

Abstract
Purpose
To determine whether there is a correlation among mutations in the cytochrome P450 1B1 gene (CYP1B1), the degree of angle dysgenesis observed histologically, and disease severity in congenital glaucoma.
Methods
Direct DNA sequencing was utilized to screen six unrelated children with congenital glaucoma, each set of parents, and all siblings for CYP1B1 mutations. Specimens of the anterior chamber angle obtained at trabeculectomy were examined histologically to identify abnormalities of the aqueous outflow pathway. CYP1B1 mutations were correlated with both the degree of angle dysgenesis and the patient’s disease severity (age at diagnosis, difficulty in achieving intraocular pressure control).
Results
Four of the six patients (66.7%) were compound heterozygotes for mutations in the CYP1B1 gene. Seven of the eight CYP1B1 mutations were identified, including two novel mutations (R117P, C209R) and five others previously described (G61E, R368H, R390H, E229K, 4340delG). The cases were divided based on histological phenotype into categories of (1) severe goniodysgenesis highlighted by the agenesis of Schlemm’s canal (two patients), (2) moderate goniodysgenesis characterized by the presence of a band of collagenous tissue in the trabecular meshwork and/or the juxtacanalicular tissues (three patients), and (3) mild goniodysgenesis with deposition of a mucopolysaccharide material in the juxtacanalicular tissue (one patient). CYP1B1 mutations were identified in both cases of severe angle dysgenesis and two of three cases of moderate dysgenesis. Disease severity closely correlated with the degree of angle dysgenesis.
Conclusion
The majority of cases in the cohort had compound heterozygous CYP1B1 mutations. Specific CYP1B1 mutations may be associated with severe or moderate angle abnormalities.
INTRODUCTION
Congenital glaucoma, which presents in neonates and during the infantile period, is most often without a manifest etiology. Patients may present with tearing, photophobia, corneal clouding, and buphthalmos as a result of high intraocular pressure (IOP). The incidence of congenital glaucoma varies among different geographic locations and ethnic groups, with the highest recorded incidence found in the Gypsy population of Slovakia (1:1,250), followed by the general populations of the Middle East (1:2,500) and the Western nations (1:10,000).1–4 The inheritance pattern for congenital glaucoma is most commonly autosomal recessive with incomplete penetrance.1,5,6
Linkage studies have shown genetic heterogeneity and have mapped three loci for primary congenital glaucoma to chromosomes 2p21 (GLC3A),7 1p36 (GLC3B),8 and 14q24 (GLC3C) (Stoilov IR, ARVO Meeting, 2002, Abstract). Molecular screening of the primary congenital glaucoma families linked to the 2p21 locus has determined that mutations in the cytochrome P450 1B1 (CYP1B1) gene are responsible for this phenotype. Cytochrome P450 1B1, which is a membrane-bound protein that functions as a monooxygenase enzyme, has been identified in 15 different nonocular human tissues in addition to its expression in cells of the trabecular meshwork, iris, ciliary body, and retina.9–11 However, the exact role of this gene in anterior segment development has yet to be determined. Mutations in the CYP1B1 gene occur in more than 85% of familial and 25% of sporadic cases of primary congenital glaucoma.11–13
Alterations in enzymatic stability and activity as a result of mutations in CYP1B1 have been demonstrated in vitro in prior studies.14 Within the CYP1B1 gene, a transmembrane domain is present at the amino (N) terminal, whereas the highly conserved j-helix, k-helix, and heme-binding regions are present at the carboxy (C) terminal.15 A proline-rich hinge region near the N-terminal permits flexibility in the overall protein structure. The highly conserved nature of these regions in the enzyme underscores their importance to overall function, and mutations in the conserved regions are presumed to lead to more severe changes in phenotype secondary to a greater impact on enzyme function.11,16
Prior morphological studies on congenital glaucoma patients have demonstrated various alterations affecting the aqueous outflow pathway, which are not apparent clinically during gonioscopy.17–20 The correlation between the outflow pathway abnormalities in congenital glaucoma and the different CYP1B1 mutations has yet to be described. This study represents the initial report correlating genotypes, namely CYP1B1 mutations, in a small cohort of congenital glaucoma patients with phenotypes, in terms of both the extent of goniodysgenesis detected histologically and the disease severity observed clinically.
METHODS
After obtaining permission from the institutional review board, trabeculectomy specimens, obtained during the course of management of patients with congenital glaucoma, were examined. The patients represented a consecutive series of children who presented at the University of California–San Francisco (UCSF) (J.A.A.) with a glaucomatous condition and required trabeculectomy in at least one eye for IOP control. Elements on examination necessary for inclusion included the presence of an open angle on gonioscopy, as well as factors leading to the diagnosis of glaucoma, namely elevated IOP, corneal enlargement, Haab’s striae, and optic nerve head cupping.
Peripheral blood samples were obtained from each of the patients, both of their parents, and all siblings for genetic testing (described in following section). Exclusion criteria included (1) patients with a known ocular or systemic syndrome associated with glaucoma, (2) patients whose parents were unavailable for peripheral blood screening and genetic analysis, and (3) patients for whom tissue specimens were unavailable.
MUTATION SCREENING
Peripheral blood samples were obtained from each patient, the parents, and all siblings. Genomic DNA was prepared from peripheral blood (MS, IS). The translated regions of the CYP1B1 gene were amplified through RT-PCR using three primer fragment pairs. Set 1:1F, 5′ (nt3676)- TCTCCAGAGAGTCAGCTCCG -3′ and 1R, 5′ (nt4461)-GGGTCGTCGTGGCTGTAG-3′ [786-bp]; Set 2: 2F, 5′ (nt4199)-ATGGCTTTCGGCCACTACT-3′ and 2R, 5′ (nt4985)-GATCTTGGTTTTGAGGGGTG-3′ [787-bp]; Set 3: 3F, 5′ (nt7740)-TCCCAGAAATATTAATTTAGTCACTG-3′ and 3R, 5′ (nt8624)-TATGGAGCACACCTCACCTG-3′ [885-bp].
PCR amplification was performed in a 50 μL volume containing 1.0 μL of 20 μM stock solution of each primer oligonucleotide and 100 ng genomic DNA, 50 mM KCl, 10 mM Tris-HCl (pH 9.3), 1.5 mM MgCl2, 0.2 mM of each dNP, 1U TaqDNA polymerase (platinum Taq; GibcoBRL, Grand Island, New York), and 10% dimethyl sulfoxide (DMSO, fragments sets 1 and 2). Thermocycling was performed (Gene Amp PCR Thermocycler, 9700; PE Applied Biosystems, Foster City, California) with initial denaturing at 94°C for 2 minutes. This was followed by 35 cycles each consisting of 30 seconds denaturation at 94°C, 30 seconds annealing at 55°C, and 1 minute extension at 72°C. The process was concluded with a final extension of 7 minutes at 72°C. Column purification of the PCR fragment was performed with a Qiaquick kit (Qiagen, Valencia, California) according to the manufacture’s protocol. Dye terminator sequencing (Big Dye Terminator kit; PE Applied Biosystems) was also performed according to the manufacturer’s recommendations. The analysis of the sequencing was performed using an automated DNA sequencer (ABI-377; PE Applied Biosystems) with the assistance of computer software (Sequencher, ver. 3.11; Gene Codes Corp, Ann Arbor; Michigan).
MORPHOLOGIC ANALYSES OF TRABECULECTOMY SPECIMENS
The tissues at the anterior chamber angle were dissected and excised during a routine trabeculectomy, using angled-Vannas scissors and Colibri forceps. Each tissue sample contained the anterior chamber angle including the iris root, trabecular meshwork, peripheral cornea at Schwalbe’s line, and the region where Schlemm’s canal is located. Each specimen was fixed within seconds after excision in a standard 1:1 mixture of 4% paraformaldehyde and 2% glutaraldehyde buffered in sodium cacodylate at pH 7.3. Specimens were processed for light and electron microscopy with dehydration in graded alcohol solution, post-fixed in osmium tetraoxide, and embedded in Araldite for sectioning and viewing, according to methods used routinely in our laboratory.21,22 For scanning electron microscopy, smaller pieces were also cut from each specimen, and after undergoing critical point drying and sputter coating with Au/Pd, the specimens were photographed using a JEOL JSM-840A instrument.
RESULTS
Six patients (three males and three females) met the criteria to be included in the study. The parents of each of the probands were without known consanguinity. The probands, parents, and all siblings were screened for CYP1B1 mutations (Table 1). The siblings were unaffected by glaucoma or by CYP1B1 mutations in all but one family. In this family (case 2), the three children, including the proband, carried the same CYP1B1 mutations. However, only two of the three children developed congenital glaucoma. Each of the patients in the study was diagnosed with bilateral disease and open angles upon examination at UCSF, and each has been followed closely for several years during the postoperative period (J.A.A.) with target IOP control achieved in all cases.
TABLE 1.
CYP1B1 MUTATIONS IN CONGENITAL GLAUCOMA PATIENTS AND THEIR FAMILY MEMBERS
| CASE | ETHNICITY | LOCATION OF CYP1B1 MUTATIONS | REGION | FATHER | MOTHER | SIBLINGS |
|---|---|---|---|---|---|---|
| 1 | Asian | 1. R117P | 1. Exon II | 1. wt | 1. R117P | Sister: wt/wt |
| 2. R390H | 2. K-Helix | 2. R390H | 2. wt | Sister: wt/wt | ||
| 2 | Hispanic | 1. C209R | 1. Exon II | 1. wt | 1. C209R | Brother*: C209R/4340delG |
| 2. 4340delG | 2. upstream H-B | 2. 4340delG | 2. wt | Brother: C209R/4340delG | ||
| 3 | Middle Eastern | 1. R368H | 1. J-Helix | 1. wt | 1. R368H | No siblings |
| 2. G61E | 2. Hinge | 2. G61E | 2. wt | |||
| 4 | Caucasian-Asian | 1. wt | NA | 1. wt | 1. wt | Brother: wt/wt |
| 2. wt | 2. wt | 2. wt | ||||
| 5 | Caucasian | 1. E229K | 1. Exon II | 1. E229K | 1. wt | Brother: wt/wt |
| 2. Undetermined | 2. Undetermined | 2. wt | 2. Undetermined | |||
| 6 | Caucasian | 1. wt | NA | 1. wt | 1. wt | Sister: wt/wt |
| 2. wt | 2. wt | 2. wt | Sister: wt/wt |
Amino acids: C = cysteine, E = glutamate, G = glycine, H = histidine, K = lysine, P = proline, R = arginine; H-B = heme binding region; wt = wild-type.
Indicates sibling with congenital glaucoma.
GENOTYPE EXPRESSION: CYP1B1 MUTATIONS
CYP1B1 mutations were present in four of six patients (66.7%), and the mutations identified in the proband and their immediate family members are listed in Table 1. Given the autosomal recessive nature of the disease, the four affected individuals would be expected to have two different mutations in compound heterozygous cases or two identical mutations in homozygous cases. In three of four patients (cases 1, 2, and 3), each of the two mutations was identified. The corresponding single mutation and a normal allele were detected in each of the three corresponding sets of unaffected parents. In one patient (case 5), only one mutation was detected. A second mutation (presumed maternal origin) was not identified in either the patient or the patient’s mother, whereas the one identified mutation was present in both the patient and the patient’s unaffected father.
The locations of the CYP1B1 mutations within the coding regions of either exons II or III are listed in Table 1, with the mutations detected most commonly affecting highly conserved regions of the gene. Of the seven mutations detected, six (85.7%) represented missense mutations and one (14.3%) was a base pair deletion resulting in a frameshift. The identified CYP1B1 mutations included two novel mutations, R117P and C209R, and five previously reported mutations, R390H, R368H, G61E, E229K, and 4340delG.
The patients in this cohort represented a diverse ethnic group, and CYP1B1 mutations were detected in patients of Hispanic, Asian, Middle Eastern, and Caucasian origin (Table 1). No CYP1B1 mutations were found in unaffected siblings of cases 1, 4, 5, and 6. Case 2 is unusual in that all three children had the same two CYP1B1 mutations, C209R and 4340delG, yet only two of three siblings developed congenital glaucoma.
PHENOTYPE EXPRESSION: GONIODYSGENESIS
Distinct angle anomalies (goniodysgenesis) were identified in all six cases, and the degree of goniodysgenesis closely correlated with the severity of glaucoma in each case (Table 2). The patients were divided into three categories based on the extent of goniodysgenesis observed histologically.
TABLE 2.
CORRELATION OF SLIT-LAMP FINDINGS, ANGLE ANOMALIES, TREATMENTS, AND OUTCOMES OF THE CONGENITAL GLAUCOMA PATIENTS
| CASE | SEX | AGE AT DIAGNOSIS | SLIT-LAMP FINDINGS | GONIOSCOPY | ANGLE ANOMALIES | TREATMENTS | OUTCOMES |
|---|---|---|---|---|---|---|---|
| 1 | F | At birth | Corneal edema OU | Open, flat iris insertion, view limited by edema | SC agenesis, prominent uveoscleral pathway | Failed trabs OU, Ahmed OU | Stable, meds OU |
| 2 | F | At birth | Corneal edema OU | Open, view limited by corneal edema | SC agenesis, prominent uveoscleral pathway | Failed trabs OU, Ahmed OU | Stable, meds OU |
| 3 | M | 6 months | Corneal enlargement, edema, Haab’s striae OU | Open, flat iris insertion | Connective tissue band in JXT | Trab OU | Stable, no meds OU |
| 4 | M | 8 months | Corneal edema and enlargement OU | Open, view limited by corneal edema | Connective tissue band in JXT | OD: meds OS: failed Ahmed, Trab | Stable, meds OU |
| 5 | F | 7 months | Corneal edema OS, Haab’s striae OU | Open, flat iris insertion, iris hypoplasia | Connective tissue band in C-S | OD: Trab OS: Ahmed | Stable, no meds OU |
| 6 | M | 4 months | Corneal enlargement OU, Haab’s striae OS | Open, flat iris insertion | Amorphous material in JXT | OD: meds OS: Trab (closed tightly) | Stable, no meds OU |
C-S = corneoscleral meshwork; JXT =juxtacanalicular tissue; SC = Schlemm’s canal; Trab = trabeculectomy.
Type I: Severe Goniodysgenesis
Case 1
The anterior chamber angle of this patient is shown in Figure 1, in which the salient feature is the absence of Schlemm’s canal. Other abnormalities include a relative hypoplasia of the trabecular meshwork, demonstrated by the paucity in the number of both uveal cords (arrows) and corneoscleral bands (arrowheads). In the absence of Schlemm’s canal, the uveoscleral pathway (asterisks) is markedly enlarged and contains numerous channels filled with aqueous.
FIGURE 1.

Case 1. Severe goniodysgenesis (type I). Low-magnification light micrograph of trabeculectomy specimen showing the anterior chamber angle of the right eye. Only a few elements of the uveal cords (arrows) and the corneal scleral meshwork (arrowheads) are present. The ciliary muscle (CM) is prominent and separated from the sclera by a distended uveoscleral pathway (asterisks). There is no Schlemm’s canal apparent along the deep limboscleral tissues (×100).
Case 2
The anterior chamber angle in one eye of this patient is shown histologically in Figure 2. (The iris root was dragged forward during the embedding procedure, when the tissue is rotated continuously in a viscous epoxy embedding material. The iris came to rest near the trabecular meshwork tissues.) Inspection of the tissues within which Schlemm’s canal is usually located reveals that there is no perceptible vessel representing this canal anywhere within the deep corneolimbal tissues. The uveoscleral pathway in this second case of agenesis of Schlemm’s canal displays features similar to those described in case 1, consisting of a prominent pathway formed by numerous distended channels (Figure 2).
FIGURE 2.

Case 2. Severe goniodysgenesis (type I). Low-magnification light micrograph. The iris epithelia (arrows) and stroma (arrowheads) are apparent on the left side of this figure. The peripheral iris tissues came to rest against Schwalbe’s line (SL) during specimen preparation. The ciliary muscle (CM) is pointed out at two locations. The major abnormality consists of the lack of any structure that could be identified as representing Schlemm’s canal along the limboscleral tissues (×100).
CORRELATION OF PHENOTYPES AND GENOTYPES FOR TYPE I CASES
Agenesis of Schlemm’s canal and the presence of a hypertrophied uveoscleral pathway are the common histological features of cases 1 and 2. This phenotype is associated with a clinical course that is distinct from that manifested by each of the other four patients included in this study. Both type I patients presented at birth with bilateral corneal edema and very high IOPs (>40 mm Hg while on medical therapy). These two cases proved to be the most recalcitrant, requiring the performance of several operations in each eye, as well as the use of medical glaucoma therapy before achieving IOP control. The corneal edema persisted for months even after the IOP had been controlled surgically.
The genotypes of both of these patients consist of one mutation affecting a highly conserved region of the CYP1B1 gene as well as a novel mutation in exon II. In case 1, an R390H mutation (G8006A transition) is present, in which an arginine is replaced by a histidine in the conserved k-helix coding region. The second mutation, R117P (G4155C transversion), is novel and results in the substitution of a hydrophobic, neutral proline for a basic, positively charged arginine in the N-terminal region, which could have a significant impact on three-dimensional protein folding. In case 2, a frameshift mutation (4340del G) is present, which results in a premature stop codon upstream from the coding sequences for several conserved regions, most notably the heme-binding region. The C209R (T4430C transition) is the novel mutation in this patient leading to the replacement of an uncharged cysteine, containing a reactive sulfhydryl group, with a basic, charged arginine.
TYPE II: MODERATE GONIODYSGENESIS
Case 3
The histological appearance of the anterior chamber angle in this patient is shown in Figure 3, left. The key anomaly is the presence of a thickened connective tissue band extending along the juxtacanalicular tissues. This band (arrowheads) is shown at higher magnification in Figure 3, right. Note that the staining property of this band is similar to that of the adjacent corneoscleral tissues, as they are both formed by a relatively dense collagenous tissue. Additional outflow pathway anomalies include the presence of denuded and fused corneoscleral bands shown in Figure 3, right (asterisks). Schlemm’s canal is identifiable in both figures (arrows).
FIGURE 3.

Case 3. Moderate goniodysgenesis (type II). Left, Low-magnification light micrograph. A view of the trabeculectomy specimen is shown, with Schwalbe’s line (SL) pointed out anteriorly and a prominent ciliary muscle (CM) posteriorly. Note the presence of a very well-developed Schlemm’s canal (arrows). The trabecular meshwork is disorganized, and the juxtacanalicular tissues have a thickened tissue pointed out with asterisks (×100 magnification). Right, High-magnification light micrograph. The juxtacanalicular tissue (JXT) is pointed out with arrowheads and the lumen of Schlemm’s canal with arrows. The JXT is filled with a dense collagenous tissue (arrowheads), which closely resembles in its density those of the deep corneolimbal region, seen on the right side of Schlemm’s canal. The bands of the corneoscleral meshwork are disorganized and without an endothelial lining (asterisks) (×400).
Case 4
The dysgenesis detected in this case closely resembles that just described for case 3. Figure 4, left, is a higher-magnification view showing Schlemm’s canal (arrows), the associated juxtacanalicular meshwork (arrowheads), and a connective-tissue band (white asterisks) within the juxtacanalicular tissues. The osmiophilic central core tissues (black asterisks) that normally make up each of the corneoscleral bands are still discernible within this relatively acellular connective tissue band. By transmission electron microscopy (Figure 4, right), the collagenous nature of the band of tissue (asterisks) is apparent.
FIGURE 4.

Case 4. Moderate goniodysgenesis (type II). Left, High-magnification light micrograph. A view of the trabeculectomy specimen is shown. Note the lumen of Schlemm’s canal (arrows), the associated juxtacanalicular meshwork (arrowheads), and several bands of the outer corneoscleral meshwork, which are fused together (white asterisks). The corneoscleral bands are identifiable by the presence of a characteristic dense, osmiophillic tissue within the core of each band (black asterisks) (×400). Right, Transmission electron micrograph. This figure shows that the connective tissue seen in Figure 4, left, is made up of tightly packed collagen fibrils that form a dense extracellular environment (asterisks) (×4,000).
Case 5
The gonioscopic appearance of the anterior chamber angle in this patient is shown in Figure 5, left. The peripheral iris forms a darkened “scalloped” structure as it inserts into the ciliary muscle band. The major arterial circle of the iris can be seen coursing circumferentially along the ciliary body band (arrows). An occasional radial iris vessel can be seen extending toward the pupillary margin (arrowheads). Figure 5, center, shows the histological appearance of the anterior chamber angle in this patient. Schlemm’s canal is readily apparent (arrowheads), and a continuous band of a connective tissue is present (asterisks) in this case as well. However, its location, in contrast to cases 3 and 4, is within the external corneoscleral bands. Electron microscopy shown in Figure 5, right, demonstrates that this material is granular in nature (asterisks), in contrast to the fibrillar appearance observed in cases 3 and 4.
FIGURE 5.

Case 5. Moderate goniodysgenesis (type II). Left, Goniophotograph. There is marked iris hypoplasia as observed gonioscopically. Note that the iris forms a scalloping arrangement as it inserts into the ciliary muscle (white arrows). Elements of the major arterial circle of the iris (arrowheads) can be seen within the ciliary body muscle. Some radial iris vessels are observed extending toward the minor arterial incomplete circle of the iris at the pupillary margin (asterisks). Center, low-magnification light micrograph. Anterior chamber angle of the specimen obtained at trabeculectomy. The histology is well correlated with the landmarks shown in the corresponding goniophotograph. The insertion of the iris into the ciliary muscle (CM) is very well demonstrated, with the specimen containing a portion of the iris epithelia (arrows). The lumen of Schlemm’s canal is indicated (arrowheads), and the salient feature, consisting of a connective tissue extending through the full length of the corneoscleral meshwork, is shown with asterisks (×100). SL = Schwalbe’s line. Right, electron micrograph. The granular tissues seen in Figure 5, center, are outlined with black ink markings and pointed out with asterisks (×3,700).
CORRELATION OF PHENOTYPES AND GENOTYPES FOR TYPE II CASES
Histologically, the salient feature common to each of the three patients with type II goniodysgenesis is the presence of an abnormal juxtacanalicular tissue, or the presence of an abnormal connective tissue band in the outer trabecular meshwork tissues near the juxtacanalicular tissues. Clinically, these three patients presented 6 to 8 months after birth with mild corneal haze, moderate IOP elevation, some corneal enlargement, and cupping of both optic nerves. The IOP elevation could be controlled in five of six eyes in the three patients either medically or with a single surgical procedure. In case 3, the IOP was controlled following bilateral trabeculectomies. In case 4, the patient was treated with a trabeculectomy in the left eye following a failed Ahmed valve and topical medical therapy alone in his right eye. In case 5, an Ahmed valve was placed in the patient’s left eye with a subsequent trabeculectomy in her right eye. Six years later, she remains with well-controlled IOPs bilaterally off glaucoma medications.
Case 3 was compound heterozygous for CYP1B1 mutations in highly conserved coding regions involving G61E and R368H mutations. The G61E mutation (G3987A transition) results in a negatively charged glutamate substituted for the uncharged glycine in the conserved hinge region. The R368H mutation (G7940A transition) leads to the substitution of basic, charged histidine for the basic, charged arginine at a conserved region in the J-helix coding region. In case 4, no CYP1B1 mutations were identified in the proband, his parents, or his unaffected sibling. In case 5, only a single E229K mutation (G4490A transition) was identified in this patient and the patient’s unaffected father. In this mutation, a basic, positively charged lysine replaces an acidic, negatively charged glutamate in exon II within a key region contributing to the three-dimensional structure of the protein. The presumed second CYP1B1 mutation in this patient was not identified in either exon II or exon III.
TYPE III: MILD GONIODYSGENESIS
Case 6
This patient had only a relatively mild angle anomaly consisting of the deposition of an extracellular substance within the juxtacanalicular tissue (Figure 6, asterisks), presumably representing glycosaminoglycans. This material has distinct staining properties compared to the nearby collagenous tissues of the cornea and trabecular meshwork (Figure 6).
FIGURE 6.
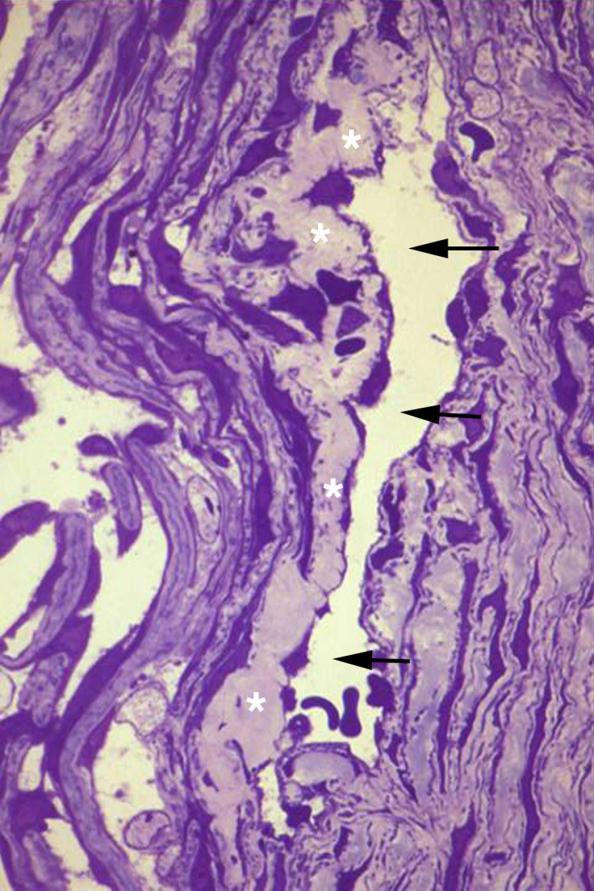
Case 6. Mild goniodysgenesis (type III). High-magnification light micrograph showing Schlemm’s canal (arrows) and the juxtacanalicular tissues (JXT) (asterisks). This specimen is different from those of the other five cases in that the only apparent abnormality consisted of the presence of a relatively dense granular tissue within the JXT (×400).
CORRELATION OF PHENOTYPE AND GENOTYPE FOR TYPE III CASE
Clinically, the only patient classified with this phenotype expression presented at the age of 4 months with an enlarged left cornea and bilateral elevated IOPs. A trabeculectomy was performed on the left eye and was closed tightly pending the expedited examination of the trabeculectomy specimen. The decision was made to leave the flap sutured tightly as no major abnormalities were apparent. The patient was treated with topical medications bilaterally for 2 years, after which time the progressive lowering of the IOP led us to discontinue all glaucoma medications. He is now 9 years old with normal IOPs and healthy optic nerves, though he has developed sensorineural hearing loss. No CYP1B1 mutations were present in this patient, his parents, or his unaffected siblings.
DISCUSSION
Underlying this study is the hypothesis that different combinations of CYP1B1 mutations may produce a range of structural alterations in the tissues of the conventional aqueous outflow pathway, including the trabecular meshwork and Schlemm’s canal. A corollary to this idea is that the degree of angle dysgenesis is congruous with the severity of the disease process in congenital glaucoma. The significant percentage of cases with genetic mutations and phenotypic abnormalities identified in this cohort perhaps compensates for the limited number of patients in this study. Detecting CYP1B1 mutations in two thirds of the cohort supports the concept that CYP1B1 mutations are responsible for the development of glaucoma in many children with congenital glaucoma. In addition, the goniodysgenesis in each of the six cases was readily apparent histologically and could account for the obstruction of aqueous outflow and the development of the glaucomatous process.
The phenotypes could be separated into three distinct categories based on histological features. Furthermore, the degree of dysgenesis observed histologically closely correlated with disease severity and the difficulty in achieving IOP control. In the type I category, with the absence of Schlemm’s canal, both patients presented at birth with congenital glaucoma. In addition, multiple surgeries and topical glaucoma medications were necessary to achieve and maintain IOP control. The distention of the uveoscleral outflow pathway in both cases with this phenotype might be expected, as this is the only route for aqueous outflow in patients lacking Schlemm’s canal. In the type II phenotype, in which a connective-tissue band is present within either the juxtacanalicular tissues or the outer trabecular meshwork, patients presented with congenital glaucoma between 6 and 8 months of age. IOP control was achieved with medications alone or a single operation in five of six (83.3%) of the eyes of the three patients in this group, and several years later four (66.7%) of six eyes are no longer using any topical medications. In the type III category, a single patient had a relatively normal outflow pathway with the exception of a mucopolysaccharide-type substance deposited in the juxtacanalicular tissue. The original trabeculectomy flap remained tightly sutured, and both eyes were treated for 2 years with topical medications alone. For the last several years, the patient has maintained normal IOP in both eyes despite being taken off all glaucoma medications. This patient provides a histological explanation for a previously described group of congenital glaucoma patients who experience spontaneous resolution of the glaucomatous process, potentially secondary to similar minor anomalies.23,24
The histological phenotypes identified here are consistent with those previously reported, though never systematically categorized. In trabeculectomy specimens from three patients, Anderson19 observed a “broad layer of collagen and amorphous material in the juxtacanalicular connective tissue,” consistent with the type II phenotype described here. Similarly, the majority of specimens from eight patients examined by Tawara and Inomata25 also had a deposition of fibrillar material containing bundles of collagen fibers within the outer trabecular meshwork. In their study, Schlemm’s canal was absent in one patient who developed early-onset glaucoma bilaterally, characteristic of type I cases. In addition, the electron micrograph of another case in this study of late-onset congenital glaucoma demonstrated a “homogeneous ground substance” in the outer meshwork, which closely resembles the type III phenotype.25 Of note, Barkan’s membrane was not present in any of the six cases described here, similar to the studies of both Anderson and Tawara. However, we have had the opportunity to correlate the gonioscopic appearance with the histological phenotype in patients with Barkan’s membrane (J.A.A., unpublished observations).
This study has demonstrated that multiple CYP1B1 mutations can be found in a heterogeneous group of patients with primary congenital glaucoma. The majority of the detected CYP1B1 mutations impacted highly conserved regions of the gene, and CYP1B1 mutations were identified in both of the cases of severe goniodysgenesis (type I) and two of the three cases of moderate dysgenesis (type II). The R390H mutation detected in case 1 lies within the k-helix core and makes up part of the Glu-X-X-Arg (387–390) motif, a highly conserved sequence among all members of the cytochrome P-450 family.11 Similarly, the frameshift mutation (4340 delG) in case 2 results in a premature stop codon upstream from the critical heme-binding region.26 The novel mutations identified in cases 1 and 2 were located in exon II and may possibly lead to changes in three-dimensional folding based on the different chemical properties of the substituted amino acids. The G61E mutation and R368H mutations in case 3 are also both within highly conserved regions, affecting the hinge region11,27 and J-helix,11 respectively. The E229K mutation found in patient 5 is located in exon II and, on the basis of prior reports, may also disrupt the three-dimensional helical structure.28,29
Several interesting observations emerged from the genetic analysis of this cohort. Incomplete penetrance was observed in the family of case 2 as each of the three children was compound heterozygous for both mutations (ie, each had identical CYP1B1 mutations), yet only two of the three siblings were affected (Figure 7). This incomplete penetrance, previously observed in Saudi Arabian and Indian populations, suggests the existence of a modifier locus, which may be an autosomal dominant gene separately inherited.12,16 The screening in this study was limited to the coding regions of the gene (exons II and III), and mutations of the promoter region may not have been identified. Also of note, the presumed second mutant allele in case 5, based on the autosomal recessive mode of inheritance, was not detected and may be located in either the promoter region of the CYP1B1 gene or in a separate gene, as suggested in other reports.26,29,30
FIGURE 7.

Pedigree of case 2. Corresponding DNA sequencing demonstrating the same two CYP1B1 mutations present in each of three offspring, only two of which developed congenital glaucoma.
The major weakness of this study is the small number of patients in this cohort, which makes direct genotype to phenotype correlations somewhat tenuous. Nevertheless, the observed phenotypic angle anomalies corresponding to particular CYP1B1 mutations were highly predictive of disease severity. Furthermore, the clinical course of some patients in this cohort corresponded to that described in prior studies of patients with the same mutations. For example, 11 (91.7%) of 12 patients previously reported with the 4340delG frameshift mutation (case 2) presented in the first month of life, each with severe, recalcitrant glaucoma.26 Additional studies examining different combinations of CYP1B1 mutations are ultimately necessary to provide an even better prediction of phenotype.
The cases in the series reported here demonstrate an anatomic basis for the elevation of IOP, indicating that in congenital glaucoma, it is the obstruction of the outflow pathway that leads to a pressure elevation and a secondary optic neuropathy. Similar obstruction of the outflow pathway has yet to be demonstrated for primary open-angle glaucoma. For this and other reasons, the prevailing notion among glaucoma specialists is that primary open-angle glaucoma represents a primary optic neuropathy in which IOP elevation serves as a major risk factor.31,32
An understanding of phenotype may provide a guide to glaucoma management in the future. For example, type I congenital glaucoma patients lacking Schlemm’s canal may require a filtration procedure at the outset of management. In contrast, in type II cases with a connective tissue band, a goniotomy knife may successfully disrupt this barrier and improve aqueous outflow. From a clinical point of view, the glaucoma specialist may be best able to determine a surgical intervention if the status of Schlemm’s canal is known. Currently, high-resolution ultrasound devices and spectral ocular coherence tomography devices are being tested to determine if they will allow one to image the lumen of Schlemm’s canal. Should these imaging devices prove to be both sensitive and specific, the demonstration of the presence or absence of Schlemm’s canal could be critical to surgical planning. In the future, genetic analysis may provide the glaucoma specialist with a useful tool to aid in the diagnosis of congenital glaucoma, the prediction of clinical course, and the development of tailored treatments.
PEER DISCUSSION
DR EDWIN M. STONE
I wish to congratulate Dr. Hollander and his co-workers on a very interesting study. This study illustrates both the overall importance of CYP1B1 in the pathogenesis of congenital glaucoma (two-thirds of their 6 patients were found to have mutations in this gene) as well as a number of important issues that face clinician scientists who wish to identify specific clinical features associated with specific genetic variations.
It may be helpful to begin by reviewing some ways that molecular diagnosis may prove useful for a patient with a frequently genetic condition like congenital glaucoma. First, molecular testing may allow a more specific diagnosis to be made and in some cases may also allow a more specific prognosis to be given. Also, as the knowledge of specific disease genes evolves, this knowledge may lead to a better understanding of disease mechanisms which in turn may allow specific types of treatment to be devised and matched to specific patients. Finally, molecular testing raises the possibility of presymptomatic treatment including various forms of prenatal and preimplantation intervention.
In a recent review of congenital glaucoma Drs. Sarfarazi and Stoilov1 summarized the evidence that 10–40% of congenital glaucoma is heritable in an autosomal recessive fashion. Thus, without molecular screening, parents with one affected child would have at most a (.4 × .25) = 10% chance of having another affected child with their next pregnancy. Since 87% of primary familial congenital glaucoma is associated with detectable mutations in the CYP1B1 gene1, if the first affected child in a family is screened for mutations and none are found, there would be at most a 2% chance of an affected child resulting from the next pregnancy. However, if two mutations with a high pathogenic probability are found in the proper configuration (i.e. one inherited from each parent) the risk can be as much as 12 times higher (25%).
A method for estimating the pathogenic probability of human mutations has been previously presented in some detail to this society.2 In the present context, it is perhaps sufficient to note that data from at least 96 ethnically matched control individuals are needed to be sure that a given variation observed in a patient is sufficiently rare to be a plausible explanation for a disease like primary congenital glaucoma that has a population prevalence of one in 10,000 people. It can also be useful to use data from studies of protein evolution summarized in a so-called blosum substitution matrix – to help decide whether mutations are more or less likely to be disease-causing.3 The majority of the mutations discussed in the present work are plausibly disease-causing by these criteria.
In this paper, Hollander and colleagues have begun the process of exploring the genotype-phenotype correlation of CYP1B1 and have provided preliminary data that specific variations may be associated with more severe anatomic features including the total absence of Schlemm’s canal. Although quite a bit of additional study will be necessary before this hypothesis can be fully tested, (at a minimum, more than one individual with a given genotype will need to be found to share a phenotype) the authors are to be congratulated for their great contributions to our understanding of the molecular genetics of glaucoma, and especially to our understanding of the present subject, congenital glaucoma.
REFERENCES
- 1.Sarfarazi M, Stoilov I. Molecular genetics of primary congenital glaucoma. Eye. 2000;14:422–428. doi: 10.1038/eye.2000.126. [DOI] [PubMed] [Google Scholar]
- 2.Stone EM. Finding and interpreting genetic variations that are important to ophthalmologists. Trans Am Ophthalmol Soc. 2003;101:437–484. [PMC free article] [PubMed] [Google Scholar]
- 3.Henikoff S. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
DR ALBERT W. BIGLAN
About 35 years ago Frank Costenbader, one of our members, made the observation that patients with cloudy corneas and congenital or primary infantile glaucoma in the first month of life were very difficult to manage. Your observations that patients have an absence of Schlemm’s canal fits our clinical experience. The histopathology and pathologic evaluation of your samples underscores the need for the ophthalmic pathologist. It is a shame that they are a diminishing resource. Ophthalmic pathologists are crucial in assisting us to better understand the disease and pathology causing the glaucoma.
DR VICTOR M. ELNER
Your histopathologic findings demonstrate clear-cut absence of Schlemm’s canal in some cases. I am concerned that some of the other findings may be due to artifacts that are present in all small biopsies. Splaying of muscle fibers or collagen fibers, or even the apparent density of elements within these structures, can vary depending on surgical trauma, fixation, osmolality of solutions, and other considerations. How did you address the issue of artifacts to ensure that the changes you report are not due to artifact, but rather true disease-associated alterations of the tissue?
DR JOSEPH CAPRIOLI
You indicated that the angle biopsy was done through the performance of trabeculectomy. Was trabeculectomy performed as the primary procedure here, and if so, why was that chosen instead of goniotomy or trabeculotomy? If it wasn’t the primary procedure, could previous procedures, such as angle surgeries, have some affect on your finding?
DR EDWARD L. RAAB
If you were able to make these genotypic correlations, would you then, for some cases, elect other than drainage procedures?
DR MARILYN B. METS
When I look at your histopathology, you have cornea, angle, iris, and ciliary body. How did you do this? What is the post-operative astigmatism in these children since it looks like you took out a sizeable piece of tissue?
DR HANS E. GROSSNIKLAUS
You need to take it to the next level by doing protein extraction, mass spectroscopy and laser capture microscopy looking at the mRNA levels, etc of these specimens. Are you considering those techniques in the future?
DR ALLAN J. FLACH
Do you think, after what you’ve seen there, that you might be able to take these children and do fluorophotometry and/or tonography, and anticipate what percentage of Schlemm’s canal and what percentage of uveoscleral flow might exist in a child without doing anything invasive. Might those physiological measurements enable your to choose a drug or procedure of choice?
DR DAVID A. HOLLANDER
We thank Dr Stone for his kind remarks and for his insightful analysis of our study. We hope to encourage other investigators to conduct studies in the future correlating histological angle anomalies with particular genetic mutations in patients with congenital glaucoma, as we believe this is a promising endeavor.
CYP1B1 mutations occurred in 4 out of 6 patients in our study, strongly supporting the concept that this gene plays a critical role in the development of congenital glaucoma. The severity and time of onset of the glaucomatous disease process is well correlated with the presence of anomalies, which are of a major nature as well. For example, a particular frameshift deletion mutation was detected in Patient # 2, who was afflicted with a major derangement involving the agenesis of Schlemm’s canal. This absence of Schlemm’s canal is a significant defect as it effectively prevents aqueous humor from reaching the episcleral venous circulation to exit the eye. In addition, the emissary episcleral venous system was also absent. It is important to note that the same frameshift deletion affecting our Patient # 2 has been described in 12 other cases in the literature. In 11 of these 12 cases, the disease process was severe, leading to presentation within the first month of life.1 Future studies involving this particular mutation may dispense with cumbersome histological evaluations as the agenesis of Schlemm’s canal can be documented, as we have shown in the oral presentation of this research, using high resolution ultrasound biomicroscopy.
Dr Biglan has underscored how he and Dr. Costenbader pointed out many decades ago that the time of disease onset is often indicative of disease severity in congenital glaucoma.2 The absence of Schlemm’s canal was observed in two patients in this study, though we have observed this finding in others since the study concluded. Both patients had mutations affecting highly conserved regions of the CYP1B1 gene.3 These patients appear to have a more severe phenotype than seen with other genotypes. Each presented in the first day of life with cloudy corneas, had disease that was recalcitrant to medications and required multiple surgeries to achieve IOP control, and had corneal edema which persisted for several months even after achieving IOP control.
Dr Elner is concerned about the possibility that the absence of Schlemm’s canal in the two cases presented may have resulted artifacticiously due to “splaying of muscle” “or collagen fibers.” As mentioned above, we found that the agenesis of Schlemm’s canal was associated with the absence of any episcleral venous channels. These channels are normally located in the vicinity of Schlemm’s canal at the limbus.4 Another possibility is that the canal was located rather posterior and outside of the surgical field from which the trabeculectomy specimen had been collected. In our paper, we point out that the agenesis of Schlemm’s canal has been demonstrated in previous studies for which the entire eyeball was available for study.5 We have also noted in other congenital glaucoma patients, including infants with aniridia and other conditions presenting with severe glaucoma at birth, the absence of Schlemm’s canal. In one case, the agenesis was present bilaterally as demonstrated in trabeculectomy specimens. Nevertheless, we made special arrangements to use a high-resolution ultrasound biomicroscopy instrument not yet available commercially to obtain multiple images from the limbus of one of the two cases with agenesis of Schlemm’s canal presented here. These studies confirmed our histological findings regarding agenesis of the canal of Schlemm, while showing the presence of the canal in other infants studied as normal controls.
To address the questions of Dr Caprioli, the primary surgery in these cases varied according to the status of the cornea and our ability to visualize the anterior chamber angle structures. In cases presenting with relatively clear corneas, we often perform a goniotomy in one eye and a filtration procedure in the other. In patients presenting with opaque corneas, we typically place an Ahmed valve in one eye and perform a trabeculectomy in the other. The cases in this series consisted of the one eye that underwent a trabeculectomy. Therefore, in none of the eyes were artifacts from prior surgical interventions considered to be responsible for the histological findings.
To answer Dr Raab, we believe that certain histological phenotypes may lend themselves to particular surgeries. For example, a filtration procedure perhaps should be the primary procedure performed in patients lacking Schlemm’s canal. Histological abnormalities, such as described with Type 2 with a connective tissue band, may be more amenable to goniotomy, though this remains to be determined in future work. In the future, being able to correlate genotypes with histological phenotypes may help guide the glaucoma specialist when planning surgical interventions for congenital glaucoma patients.
To answer Dr Metz, specimens were obtained at trabeculectomy, modified to permit the acquisition of samples containing tissues of the peripheral anterior chamber angle. Following a limbal-based conjunctival peritomy, an equilateral triangle based at the limbus and measuring 4 mm on each side was outlined in the episclera. A 67-Beaver blade was then used to dissect a partial thickness triangular scleral flap into clear cornea. The 67-Beaver blade was then used to outline a rectangular trabeculectomy block, with the long anterior side measuring roughly 3 mm. The short ends were then marked to connect with the sides of the triangle. After filling the anterior chamber with a viscoelastic material, a “Super-sharp” blade was passed along the long anterior side of the rectangle. An iridotomy was made with angled-Vannas scissors. The wound was then extended using the scissors along the short right side of the trabeculectomy block posteriorly until the iris root was visualized. The iridotomy incision was then extended in both directions, and the blade of the angled-Vannas scissors was placed behind the iris into the posterior chamber sulcus. This enables one to cut the iris root and the anterior tip of the ciliary muscle and produce a “goniectomy” specimen measuring roughly 3 × 2 mm. The flap was closed with 3 interrupted 10-0 Nylon sutures and the conjunctival incision was closed with 3 overlapping, interrupted 10-0 Nylon mattress sutures. The excised trabeculectomy block typically contained the anterior chamber angle including the iris root, trabecular meshwork, peripheral cornea at Schwalbe’s line, and the region normally occupied by Schlemm’s canal. Each of the patients has been managed in conjunction with our coauthor, Dr. D. Frederick, who is a pediatric ophthalmologist, and post-operative astigmatism has not been an issue with any of these patients. In fact, from a visual standpoint, most of these patients have done remarkably well. However, Dr. Metz’s point is well taken that unless the surgeon takes meticulous care to close the scleral flap well, in particular by placing the apical suture tightly, astigmatism is a potential drawback associated with trabeculectomy procedures.
In regards to Dr. Grossniklaus’ questions about using more specific protein extraction techniques, this is a very interesting suggestion and we will take that into consideration for future studies.
To address Dr Flach’s point about utilizing non-invasive techniques, the goal of such genotype-phenotype correlations is to eliminate the need for certain invasive modalities in the future. Some of the studies Dr. Flach suggests would be extremely difficult to perform in children. In this study, all samples for genetic analysis were obtained from peripheral blood. Buccal mucosal swabs are growing in popularity and are typically easier to perform in children for future studies.6 Some of the high-resolution ultrasound and OCT studies which we have described may also play a role in the future.
REFERENCES
- 1.Stoilov IR, Costa VP, Vasconcellos JP, et al. Molecular genetics of primary congenital glaucoma in Brazil. Invest Ophthalmol Vis Sci. 2002;43:1820–1827. [PubMed] [Google Scholar]
- 2.Costenbader FD, Kwitko ML. Congenital glaucoma. Clin Proc Child Hosp Dist Columbia. 1961;17:100–109. [PubMed] [Google Scholar]
- 3.Stoilov I, Akarsu AN, Alozie I, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am J Hum Genet. 1998;62:573–584. doi: 10.1086/301764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hogan MJ, Alvarado JA, Weddell JE. Histology of the Human Eye Philadelphia: W.B. Saunders Company, 1971:112–259.
- 5.Tawara A, Inomata H. Developmental immaturity of the trabecular meshwork in congenital glaucoma. Am J Ophthalmol. 1981;92:508–525. doi: 10.1016/0002-9394(81)90644-9. [DOI] [PubMed] [Google Scholar]
REFERENCES
- 1.Francois J. Congenital glaucoma and its inheritance. Ophthalmologica. 1980;181:61–73. doi: 10.1159/000309028. [DOI] [PubMed] [Google Scholar]
- 2.Gencik A, Gencikova A, Ferak V. Population genetical aspects of primary congenital glaucoma. I. Incidence, prevalence, gene frequency, and age of onset. Hum Genet. 1982;61:193–197. doi: 10.1007/BF00296440. [DOI] [PubMed] [Google Scholar]
- 3.Jaffar MS. Care of the infantile glaucoma patient. In: Reineck RD, ed. Ophthalmology Annual New York: Raven Press; 1988:15.
- 4.Wagner RS. Glaucoma in children. Pediatr Clin North Am. 1993;40:855–867. doi: 10.1016/s0031-3955(16)38592-3. [DOI] [PubMed] [Google Scholar]
- 5.Gencik A. Epidemiology and genetics of primary congenital glaucoma in Slovakia. Description of a form of primary congenital glaucoma in gypsies with autosomal-recessive inheritance and complete penetrance. Dev Ophthalmol. 1989;16:76–115. [PubMed] [Google Scholar]
- 6.Bejjani BA, Lewis RA, Tomey KF, et al. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. Am J Hum Genet. 1998;62:325–333. doi: 10.1086/301725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarfarazi M, Akarsu AN, Hossain A, et al. Assignment of a locus (GLC3A) for primary congenital glaucoma (buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics. 1995;30:171–177. doi: 10.1006/geno.1995.9888. [DOI] [PubMed] [Google Scholar]
- 8.Akarsu AN, Turacli ME, Aktan SG, et al. A second locus (GLC3B) for primary congenital glaucoma (buphthalmos) maps to the 1p36 region. Hum Mol Genet. 1996;5:1199–1203. doi: 10.1093/hmg/5.8.1199. [DOI] [PubMed] [Google Scholar]
- 9.Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (buphthalmos) in families linked to GLC3A locus on chromosome 2p21. Hum Mol Genet. 1997;6:641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- 10.Muskhelishvili L, Thompson PA, Kusewitt DF, et al. In situ hybridization and immunohistochemical analysis of cytochrome P450 1B1 expression in human normal tissues. J Histochem Cytochem. 2001;49:229–236. doi: 10.1177/002215540104900210. [DOI] [PubMed] [Google Scholar]
- 11.Stoilov I, Akarsu AN, Alozie I, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am J Hum Genet. 1998;62:573–584. doi: 10.1086/301764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bejjani BA, Stockton DW, Lewis RA, et al. Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Hum Mol Genet. 2000;9:367–374. doi: 10.1093/hmg/9.3.367. [DOI] [PubMed] [Google Scholar]
- 13.Sarfarazi M, Stoilov I. Molecular genetics of primary congenital glaucoma. Eye. 2000;14:422–428. doi: 10.1038/eye.2000.126. [DOI] [PubMed] [Google Scholar]
- 14.Jansson I, Stoilov I, Sarfarazi M, et al. Effect of two mutations of human CYP1B1, G61E and R469W, on stability and endogenous steroid substrate metabolism. Pharmacogenetics. 2001;11:793–801. doi: 10.1097/00008571-200112000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Graham-Lorence SE, Peterson JA. Structural alignments of P450s and extrapolations to the unknown. Methods Enzymol. 1996;272:315–326. doi: 10.1016/s0076-6879(96)72037-2. [DOI] [PubMed] [Google Scholar]
- 16.Panicker SG, Mandal AK, Reddy AB, et al. Correlations of genotype with phenotype in Indian patients with primary congenital glaucoma. Invest Ophthalmol Vis Sci. 2004;45:1149–1156. doi: 10.1167/iovs.03-0404. [DOI] [PubMed] [Google Scholar]
- 17.Maumenee AE. The pathogenesis of congenital glaucoma: a new theory. Trans Am Ophthalmol Soc. 1958;56:507–570. [PMC free article] [PubMed] [Google Scholar]
- 18.Maul E, Strozzi L, Munoz C, et al. The outflow pathway in congenital glaucoma. Am J Ophthalmol. 1980;89:667–673. doi: 10.1016/0002-9394(80)90286-x. [DOI] [PubMed] [Google Scholar]
- 19.Anderson DR. The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. Trans Am Ophthalmol Soc. 1981;79:458–485. [PMC free article] [PubMed] [Google Scholar]
- 20.Cibis GW, Tripathi RC, Tripathi BJ. Glaucoma in Sturge-Weber syndrome. Ophthalmology. 1984;91:1061–1071. doi: 10.1016/s0161-6420(84)34194-x. [DOI] [PubMed] [Google Scholar]
- 21.Alvarado JA, Betanzos A, Franse-Carman L, et al. Endothelia of Schlemm’s canal and trabecular meshwork: distinct molecular, functional, and anatomic features. Am J Physiol Cell Physiol. 2004;286:C621–634. doi: 10.1152/ajpcell.00108.2003. [DOI] [PubMed] [Google Scholar]
- 22.Alvarado J, Murphy C, Juster R. Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology. 1984;91:564–579. doi: 10.1016/s0161-6420(84)34248-8. [DOI] [PubMed] [Google Scholar]
- 23.Lockie P, Elder J. Spontaneous resolution of primary congenital glaucoma. Aust N Z J Ophthalmol. 1989;17:75–77. doi: 10.1111/j.1442-9071.1989.tb00490.x. [DOI] [PubMed] [Google Scholar]
- 24.Grehn F, Mackensen G. Riegers’ anomaly with signs of hydrophthalmia and spontaneous pressure regulation. Klin Monatsbl Augenheilkd. 1982;181:197–201. doi: 10.1055/s-2008-1055199. [DOI] [PubMed] [Google Scholar]
- 25.Tawara A, Inomata H. Developmental immaturity of the trabecular meshwork in congenital glaucoma. Am J Ophthalmol. 1981;92:508–525. doi: 10.1016/0002-9394(81)90644-9. [DOI] [PubMed] [Google Scholar]
- 26.Stoilov IR, Costa VP, Vasconcellos JP, et al. Molecular genetics of primary congenital glaucoma in Brazil. Invest Ophthalmol Vis Sci. 2002;43:1820–1827. [PubMed] [Google Scholar]
- 27.Yamazaki S, Sato K, Suhara K, et al. Importance of the proline-rich region following signal-anchor sequence in the formation of correct conformation of microsomal cytochrome P-450s. J Biochem (Tokyo) 1993;114:652–657. doi: 10.1093/oxfordjournals.jbchem.a124232. [DOI] [PubMed] [Google Scholar]
- 28.Michels-Rautenstrauss KG, Mardin CY, Zenker M, et al. Primary congenital glaucoma: three case reports on novel mutations and combinations of mutations in the GLC3A (CYP1B1) gene. J Glaucoma. 2001;10:354–357. doi: 10.1097/00061198-200108000-00017. [DOI] [PubMed] [Google Scholar]
- 29.Panicker SG, Reddy AB, Mandal AK, et al. Identification of novel mutations causing familial primary congenital glaucoma in Indian pedigrees. Invest Ophthalmol Vis Sci. 2002;43:1358–1366. [PubMed] [Google Scholar]
- 30.Mashima Y, Suzuki Y, Sergeev Y, et al. Novel cytochrome P4501B1 (CYP1B1) gene mutations in Japanese patients with primary congenital glaucoma. Invest Ophthalmol Vis Sci. 2001;42:2211–2216. [PubMed] [Google Scholar]
- 31.Weinreb RN. Toward understanding the optic neuropathy of glaucoma. Arch Ophthalmol. 1998;116:1102–1103. doi: 10.1001/archopht.116.8.1102. [DOI] [PubMed] [Google Scholar]
- 32.Weinreb RN, Levin LA. Is neuroprotection a viable therapy for glaucoma? Arch Ophthalmol. 1999;117:1540–1544. doi: 10.1001/archopht.117.11.1540. [DOI] [PubMed] [Google Scholar]