int in higher-order complexes with substrates and regulatory DNAs representing different intermediates along the reaction pathway. The structures show how the simultaneous binding of two separate domains of λ -int to DNA facilitates synapsis and can specify the order of DNA strand cleavage and exchange. An intertwined layer of amino-terminal domains bound to accessory (arm) DNAs shapes the recombination complex in a way that suggests how arm binding shifts the reaction equilibrium in favour of recombinant products.
λ -int catalyses an ordered, pair-wise exchange of four DNA strands between two different pairs of recombination substrates1,2. During integration, λ -int aligns the bacteriophage attachment site attP with the bacterial attachment site attB and recombines these sequences to generate the recombination joints attL and attR flanking the integrated prophage (Fig. 1a, b). During the transition to lytic growth, the bacteriophage DNA is excised to regenerate attP and attB. In both reactions, the analogous pair of DNA strands (‘top’ strands) is exchanged first3,4 to form a branched, four-way DNA intermediate known as a Holliday junction. Subsequent exchange of ‘bottom’ strands resolves the Holliday junction into linear recombinant products2. Although integration and excision might appear to be reciprocal reactions (Fig. 1), they involve different substrates and are effectively irreversible5. The recombination machinery is configured differently during integration or excision by two different overlapping subsets of accessory factors and binding sites that bend the DNA arms flanking the core sites of strand exchange2,6. DNA bending is a prerequisite for the simultaneous interactions with arm and core sites6–9 that deliver λ -int to lower-affinity core sites10. Arm-binding interactions allosterically enhance the fidelity of DNA strand exchange11 and bias the outcome of Holliday junction resolution in favour of the recombined products12.
Figure 1. Pathways for integrative and excisive recombination.

a, λ-int recombines bacteriophage attP and bacterial attB attachment sites during integration, and attL and attR during excision. Different sets of DNA bending proteins (IHF, Xis and Fis) uniquely configure the recombinogenic complexes for these reactions and facilitate simultaneous interaction of the heterobivalent λ -int with the core and arm sites1 (see Fig. 5). The occupied (black labels) and unoccupied (grey labels) sites during each recombination process are shown for λ -int, IHF (cyan), Xis (brown) and Fis (orange). b, Conformation of the core DNAs and interactions of the N domains (small ellipsoids) with the arm DNAs (green arrows) during a recombination reaction. c–e, Experimental electron densities of λ -int complexes after phase modification (top). DNA substrates used for crystallization (see Fig. 3) along with the alternative isomer of the core DNA shown in the inset of d (bottom; see Methods).
Two related tyrosine recombinases—the bacteriophage P1 Cre and yeast Flp proteins—lack the regulatory arm-binding functions of λ -int and catalyse a reversible exchange of DNA strands between identical sites13,14. Crystal structures of Cre15,16 and Flp17 show that a protein tetramer mediates the synapse of two strongly bent antiparallel DNAs closely resembling a Holliday junction. These structures suggest that only a small shift in subunit packing is sufficient to redirect DNA cleavage and exchange activities from one pair of strands to the other, in order to resolve the Holliday junction into products13,14. However, the molecular mechanism of isomerization is poorly understood.
In contrast to Cre and Flp, recombination by λ -int depends absolutely on secondary interactions with the DNA arms flanking the site of strand exchange. Arm binding by λ -int provides a control point for site-specific recombination and facilitates crystallization of trapped intermediates from the multi-step reaction. We have determined crystal structures of λ -int tetramers complexed to arm and core DNAs in different conformations that provide a series of views along the recombination pathway (Fig. 1c–e). The two distinct DNA-binding activities of λ -int dictate a specific arrangement of core and arm sites that facilitates synapsis and explains the order of strand cleavage and exchange. Arm binding constrains the topology of the bound DNA and promotes isomerization towards a conformation that is proficient for the second exchange of DNA strands to complete recombination. This allosteric control makes λ recombination a conditional, effectively irreversible molecular switch during the bacteriophage lifecycle.
Assembly of a recombinogenic complex
λ -int was crystallized in three different complexes with either a Holliday junction18 or a COC′ DNA substrate19 in the presence and absence of oligonucleotides containing tandem P′ 1–P′ 2 binding sites derived from attP11 (Fig. 1). The crystal structures of these recombination complexes were determined by isomorphous replacement methods (see Methods and Supplementary Table S1), and they capture different steps of recombination. A synaptic complex consisting of an N-terminally truncated λ -int(75–356) tetramer bound to two COC′ core sites represents an early step after the initial DNA cleavage but before strand exchange (Fig. 1c). In the complex with full-length λ -int and arm DNAs, the cleaved DNA strands have exchanged but ligation is prevented by a modification of the substrate DNA19 (Fig. 1d). The synaptic and post-exchange complexes crystallized in different space groups with a crystallographic two-fold axis relating two λ -int dimers and generating a two-fold symmetric tetramer. A Holliday junction intermediate (Fig. 1e) together with arm DNAs was crystallized in a complex with the λ -int(Tyr342Phe) mutant that is unable to cleave DNA and resolve the Holliday junction into products. This complex has a roughly two-fold symmetry and is configured for the second strand exchange reaction.
The Holliday junction complex exemplifies the overall organization of λ recombination complexes. Upon DNA binding, the three functional domains of λ -int20,21 assemble into the three distinct layers of the tetramer. The N-terminal domain (N domain; residues 1–63) that binds to arm sites is connected to the core-binding domain (CB domain; residues 75–175) by a short, α -helical coupler (residues 64–74). The CB domain is, in turn, joined to the C-terminal catalytic domain (residues 176–356) through another extended segment (residues 160–176; Fig. 2)19. All three domains of λ -int collaborate to engage the core and arm DNAs, establishing a tightly knit but flexible recombinogenic complex.
Figure 2. Structure of the λ -int tetramer bound to a Holliday junction and arm DNAs.

a, The domains of λ -int pack together as three stacked layers, with the N domains cyclically swapped onto neighbouring subunits. The N-domain layer embraced by two anti-parallel arm DNAs is linked through short α -helical couplers to the CB and catalytic domains, which encircle the branches of the Holliday junction. The active subunits are coloured red/green and inactive subunits are blue/yellow. b, The two-fold symmetry of the N-domain layer is reflected in the skewed arrangement of CB domains and the shape of the four-way junction (thick dark grey lines) in the bottom strands reactive isomer. c, d, Active (c) and inactive (d) interfaces are shown, with the area around Tyr 342 nucleophile marked by dashed circles (see Fig. 4).
The λ -int tetramer has a cyclically permuted topology in which each N domain packs on top of the neighbouring CB domain (Fig. 2a). Fluorescence resonance energy transfer studies of similar complexes also show this permuted arrangement22. The N domains of two adjacent subunits adopt different conformations that orient pairs of DNA-reading heads (residues 12–55) for interactions with tandem P′ 1–P′ 2 arm sites (Fig. 2b–d). The convoluted packing arrangement of the four N domains buries 2,900 Å2 of protein surface area, suggestive of a stable structure once the arms are engaged20. The structure of the N-domain layer and its interactions with arm DNAs are very similar in both post-exchange and Holliday junction complexes of λ -int despite different crystal-packing environments (Supplementary Table S1), a further indication of a fairly rigid structure. The two-fold symmetry of the assembled N-domain layer is reflected to varying extents in the skewed arrangement of catalytic domains and associated core DNAs (Figs 2b and 3d, g). Our results suggest that interactions with the arm DNAs bias the conformation of the λ -int tetramer to drive the reaction to completion.
Figure 3. Three different conformations of λ -int tetramers representing distinct steps of the recombination reaction.

The core DNAs within the λ -int(75–356) synaptic complex (a, b), the λ -int post-strand exchange complex (d, e) and the λ -int Holliday junction complex (g, h) are shown along with schematic diagrams illustrating the inter-branch angles and position of branch points. The pair of λ -int subunits in the active conformation (orange) is positioned closer to the centre of each complex, whereas the inactive pair of subunits (cyan) is further apart. Scissile phosphates (spheres) activated for cleavage are coloured in red. A sharp kink in the bottom strand located two (c; synaptic) or four (f; post-exchange) nucleotides from the site of top strand cleavage is marked with arrows.
Four N domains are embraced by two anti-parallel arm DNAs that bend towards one another (13° bend), although not as strongly as inferred from solution studies11,23. Pairs of N domains engage adjacent binding sites (5′ -GTCA-3′ ) that are separated by 10 bp (Fig. 2b, c). Each reading head is positioned in the major groove by interactions with the phosphosugar backbone involving side chains on the ends of strand β 1 (Asn 15, Asn 20) and main chain nitrogens from strand β 3 (Glu 34, Gly 36; Supplementary Fig. S1). Side chains from β 1 (Tyr 17) and the C-terminal end of β 2 (Arg 27) make sequence-specific polar interactions with the base pairs of the P′ 1 and P′ 2 arm sites. A basic N-terminal segment (residues 2–10) that is required for recombination activity24 dips into the minor groove adjacent to the 3′ side of the consensus arm binding sequence. Patchy electron density indicates that this segment is not well ordered in the presence of arm DNA, and it was entirely disordered in an NMR structure of the unliganded N domain24.
The CB and catalytic domains of λ -int together constitute a minimal recombinase that is structurally analogous to the full-length Cre15, Flp17 and XerC/D25 tyrosine recombinases21. The active site of λ -int resembles that of other tyrosine recombinases19,26, with conserved residues (Arg 212, Lys 235, His 308, Arg 311, His 333) that engage the scissile phosphate and a tyrosine nucleophile (Tyr 342) that forms a 3′ phosphotyrosine linkage with the cleaved DNA14. The catalytic domain of λ -int binds to core sites with low affinity10 and has significant DNA cleavage activity only when linked to the CB domain, which contributes binding affinity and specificity for core sites21.
Although the CB domain of λ -int resembles the N-terminal domain of Cre, it lacks a helix ( α E) that contributes many inter-subunit interactions in the Cre tetramer15. Without this helix, the loosely packed CB domains of the λ -int tetramer rotate against one another by as much as 30° in the different isomers that were crystallized (Fig. 3). The N domains do not rotate and the α -helical coupler connecting the N-domain layer to the CB domains (Fig. 2a, c) appears to restrict the range of subunit rotation. The coupler is essential for cooperative binding to arm sites20,27, and it is well positioned to transmit the allosteric effects of arm binding to the catalytic core.
Isomerization and control of catalytic activity
The complex between λ -int and its DNA substrates isomerizes halfway through the recombination reaction, redirecting DNA cleavage activity from one pair of strands to the other pair for resolution of the Holliday junction (Fig. 1b). The three independent crystal structures represent different steps of isomerization: a synaptic complex (Figs 1c and 3a, b), a post-strand exchange complex (Figs 1d and 3d, e) and a Holliday junction complex (Figs 1e and 3g, h). Each step reveals a different conformation of the core DNAs and different subunit packing interactions. Conformational transitions between steps require three interrelated modes of motion: a migration of the DNA bend that generates different base-pairing combinations at the centre of the crossover region of the four-way junction, a scissoring motion of the DNA branches extending from the junction, and a sliding of the two halves of the complex past one another that skews its shape away from four-fold symmetry (Fig. 3). These snapshots of the reaction pathway suggest how DNA cleavage is regulated and reveal the molecular features promoting strand exchange.
λ -int’s catalytic activity is controlled by the quaternary structure of the tetramer, resulting in two active and two inactive subunits that switch roles during isomerization (Fig. 1b). The experimentally phased electron density, even at the limited resolution of these structures (Fig. 1c–e; see also Supplementary Table S1), shows clear evidence of structural differences among the subunits of the λ -int tetramer, corresponding to active (red subunit in Fig. 2c) and inactive (yellow subunit in Fig. 2d) conformations of the helix containing the Tyr 342 nucleophile. These two states are most clearly illustrated by the 2.8 Å structure of the synaptic complex (Fig. 4). The catalytic domains interact through a C-terminal extended segment (residues 350–356, including β 9) that packs in trans against the neighbouring subunits (Figs 2c, d and 4a) in a cyclically permuted arrangement similar to that of the N domains (Fig. 2a). The trans interactions of λ -int’s catalytic domains are functionally analogous to the trans packing of active site control elements of Cre and Flp recombinases13,14. The skewed packing of subunits gives rise to two very different subunit interfaces harbouring active or inactive catalytic sites (Figs 2c, d and 4). In the active conformation the Tyr 342 helix is well ordered, stabilized by electrostatic interactions involving the catalytically essential residues Arg 346 and Arg 348 (ref. 28) (Fig. 4b). The position of β 9 in the inactive subunit is incompatible with these stabilizing interactions and the region around Tyr 342 is correspondingly disordered (Fig. 4c). The active and inactive subunit interfaces (Fig. 2d) also differ by a significant rotation (up to 37° ) of the apposing subunits.
Figure 4. Control of catalytic activity by trans cyclic interactions of the C-terminal tail.
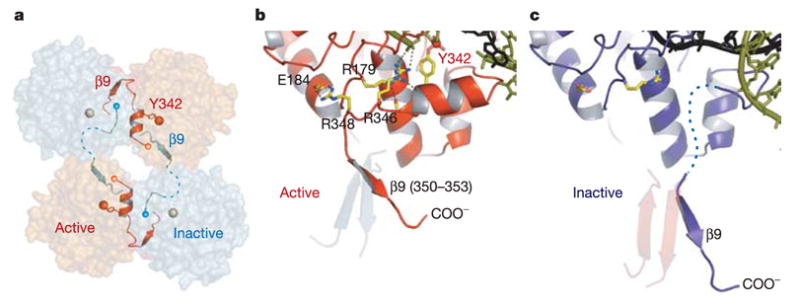
a, The assembly of active (red) and inactive (cyan) catalytic sites results from a skewed packing arrangement of λ -int(75–356) subunits in the tetramer. The scissile phosphates bound by active and inactive subunits are shown as red and grey spheres, respectively. b, In the active conformation, the tail is oriented to permit stabilizing interactions with residues Arg 346 and Arg 348 that result in a well-ordered segment (residues 339–348) containing the Tyr 342 nucleophile. c, In the inactive molecule, a large shift of β 9 causes this segment to become disordered.
The α -helical conformation around Tyr 342 differs from the active site conformation previously reported for a crystal structure of a λ -int(75–356) monomer using a similar flap-type DNA substrate with a phosphorylated 5′ end19 (Fig. 1d). To remove the influence of the added 5′ phosphate we determined an additional structure of λ -int(75–356) complexed to a nicked COC′ DNA with an unmodified 5′ -OH end and orthovanadate bridging between Tyr 342 and the 3′ -OH of the nicked DNA29,30 (Supplementary Table S1). The active subunits of the vanadate complex adopt the α -helical conformation around Tyr 342 (not shown), suggesting that this is the true active conformation.
DNA bending and strand exchange
The synaptic complex of λ -int(75–356) without arm DNAs represents an early stage of recombination with the 5′ ends of the cleaved DNA strands unexchanged (Fig. 3a, b). This synaptic complex deviates strongly from four-fold symmetry, with the scissile phosphates (represented as the corners of a parallelogram) of the cleaved strands located 39 Å apart and those of the uncleaved strands 50 Å apart (Fig. 3a, b). This skewed arrangement of subunits and cleavage sites produces a translational offset between the synapsed DNAs that brings the cleaved 5′ ends closer to the 3′ phosphotyrosine of the synapsing partner to facilitate strand exchange and ligation. The core DNAs are bent asymmetrically in the overlap region between the sites of DNA cleavage (Fig. 3b, c). A kink in the uncleaved bottom strand located two base pairs away from the cleaved site causes an unstacking of two adjacent adenines, generating a 109° angle between the C and C′ binding sites. A break in the DNA backbone facilitates the localized kinking of DNA that disrupts base pairing interactions with two nucleotides at the 5′ end of the cleaved top strand (Fig. 3c). Thus, the sharp kinking of the DNA substrate may facilitate strand exchange and prevent religation.
In the complex with arm DNAs, the cleaved 5′ ends of the core DNAs have been exchanged and the core DNAs resemble a Holliday junction with approximate four-fold symmetry (Fig. 3d, e). Ligation of the exchanged strands is prevented by an added 5′ phosphate19, and this may have prevented further isomerization of the complex to a conformer that is proficient for the second strand exchange reaction. In this post-exchange complex, the kink in the overlap region has shifted to a more central location 4 bp away from the cleaved site (Fig. 3d, e). The angle between the C and C′ binding sites has also changed (Fig. 3b, e), switching base pairing partners to promote exchange of the 5′ ends. This remodelling occurs by local adjustments to the DNA backbone without requiring a significant rotation of the DNA branches radiating from the centre of the four-way junction. The central position of the DNA bend in the post-exchange complex brings the cleavage sites on the bottom strands closer together (44 Å), and this may help prevent the reversal of top strand cleavage. Strand exchange appears to be promoted by a reshaping of the DNA substrate brought about by the binding of the arm DNAs (Figs 1b and 4). There are few interactions with the 5′ ends of the cleaved DNA to provide a more directed mechanism.
The recombination complex with a Holliday junction and arm DNAs (Fig. 3g, h) could, in principle, be resolved by the exchange of either pair of DNA strands, and this choice is strongly influenced by the position of the crossover junction within the overlap region18,31. The Holliday junction used for crystallization (Fig. 1e) is immobile, with its crossover point fixed three nucleotides from one pair of cleavage sites and, consequently, these sites are used preferentially for resolution18. The complex is highly skewed, and the scissile phosphates bound by active subunits are brought close together (38 Å) after the initial pair of strands have exchanged (Fig. 3g, h). For reasons described below, we propose that this complex represents an isomer poised for the second strand exchange that would resolve the Holliday junction into recombinant products.
Because recombination involves two analogous strand exchange reactions18 (Fig. 1b), we could reasonably expect that complexes catalysing the first and second strand exchanges will have similar conformations and be distinguished only by the identities of the exchanging strands. The arm DNAs bind in a unique orientation (Fig. 2c, d; see Methods). As a result, the missing connections with the core sites can be readily modelled to establish which core sites are bound by active and inactive subunits (Fig. 5; see also Supplementary Information). On the basis of the proximity to the P′ 1 arm sites, we propose that the C′ sites (sites of bottom strand cleavage)3,4 are bound by subunits in the active conformation in both complexes with arms (Fig. 2d). Correspondingly, biochemical assays have shown that interactions with arm DNAs stimulate Holliday junction resolution by bottom strand exchange22.
Figure 5. Models showing the topology of DNA during λ integration and excision.

a, A schematic representation of the integrative complex. The P′ arm engages the N domains of subunits bound to C and B′ core sites. The P arm is positioned over the N domains of subunits on the C′ and B core sites by a compound bend induced by IHF. d, During excision, the P′ arm is oriented similarly as in integration, whereas the P arm could form a more compact loop upon binding to Xis and/or Fis (see text). b, c, e, f, Docking models of the hypothetical integrative (b, c) and excisive (e, f) complexes based on our crystal structures and that of an IHF–DNA complex36 are shown in front and back views.
Although the positions of active and inactive subunits are defined with respect to the arms, the COC′ core DNAs freely isomerize, and X-ray experiments with iodinated DNAs reveal two isomers of the cleaved COC′ DNAs bound in different orientations (Fig. 1d). The majority of the DNA molecules have isomerized so that the cleaved DNA strands are bound by subunits now in an inactive conformation. The existence of both isomers in the same crystal highlights the reciprocal nature of the isomerization process when the core and arm-binding sites are supplied on separate DNAs. Nonetheless, the unique binding orientation of the arm DNAs specifies a topological linkage to the core sites of bottom strand cleavage, which are bound by subunits in the active conformation.
Allosteric control of recombination
How do interactions with the arms promote the exchange of bottom strands that completes λ integration and excision3,4 (Fig. 1b, step 5)? The similarly skewed geometry of the N- and C-terminal halves of λ recombination complexes raises the possibility that arm-binding interactions may restrain the quaternary structure of the λ -int tetramer. The N domains in complex with arm sites are arranged in a parallelogram (Fig. 2b), mirroring the skewed arrangement of scissile phosphates configured for bottom strand exchange (Fig. 3g). One pair of N domains lies near the two-fold axis of symmetry and the other pair lies further away, reflecting the arrangement of active and inactive subunits. It appears that the α -helical coupler joining the N domain with the CB domain (Fig. 2c) functions as a short tether that imposes the skewed, two-fold symmetry of the N-domain layer onto the four CB domains and associated core DNAs.
This is a type of allosteric control that positions the bottom strands for cleavage and exchange (Fig. 1b), effectively shifting the reaction equilibrium in favour of recombinant products. Despite the conformational bias favouring bottom strand exchange, the exchange of top strands is a prerequisite that removes a physical barrier from the path of bottom strand exchange13. It is therefore notable that arm binding also establishes an orientation of core DNAs that is compatible with top strand cleavage in the initial synaptic complex. Cleavage of the top strands may render the DNAs more flexible, allowing an inter-conversion to the bottom strands reactive isomer that is stabilized by the N-domain layer.
Interactions with the arms of attP
Although the basic features of the DNA cleavage and strand exchange reactions in the λ recombination system are similar to those of other tyrosine recombinases13,14,32, the arms of the bacteriophage λ att sites provide a level of regulation and unidirectionality that is not available to simpler recombinases33. The decision to insert bacteriophage λ DNA into the Escherichia coli chromosome or to excise it is made by committees of DNA bending proteins that wrap the P and P′ arms around the integrative and excisive complexes in different ways1,34,35. Our crystal structures of λ -int tetramers reveal the spatial relationship between arm and core sites that together with the crystal structure of IHF bound to DNA36 can be used to model the topology of the arms in both complexes (Fig. 5).
The conformations of the P′ arm are readily explained by simple docking models that are similar for both integrative and excisive complexes (Figs 1 and 5a). IHF binding to the H′ site (Fig. 1a) creates an approximately 180° bend that redirects the P′ arm over the λ -int tetramer for interactions with two N domains located on opposing sides of the synaptic interface36,37. Only a minor change in the bending angle around H′ is required to alternatively engage P′ 2–P′ 3 (integration) or P′ 1–P′ 2 (excision) binding sites8 (Fig. 5b, e).
The P arm of the phage attachment site is configured differently during integration and excision8,38, engaging either the P1 or P2 site, respectively (Fig. 1a). Different pairs of attachment sites are used for integration (attP and attB) or excision (attL and attR) with resultant differences in the relative orientations of P and P′ arms (Fig. 5a, d). Our modelling suggests that during integration IHF bends the P arm at H2 and directs it under the λ -int tetramer. A second IHF-induced bend at the distal H1 site would position the P1 site over one N domain, and the adjacent N domain binds nonspecifically to the flanking DNA (Fig. 5a–c). During excision, IHF is displaced from H1 by the DNA-bending factors Xis and Fis that bind nearby sites X1 and X2 or F1,6. Presumably, a sharp bend in the P arm, induced by Xis23 binding to DNA and its interaction with the N domain39, is required to bring the P2 site into position. We have modelled one such conformation of P arm in the excisive complex (Fig. 5d–f). In both reactions, we predict that the P arm wraps around the complex with a left-handed curvature that would be stabilized in underwound DNA. Negative supercoiling of attP is required for integration40, and this may compensate for the weaker interactions of the P arm with λ -int.
The crystal structures of λ -int tetramers and the topology of arm binding inferred from them indicate how the tetramer may initially assemble by the coalescence of N domains in complex with high-affinity arm sites (Fig. 2) to enable the low-affinity interactions with core sites41. During integration, the interlocked assembly of N domains and arm DNAs positions two subunits on the attP core and establishes a preferred bending direction for top strand cleavage. Two remaining subunits of the tetramer are then available for the capture of attB, consistent with earlier DNA footprinting studies42. During excision, the arm DNAs create a bridge between subunits across the synaptic interface of attL and attR, dictating a bend in the core DNA that again initially favours top strand cleavage before isomerization and exchange of the bottom strands. Arm binding interactions appear to bias isomerization in favour of the second half of the reaction: the exchange of bottom strands and resolution into products (Figs 1b and 3). The structures of higher-order λ recombinogenic complexes show how the simultaneous binding of λ -int to core and arm sites facilitates synapsis, and they suggest a mechanism for coordinating the order of strand exchange and driving the multi-step recombination reaction to completion.
METHODS
The preparation of λ -int proteins and the DNA complexes that were crystallized are described in the Supplementary Information. X-ray diffraction data were collected at beamlines X-25 and X-26C of National Synchrotron Light Source, and beamline 19-ID of Advanced Photon Source. X-ray data were processed with HKL2000 (ref. 43) and CCP444 program suites. The structure of the λ -int(75–356) complex (synaptic) was determined by the single-wavelength anomalous dispersion (SAD) method using a selenomethionine (SeMet)-labelled crystal. The structure of the λ -int–Holliday junction–arm DNA complex was determined by a combination of multiple-wavelength anomalous dispersion (MAD) and conventional isomorphous replacement phasing, using SeMet, Ta6Br14· 8H2O and Pt-terpyridine derivatives. Heavy atom derivatives were prepared by soaking crystals for 12 h at 20 ° C in mother liquor containing 0.4 mM tantalum bromide or 0.1 mM platinum terpyridine. The structure of the λ -int–COC′ –arm DNA complex (post-strand exchange) was determined with phase information from SeMet and Ta6Br14· 8H2O derivatives. MLPHARE44 and SHARP45 were used for heavy-atom parameter refinement and calculation of experimental phases, which were subsequently improved by density modification using DM44, RESOLVE46 and SOLOMON44 (Supplementary Table S1). Atomic models for all structures were fitted into the electron density using the program O47, starting with the refined model of a λ -int(75–356) DNA complex and the NMR solution structure24 of the N domain. The register and orientation of the core and arm DNAs in the λ -int structures were determined from X-ray experiments using DNA substrates containing 5′ -iodo-2′ -deoxyuridine substitutions at multiple thymidine positions, and the locations of covalent 3′ phosphotyrosine linkages. The λ -int(75–356) structure was refined using CNS48 without restraints of non-crystallographic symmetry (NCS) to values of Rwork/Rfree = 0.218/0.262 at 2.8 Å. The λ -int–COC′ –arm structure was initially refined using CNS with a grouped B-factor refinement, followed by REFMAC544 with positional and TLS parameter refinement to a final Rwork/Rfree = 0.248/0.296 at 3.8 Å. The λ -int–Holliday junction–arm structure was refined using REFMAC5 with the rigid body, TLS and positional refinements, resulting in Rwork/Rfree = 0.281/0.316 at 4.4 Å. Experimental phases were included as refinement standards in all cases. NCS restraints were imposed on the individual domains during refinement of the λ -int–core–arm structures, and hydrogen bonding restraints were used for protein secondary structures and DNA base pairs. The figures were produced with PyMOL (http://www.pymol.org).
Supplementary Material
Acknowledgments
X-ray data were measured at beamlines X-26C and X-25 of the National Synchrotron Light Source, station A1 of the Cornell High Energy Synchrotron Source, and beamline 19ID at the Advanced Photon Source, facilities that are supported by the US Department of Energy and the National Institutes of Health. This work was supported by research grants from the National Institutes of Health (to T.E. and A.L.) and fellowships from Jeane B. Kempner Fund (to T.B.) and the Human Frontier Science Program (to H.A.). T.E. is the Hsien Wu and Daisy Yen Wu Professor at Harvard Medical School.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Coordinates and structure factors are deposited in the Protein Data Bank under accession codes 1Z19, 1Z1B and 1Z1G. Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
References
- 1.Landy A. Dynamic, structural, and regulatory aspects of λ site-specific recombination. Annu Rev Biochem. 1989;58:913–949. doi: 10.1146/annurev.bi.58.070189.004405. [DOI] [PubMed] [Google Scholar]
- 2.Azaro MA, Landy A. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz A, editors. ASM Press; Washington DC: 2002. pp. 118–148. [Google Scholar]
- 3.Kitts PA, Nash HA. Bacteriophage lambda site-specific recombination proceeds with a defined order of strand exchanges. J Mol Biol. 1988;204:95–107. doi: 10.1016/0022-2836(88)90602-x. [DOI] [PubMed] [Google Scholar]
- 4.Nunes-Duby SE, Matsumoto L, Landy A. Site-specific recombination intermediates trapped with suicide substrates. Cell. 1987;50:779–788. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- 5.Segall AM, Nash HA. Architectural flexibility in lambda site-specific recombination: three alternate conformations channel the attL site into three distinct pathways. Genes Cells. 1996;1:453–463. doi: 10.1046/j.1365-2443.1996.d01-254.x. [DOI] [PubMed] [Google Scholar]
- 6.Nash HA, et al. Escherichia coli and Salmonella typhimurium. In: Neidhardt FC, editor. Cellular and Molecular Biology. ASM Press; Washington DC: 1996. pp. 2263–2376. [Google Scholar]
- 7.Moitoso de Vargas L, Kim S, Landy A. DNA looping generated by DNA bending protein IHF and the two domains of lambda integrase. Science. 1989;244:1457–1461. doi: 10.1126/science.2544029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Numrych TE, Gumport RI, Gardner JF. A comparison of the effects of single-base and triple-base changes in the integrase arm-type binding sites on the site-specific recombination of bacteriophage lambda. Nucleic Acids Res. 1990;18:3953–3959. doi: 10.1093/nar/18.13.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross W, Landy A. Bacteriophage λ int protein recognizes two classes of sequence in the phage att site: characterization of arm-type sites. Proc Natl Acad Sci USA. 1982;79:7724–7728. doi: 10.1073/pnas.79.24.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross W, Landy A, Kikuchi Y, Nash H. Interaction of int protein with specific sites on λ att DNA. Cell. 1979;18:297–307. doi: 10.1016/0092-8674(79)90049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radman-Livaja M, et al. Arm sequences contribute to the architecture and catalytic function of a λ integrase-Holliday junction complex. Mol Cell. 2003;11:783–794. doi: 10.1016/s1097-2765(03)00111-4. [DOI] [PubMed] [Google Scholar]
- 12.Franz B, Landy A. The Holliday junction intermediates of λ integrative and excisive recombination respond differently to the bending proteins integration host factor and excisionase. EMBO J. 1995;14:397–406. doi: 10.1002/j.1460-2075.1995.tb07014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Duyne GD. A structural view of creloxp site-specific recombination. Annu Rev Biophys Biomol Struct. 2001;30:87–104. doi: 10.1146/annurev.biophys.30.1.87. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Rice PA. New insight into site-specific recombination from Flp recombinase-DNA structures. Annu Rev Biophys Biomol Struct. 2003;32:135–159. doi: 10.1146/annurev.biophys.32.110601.141732. [DOI] [PubMed] [Google Scholar]
- 15.Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 16.Gopaul DN, Guo F, Van Duyne GD. Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J. 1998;17:4175–4187. doi: 10.1093/emboj/17.14.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell. 2000;6:885–897. [PubMed] [Google Scholar]
- 18.Azaro MA, Landy A. The isomeric preference of Holliday junctions influences resolution bias by λ integrase. EMBO J. 1997;16:3744–3755. doi: 10.1093/emboj/16.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aihara H, Kwon HJ, Nunes-Duby SE, Landy A, Ellenberger T. A conformational switch controls the DNA cleavage activity of λ integrase. Mol Cell. 2003;12:187–198. doi: 10.1016/s1097-2765(03)00268-5. [DOI] [PubMed] [Google Scholar]
- 20.Sarkar D, et al. Differential affinity and cooperativity functions of the amino-terminal 70 residues of λ integrase. J Mol Biol. 2002;324:775–789. doi: 10.1016/s0022-2836(02)01199-3. [DOI] [PubMed] [Google Scholar]
- 21.Tirumalai RS, Kwon HJ, Cardente EH, Ellenberger T, Landy A. Recognition of core-type DNA sites by λ integrase. J Mol Biol. 1998;279:513–527. doi: 10.1006/jmbi.1998.1786. [DOI] [PubMed] [Google Scholar]
- 22.Radman-Livaja M, Biswas T, Mierke D, Landy A. Architecture of recombination intermediates visualized by in-gel FRET of λ integrase-Holliday junction-arm-DNA complexes. Proc Natl Acad Sci USA. 2005;102:3913–3920. doi: 10.1073/pnas.0500844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson JF, Landy A. Empirical estimation of protein-induced DNA bending angles: applications to lambda site-specific recombination complexes. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wojciak JM, Sarkar D, Landy A, Clubb RT. Arm-site binding by λ -integrase: solution structure and functional characterization of its amino-terminal domain. Proc Natl Acad Sci USA. 2002;99:3434–3439. doi: 10.1073/pnas.052017999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanya HS, et al. Crystal structure of the site-specific recombinase, XerD. EMBO J. 1997;16:5178–5187. doi: 10.1093/emboj/16.17.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon HJ, Tirumalai R, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science. 1997;276:126–131. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren D, Lee SY, Landy A. Mutations in the amino-terminal domain of λ -integrase have differential effects on integrative and excisive recombination. Mol Microbiol. 2005;55:1104–1112. doi: 10.1111/j.1365-2958.2004.04447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kazmierczak RA, Swalla BM, Burgin AB, Gumport RI, Gardner JF. Regulation of site-specific recombination by the C-terminus of lambda integrase. Nucleic Acids Res. 2002;30:5193–5204. doi: 10.1093/nar/gkf652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies DR, Interthal H, Champoux JJ, Hol WG. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem Biol. 2003;10:139–147. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 30.Martin SS, Wachi S, Baldwin EP. Vanadate-based transition-state analog inhibitors of Cre-LoxP recombination. Biochem Biophys Res Commun. 2003;308:529–534. doi: 10.1016/s0006-291x(03)01437-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nunes-Duby SE, Azaro MA, Landy A. Swapping DNA strands and sensing homology without branch migration in λ site-specific recombination. Curr Biol. 1995;5:139–148. doi: 10.1016/s0960-9822(95)00035-2. [DOI] [PubMed] [Google Scholar]
- 32.Sherratt DJ, Wigley DB. Conserved themes but novel activities in recombinases and topoisomerases. Cell. 1998;93:149–152. doi: 10.1016/s0092-8674(00)81566-4. [DOI] [PubMed] [Google Scholar]
- 33.Bushman W, Thompson JF, Vargas L, Landy A. Control of directionality in lambda site specific recombination. Science. 1985;230:906–911. doi: 10.1126/science.2932798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crisona NJ, Weinberg RL, Peter BJ, Sumners DW, Cozzarelli NR. The topological mechanism of phage λ integrase. J Mol Biol. 1999;289:747–775. doi: 10.1006/jmbi.1999.2771. [DOI] [PubMed] [Google Scholar]
- 35.Bankhead TM, et al. Mutations at residues 282, 286, and 293 of phage λ integrase exert pathway-specific effects on synapsis and catalysis in recombination. J Bacteriol. 2003;185:2653–2666. doi: 10.1128/JB.185.8.2653-2666.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 37.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Thompson JF, de Vargas LM, Skinner SE, Landy A. Protein-protein interactions in a higher-order structure direct lambda site-specific recombination. J Mol Biol. 1987;195:481–493. doi: 10.1016/0022-2836(87)90177-x. [DOI] [PubMed] [Google Scholar]
- 39.Sam MD, Cascio D, Johnson RC, Clubb RT. Crystal structure of the excisionase-DNA complex from bacteriophage lambda. J Mol Biol. 2004;338:229–240. doi: 10.1016/j.jmb.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 40.Mizuuchi K, Gellert M, Nash HA. Involvement of supertwisted DNA in integrative recombination of bacteriophage lambda. J Mol Biol. 1978;121:375–392. doi: 10.1016/0022-2836(78)90370-4. [DOI] [PubMed] [Google Scholar]
- 41.Kim S, Landy A. Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science. 1992;256:198–203. doi: 10.1126/science.1533056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richet E, Abcarian P, Nash HA. Synapsis of attachment sites during lambda integrative recombination involves capture of a naked DNA by a protein-DNA complex. Cell. 1988;52:9–17. doi: 10.1016/0092-8674(88)90526-0. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.De LaFortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 1997;276:472–499. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 46.Terwilliger TC. Automated structure solution, density modification and model building. Acta Crystallogr D. 2002;58:1937–1940. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- 47.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.