

Abstract
Purpose
To determine the effect of rapamycin on the fate of misfolded opsin associated with retinitis pigmentosa.
Methods
Stable cell lines separately expressing WT and P23H opsins and WT and ΔF508 CFTR were used. Cells were incubated with complete media or amino acid–depleted medium or in the presence of rapamycin. At various time points thereafter, quantitative opsin and CFTR immunoblotting was performed. Immunofluorescence and electron microscopy were also performed to observe the expression and colocalization of autophagy specific marker proteins with opsin or CFTR.
Results
Upon incubation with rapamycin, the levels of P23H opsin and ΔF508 CFTR were reduced more rapidly than in untreated controls while no observable changes in the amounts of WT opsin was seen. The autophagy specific marker proteins, Atg7, Atg8 (LC3), and LAMP-1, which associate with autophagic vacuoles, colocalized with P23H opsin. A dramatic increase in the immunofluorescence signals of Atg7, LC3, and LAMP-1 was observed. All three of these proteins were found to decorate P23H opsin, suggesting that autophagy may be directly responsible for the clearance of this protein. Also, it was determined that neither the unfolded protein response nor the heat shock response was induced upon rapamycin-associated degradation of P23H opsin.
Conclusions
These data suggest that rapamycin induces the loss of P23H opsin and ΔF508 CFTR from the cell under the experimental conditions described. Concomitantly, there is increased expression and colocalization of autophagy marker proteins with P23H opsin. Immunogold electron microscopic studies demonstrate autophagic vacuoles clustered in physical proximity to the aggregates of P23H opsin, suggesting that some of the loss of P23H is related to the induction of autophagy. Thus, rapamycin may be useful to clear misfolded proteins associated with retinal degeneration.
INTRODUCTION
Retinitis pigmentosa is a debilitating neurodegenerative disease of the retina characterized initially by night blindness and progressing to severe tunnel vision or complete blindness. Retinitis pigmentosa is one of the most common inherited eye diseases, with an incidence of roughly 1:3000. Mutations in more than 30 distinct genes have been linked to the disease (www.sph.uth.tmc.edu/Retnet, www.retina-international.org). Many of these genes encode proteins unique to rod photoreceptor cells, which are required for night and peripheral vision. Production of these mutant proteins is thought to cause rod cell dysfunction and death, resulting in night blindness and tunnel vision.
In North America, the most common form of retinitis pigmentosa is caused by mutations in rhodopsin, an integral membrane glycoprotein consisting of the polypeptide opsin and a single covalently bound molecule of the chromophore, 11-cis retinal (vitamin A). Rhodopsin is a G-protein coupled receptor that functions as the primary photoreceptor molecule for dim light vision and is accordingly the most abundant protein in the rod cell (~108 molecules per cell). In this regard it is both a photoreceptor and an important structural protein of the rod cell. More than 140 rhodopsin (www.retina-international.org) mutations have been associated with retinitis pigmentosa since the opsin gene was linked to the disease in 1990.1 Previous studies demonstrate that most of the P23H opsin expressed is misfolded and retained in the cell.2,3 This protein is eventually degraded by the ubiquitin-proteasome system, one of the two major degradation pathways in the cell.4,5 If the mutant opsin is not degraded by the ubiquitin-proteasome system, then it aggregates in the cytosol.2–5
Besides the opsin mutants associated with retinitis pigmentosa, mutants of the cystic fibrosis transmembrane regulator (CFTR), another polytopic integral membrane glycoprotein, can cause a protein conformational disease. CFTR contains 12 transmembrane segments and functions as a chloride channel at the apical membrane of epithelial cells. Although many mutations have been identified, 70% of CF patients harbor the ΔF508 mutation in at least one allele.6 Similar to P23H, a major fraction of ΔF508 CFTR neither matures nor traffics to the plasma membrane. Most of it exists as a core glycosylated intermediate in the endoplasmic reticulum (ER). This misfolded protein is recognized by the ER quality control system and is targeted for degradation by the proteasome.7–10
WORKING MODEL OF RHODOPSIN RETINITIS PIGMENTOSA
Despite considerable efforts over the past 15 years to understand how these mutations cause retinitis pigmentosa, there has been relatively little progress toward developing drugs that can ameliorate the disease. A rational approach to developing therapies for retinitis pigmentosa requires a working model that explains the disease at the molecular, cellular, and whole animal level. A working model (Figure 1) is that most mutations associated with rhodopsin retinitis pigmentosa cause a protein conformational disease, in which protein misfolding results in the death of cells that produce it and the deterioration of tissues that contain these cells. Below, the essential elements of this model and the potential sites for therapeutic intervention are discussed.
FIGURE 1.
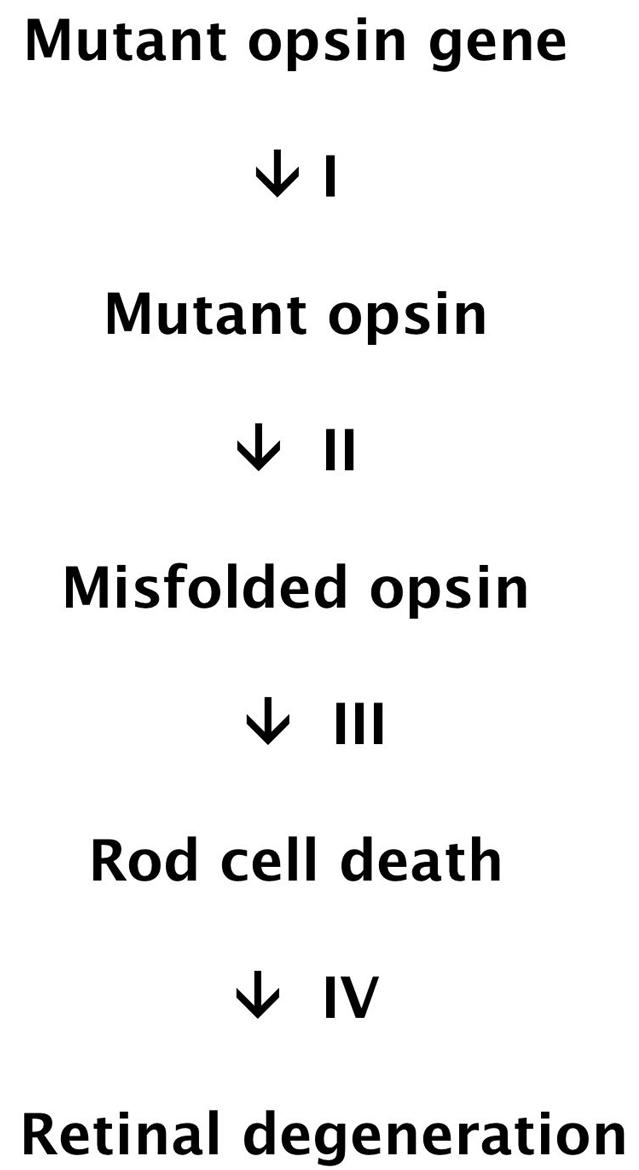
Working model of rhodopsin retinitis pigmentosa as a protein conformational disease.
STEP I. SYNTHESIS OF MUTANT OPSIN
Rhodopsin retinitis pigmentosa is typically an autosomal dominant disease, in which the mutant opsin gene is heterozygous with wild type. Two hypotheses have been proposed to explain the dominant effects of mutant opsin production. In the first, retinitis pigmentosa results from “gain-of-function” mutations in opsin that cause the protein to be toxic to rod cells. In the second, the mutant protein is not cytotoxic, but interferes with wild-type opsin production. In this “dominant-negative” model, decreasing wild-type opsin production below a threshold value results in rod cell death. Although the gain-of-function hypothesis is widely accepted, the dominant-negative model has not been ruled out.
Most rhodopsin mutations associated with retinitis pigmentosa are single amino acid substitutions (Figure 2), although frameshift mutations and in-frame deletions have also been documented. The mutations are distributed throughout the protein, suggesting that their predominant effect is on global protein structure. There are no examples of mutations in the opsin gene that affect mRNA splicing without also altering the polypeptide, or mutations in the opsin promoter that alter mRNA levels. Thus, the disease is caused by synthesis of the mutant protein rather than defects in opsin gene transcription, mRNA processing, or translation. Small molecules that target these reactions may not be specific for the mutant and most likely would also affect the levels of the WT protein as well. Thus, intervention at this step might even hasten the demise of photoreceptor cells.
FIGURE 2.

Secondary structural model of human rhodopsin based on the crystal structure of the bovine protein. Single amino acid substitutions at positions shown in yellow account for most of the rhodopsin mutations associated with retinitis pigmentosa. The approximate location of the lipid bilayer is shown in gray. Sites of glycosylation (hexagons) and palmitoylation (zig-zags) are indicated.
STEP II. OPSIN MISFOLDING
Opsin is synthesized by ribosomes docked on the ER membrane of the rod cell (Figure 3). By analogy with other integral membrane proteins, wild-type opsin folding is thought to be initiated during translation and to continue after translation is completed. The folded protein transits from the ER through the Golgi apparatus to the plasma membrane of the rod cell inner segment. The protein then crosses the connecting cilium, a bridge between the inner and outer segments, and is ultimately distributed in flattened membrane vesicles (discs) of the outer segment, where it interacts with other components of the visual transduction machinery.
FIGURE 3.

Schematic of the rod cell indicating critical sites for rhodopsin biogenesis and the arrangement of rhodopsin molecules in a rod outer segment disc. ER = endoplasmic reticulum.
Evidence for opsin misfolding in retinitis pigmentosa was first obtained from the finding that many mutant opsins expressed heterologously in cell culture are found at low levels in the plasma membrane and accumulate intracellularly at higher levels than wild-type opsin. Two classes of misfolding were distinguished: IIa mutant opsins (Class II of Khorana and coworkers) fail to bind retinal, whereas IIb (Khorana Class III) opsins bind retinal at low levels.2,3 In animal models, Class IIb opsins localize to both inner and outer segments.11,12 These mutant proteins are thought to have lower thermal stability consistent with misfolding, as evidenced by thermal bleaching studies of P23H rhodopsin.13,14 Class IIa and IIb mutants probably represent differing degrees of the same, fundamental defect: a decrease in the stability of the native, retinal-binding conformation of opsin.
Not all opsin mutations associated with retinitis pigmentosa cause misfolding. Class I mutant opsins are expressed at normal levels and bind 11-cis retinal to the same extent as wild-type opsin. In mouse and pig models of the disease, the Class I mutants P347S and Q344ter accumulate in both the rod inner and outer segments, indicating defects in transport across the connecting cilium.15–17 Class I mutations are largely clustered at the opsin C terminus, the last six residues of which constitute a post-Golgi sorting signal recognized by the transport machinery.18,19 Thus, loss of a functional site essential for opsin trafficking rather than disruption of global folding appears to explain Class I mutants. Another class corresponding to mutations at Arg135 appears to increase the affinity between opsin and its regulatory protein arrestin, resulting in increased endocytosis of the mutant proteins.20 Although these additional classes suggest that opsin misfolding is not the only path to disease, they are limited to small regions of the protein (Figure 2). Most opsin mutations probably cause retinitis pigmentosa by misfolding.
Step II represents a potential site for therapeutic intervention. One set of small molecules could serve as stabilizers of the mutant misfolded protein, ie, pharmacological chaperones. This approach was clearly demonstrated for P23H opsin by Noorwez and associates.13,14 A second set of molecules could alter the degradation environment of the cell allowing for a more favorable intracellular milieu for the folded mutant protein. if, indeed, there is excess steady-state quantities of the mutant protein, then perhaps stimulating the selective degradation of these molecules would be beneficial to the cell.
STEP III. ROD CELL DEATH
Apoptotic cell death is thought to be a common feature of retinal degenerative diseases, including rhodopsin retinitis pigmentosa. Rod cell apoptosis has been documented in mice expressing mouse opsin carrying the retinitis pigmentosa mutations P347S or Q344ter and in fruit flies expressing Drosophila Rh1P37H opsin, which carries a mutation analogous to human P23H opsin.12,21,22 Although the evidence for apoptosis in rhodopsin retinitis pigmentosa is clear, the signaling pathway(s) linking production of mutant opsins to cell death is poorly understood. One hypothesis is that rod cells die because of oxygen toxicity resulting from constitutive activation of the rhodopsin signal transduction cascade.23 However, this hypothesis is contradicted by evidence that K296E opsin, which is constitutively active in vitro, is fully bound by arrestin in vivo and thereby prevented from activating the visual transduction cascade.24
An alternative explanation, which we favor, is that opsin aggregates are responsible for cell death. As mentioned above, large cytosolic aggregates are correlated with production of mutant opsins. These aggregates may be directly toxic to the cells by sequestering essential cytosolic factors, such as components of the ubiquitin-proteosome machinery. However, recent studies suggest that similar inclusions in other protein conformational diseases are nontoxic or even cytoprotective.25 Thus, attention has shifted to the possibility that smaller aggregates of misfolded proteins may cause disease. For proteins such as rhodopsin that are synthesized in the secretory pathway, aggregation in the ER may lead to apoptosis, perhaps by signaling through the ER stress pathway.25 Identifying the form of mutant opsin that initiates apoptosis and elucidating the apoptotic signaling pathway are active areas of research. There could be a class of molecules that block apoptosis—the final common pathway that leads to photoreceptor cell death in retinal degenerations.
STEP IV. RETINAL DEGENERATION
The events following rod cell death that lead to retinal degeneration are varied and complex. Clinical findings indicate a broad range of retinitis pigmentosa severity, with some individuals showing severe loss within the first decade while others present with mild disease after the fifth or sixth decade. In some affected individuals, rod cell loss is accompanied by loss of cone photoreceptors, and in others cones are spared. These differences may be due in part to differences in the nature of the mutation, as demonstrated for P23A and P23H patients.26 However, genetic and environmental factors such as diet and light exposure undoubtedly also affect the course of the disease. Identifying the factors that affect the severity of the disease is a separate important area of research.
SUMMARY OF WORKING MODEL
Although aspects of rhodopsin retinitis pigmentosa are understood at the molecular and cellular level, there remain large gaps in our understanding. The fate of misfolded opsin in the cell and the mechanism of toxicity of the mutant proteins are major issues to be resolved.
One small molecule that may potentially be useful in affecting misfolded P23H opsin is rapamycin, a multipotent drug that inhibits the mammalian target of rapamycin (mTOR), a protein serine-threonine kinase that regulates many cell processes, including transcription, translation, cell growth, and autophagy. Besides proteasomal degradation, autophagy is the other major cellular pathway for protein degradation. It can be stimulated by a variety of stimuli, including amino acid starvation, aggregation of misfolded proteins, and accumulation of damaged organelles. Autophagy appears to be a largely nonselective process, with a few exceptions like specific removal of peroxisomes in yeast and in rat hepatocytes, under conditions where peroxisomes are functionally redundant.27–33 Other examples of selective autophagy are the elimination of smooth ER and mitochondria.34–36 Autophagy has been shown to play a role in removing aggregates of peripheral myelin protein 22 (PMP22), a short-lived Schwann cell membrane protein.37 Ravikumar and associates38 have shown the aggregate-prone polyglutamine and polyalanine expanded proteins associated with Huntington’s disease are also substrates for autophagy. Inhibition of mTOR induced autophagy and in turn reduced the toxicity of the mutant Huntington proteins in fly and mouse models of Huntington’s disease. Autophagy has also been shown to contribute to the elimination of the wild-type olfactory receptor mOREG that accumulates within the ER. When the autophagy inhibitors, 3-methyladenine (3MA) or bafilomycin A1, were added independently to cells, Lu and associates39 observed an increase in number of cells containing aggregates.
On the basis of the previously discussed observations with soluble, aggregated, misfolded proteins, this study determined if rapamycin could decrease the cellular amounts of misfolded integral membrane proteins and correlated it to the induction of autophagy.
METHODS
MAMMALIAN CELL CULTURES
HEK293 tetracycline-inducible stable cell lines expressing WT or P23H opsin have been described previously and were grown at 37°C in the presence of 5.0% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Sigma) with antibiotic-antimycotic solution (Invitrogen), blasticidin (Invitrogen), and zeocin (Invitrogen). Opsin synthesis was induced by addition of tetracycline (1 μg/mL). Baby hamster kidney (BHK) cell lines stably expressing the wild type and ΔF508 CFTR variant with a C-terminal HA epitope (CFTR-HA) were provided by Dr G. Lukacs.40 The cells were grown in DMEM/F12 (Invitrogen) 1:1 ratio containing 10% FBS at 37°C in the presence of 5.0% CO2.
INCUBATION WITH RAPAMYCIN
Autophagy was induced in cells by culturing the cells either in medium lacking amino acids and serum or in medium containing rapamycin (50 nM), or both, following tetracycline washoff. After 0, 2, 6, and 12 hours of treatment, cells were lysed in phosphate buffer containing 1% n-dodecyl-β-maltoside (Anatrace) in the presence of protease inhibitors (complete protease inhibitor mixture tablets; Roche Molecular Biochemicals) for 1 hour at 4°C. The lysates were centrifuged at 36,000 rpm in a Beckman ultracentrifuge for 10 minutes at 4°C. The supernatant was collected and immunoblotting was performed.
SDS GEL ELECTROPHORESIS AND IMMUNOBLOTTING
Cell lysates were electrophoresed on 10% SDS polyacrylamide gels and transferred onto Immobilon-NC (Millipore) nitrocellulose membranes. Membranes were incubated at room temperature for 1 hour with blocking buffer (Odyssey, Licor) diluted 1:1 in PBST (PBS with 0.1% Tween 20, pH 7.4), followed by incubation for 1 hour with the primary antibody. The blots were washed three times for 5 minutes each in PBST, and incubated for 1 hour with IRDye800-conjugated secondary antibody (Rockland, Inc). Finally, the membranes were again washed three times with PBST and scanned in an Odyssey infrared scanner (Licor). Primary antibodies included antibodies to opsin (University of British Columbia), HA-tag (Covance), phosphorylated mTOR (Upstate), calnexin, hsp70 (Stressgen), Bip (BD Pharmingen), and tubulin (Sigma).
IMMUNOFLUORESCENCE
Cells grown on glass coverslips were fixed in 4% p-formaldehyde (Electron Microscope Sciences). Following quenching with 50 mM NH4Cl, cells were washed with PBS and incubated for 1 hour with primary antibody at room temperature. Cells were washed five times in PBS and incubated with secondary antibody (TRITC- and FITC-conjugate) for 1 hour. The cells were washed again and mounted with Vectashield containing DAPI. Primary antibodies included antibodies to Atg7 (20) Atg8, lysosomal associated membrane protein 1 (LAMP-1) (27), opsin (University of British Columbia), and HA-tag (Covance). Immunofluorescence was observed using a Zeiss Axiophot microscope.
ELECTRON MICROSCOPY
Both WT and P23H opsin-expressing cells were grown on ACLAR sheets in a 24-well plate. Opsin production was induced by the addition of tetracycline for 48 hours, and autophagy was induced using starvation or rapamycin treatment for 6 hours after tetracycline removal. Following a wash with PBS, cells were fixed with 2% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 for 30 minutes at 4°C and processed for CMPase cytochemistry as previously described.41 Morphometric quantification of vacuoles (AVs) was done on 20 electron micrographs per condition using Image J software. Analysis with the t test was performed, and P value (two-tailed significance) under or equal to 0.05 was considered significant.
IMMUNOGOLD LABELING
The cells were grown in a monolayer on ACLAR sheets and fixed in 0.1% glutaraldehyde and 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 3 hours at room temperature. Fixed cells were dehydrated to 98% ethanol, embedded in LR White, and polymerized at 40°C. Ultrathin sections (60 to 70 μm) were collected on formvar-coated nickel grids. Sections were first etched with saturated sodium periodate (Sigma) at room temperature for 3 minutes. The grids were preincubated with 0.1% Tween 20 in PBS and blocked with 50 mM NH4Cl in PBS and in blocking solution. Sections were incubated with anti-opsin antibodies (1D4, B6-30, and R2-15, 1:1:1) diluted 1:500 in blocking solution at 4°C for 24 hours and washed once in PBS and twice in a mixture of 0.1% ovalbumin, 0.5% cold-water fish gelatin, 0.01% Tween 20, and 0.5 M NaCl in 10 mM phosphate buffer (pH 7.3). The sections were incubated for 2 hours with goat anti-mouse IgG conjugated to gold, diluted in 0.1% ovalbumin, 0.5% fish gelatin, 0.01% Tween 20, and 0.5 M NaCl in 10 mM phosphate buffer (pH 7.3). Washed sections were post-fixed in 2% glutaraldehyde for 10 minutes and air-dried. The grids were then washed in distilled water and stained with 2% ethanolic uranyl acetate for 10 minutes before observation by JEOL 100 CX electron microscope.
RESULTS
HEK293 cells that stably expressed either WT or P23H opsin behind a tetracycline-dependent promoter were incubated in the presence of tetracycline. After 48 hours, the tetracycline was removed to halt opsin synthesis and the cells incubated in nutrient-enriched medium (fed), amino acid and serum-depleted medium (starved), nutrient-enriched medium with rapamycin, or starved medium with rapamycin. At 0 to 12 hours, the cells were solubilized and opsin levels determined by immunoblotting. The levels of WT opsin remained essentially unchanged under these conditions (Figure 4, A and B). In contrast, the levels of P23H opsin decreased dramatically when the cells were incubated in starvation media (Figure 4C, lanes 4 through 6) or with rapamycin (Figure 4C, lanes 7 through 9) or combination of starvation with rapamycin treatment (Figure 4C, lanes 10 through 12). Although only 20% of misfolded P23H opsin was degraded upon 12 hours of starvation (Figure 4D, squares), more than 50% degradation was observed at 24 hours (data not shown). Rapamycin caused a more rapid loss of protein, that is, 50% of the protein remained after 6 hours of treatment and about 30% within 12 hours (Figure 4D, triangles). This loss was further enhanced when starvation was combined with rapamycin treatment (Figure 4D, circles), whereby almost 60% of P23H opsin was lost within 2 hours and 81% after 12 hours.
FIGURE 4.

Rapamycin or amino acid starvation lead to the preferential loss of misfolded P23H opsin. HEK293 stably expressing the WT opsin (A), P23H opsin (C), and P23H opsin rescued with 11-cis retinal (G) were either fed (lanes 1–3) or starved for amino acids and serum (lanes 4–6) or treated with 50 nM rapamycin (lanes 7–9) or starved and treated with rapamycin (lanes 10–12) and the cellular levels of opsin visualized by immunoblotting. At 48 hours prior to treatments, opsin production was induced by addition of tetracycline (lanes 1–12) or uninduced (lanes 13–14). Quantification of cellular opsin levels (see brackets) using tubulin as a loading control was performed on immunoblots using Odyssey (Licor) program. Time-course profiles show the levels of WT opsin (B), P23H opsin (D), and P23H opsin rescued with 11-cis retinal (H) in cells that have been fed (
 ) or starved (
) or starved (
 ) or treated with rapamycin (
) or treated with rapamycin (
 ) or starved and treated with rapamycin (
) or starved and treated with rapamycin (
 ). The levels of WT opsin remain essentially unchanged whereas the mutant P23H opsin is lost dramatically. Even the P23H opsin rescued with 11-cis retinal is susceptible to degradation by treatment with rapamycin. Next, HEK 293 cells stably expressing P23H opsin were fed and starved in the presence (
). The levels of WT opsin remain essentially unchanged whereas the mutant P23H opsin is lost dramatically. Even the P23H opsin rescued with 11-cis retinal is susceptible to degradation by treatment with rapamycin. Next, HEK 293 cells stably expressing P23H opsin were fed and starved in the presence (
 ) and absence (
) and absence (
 ) of 3MA, an inhibitor of autophagy (E) and the opsin levels quantified. The upper panel shows the immunoblot of P23H opsin under fed and starved conditions over a time course of 0 to 24 hours. The lower panel shows the immunoblot of P23H opsin after treatment of cells with 3MA in fed and starved medium. Quantification of immunoblot reveals an increase in the relative band intensity of opsin in presence of 3MA. However, P23H opsin-expressing cells when treated with proteasome inhibitor, MG132, under fed and starved conditions for 24 hours (F) showed that MG132 did not significantly reduce the levels of misfolded P23H opsin during starvation. The bar graph shows relative band intensities of P23H opsin under conditions of fed with MG132 (
) and starved with MG132 (
).
) of 3MA, an inhibitor of autophagy (E) and the opsin levels quantified. The upper panel shows the immunoblot of P23H opsin under fed and starved conditions over a time course of 0 to 24 hours. The lower panel shows the immunoblot of P23H opsin after treatment of cells with 3MA in fed and starved medium. Quantification of immunoblot reveals an increase in the relative band intensity of opsin in presence of 3MA. However, P23H opsin-expressing cells when treated with proteasome inhibitor, MG132, under fed and starved conditions for 24 hours (F) showed that MG132 did not significantly reduce the levels of misfolded P23H opsin during starvation. The bar graph shows relative band intensities of P23H opsin under conditions of fed with MG132 (
) and starved with MG132 (
).
The loss of P23H opsin was examined under conditions that inhibit either autophagy by incubating the cells with 3MA, or the proteasome by incubating with the inhibitor MG132. When P23H cells were incubated in starvation media containing 3MA, protein loss was negligible (Figure 4E). However, in the presence of the same media and MG132, P23H opsin degraded with similar kinetics as in the absence of the proteasome inhibitor (Figure 4F). These studies indicate that autophagy may mediate the specific reduction of misfolded P23H opsin while the role of proteasomal degradation in this pathway is limited.
Previously, it had been shown that 11-cis retinal functions as a pharmacological chaperone by assisting the folding and stabilization of P23H opsin, allowing the protein to reach the cell surface.13,14 The rescued protein does not form aggregates. Therefore, the loss of P23H opsin that was rescued with 11-cis retinal upon the onset of autophagy (Figure 4G) was examined. The degradation profile for the rescued P23H opsin under these conditions was intermediate when compared to wild-type or nonrescued P23H opsin. Contrary to our results with P23H opsin, starvation had no effect on the levels of rescued P23H (Figure 4G, lanes 4 through 6). However, the levels of the rescued protein were more sensitive to starvation and treatment with rapamycin (Figure 4, G and H). The data suggest that rapamycin can reduce the cellular amounts of even the rescued P23H opsin, presumably the form that does not form aggregates.
We then sought to determine if the unfolded protein response or the heat shock response was induced in conjunction with clearance of P23H opsin aggregates. Cellular levels of Bip and calnexin, ER chaperones associated with unfolded protein response, and Hsp70, a cytoplasmic chaperone regulating the heat shock response, remained unchanged under conditions that enhance autophagy (Figure 5). The data suggest that the induction of autophagy was exclusive of either of these two responses.
FIGURE 5.

Rapamycin-induced autophagy does not induce the unfolded protein response or heat shock response. The cellular levels of chaperones Bip, calnexin, and Hsp70 did not change when cells expressing WT opsin (A) or P23H opsin (B) were starved or treated with rapamycin for 2 to 12 hours.
Next, CFTR was studied as a second model of a misfolded polytopic membrane protein. The induction of autophagy by either amino-acid and serum starvation (Figure 6A, lanes 4 through 6) or rapamycin (Figure 3A, lanes 7 through 9) or starvation combined with rapamycin treatment (Figure 6A, lanes 10 through 12) did not affect the levels of the WT CFTR protein in BHK stable cell lines. However, the levels of ΔF508 CFTR protein rapidly decreased when cells were starved for amino acids and serum (Figure 6C, lanes 4 through 6) or treated with rapamycin (Figure 6C, lanes 7 through 9) or combined treatment with starvation and rapamycin (Figure 6C, lanes 10 through 12). Starvation caused a 34% reduction in protein levels after 12 hours (Figure 6D, triangles), whereas rapamycin treatment caused a 50% reduction of the protein after 12 hours (Figure 6D, squares). Further, there nearly a 75% loss of the mutant protein within 6 hours and 80% after 12 hours when both treatments were combined (Figure 6D, circles). These results suggest that the intracellular levels of either P23H opsin or ΔF508 CFTR can be significantly reduced by both starvation and rapamycin treatment. There appears to be selective loss of the misfolded protein, minimally affecting the WT form of these proteins.
FIGURE 6.

Preferential loss of ΔF508 cystic fibrosis transmembrane regulator (CFTR) by rapamycin. BHK cells stably expressing the WT CFTR (A) or ΔF508 CFTR (C) were either fed (lanes 1–3) or starved for amino acids and serum (lanes 4–6) or treated with 50 nM rapamycin (lanes 7–9) or starved and treated with rapamycin (lanes 10–12) for 0 to 12 hours and the cellular levels of HA-tag CFTR visualized by immunoblotting. Quantification of the immunoblots revealed a significant loss of ΔF508 CFTR in cells that had been starved (
) or treated with rapamycin (
) or starved and treated with rapamycin (
) when compared to fed cells (
). The levels of WT CFTR remain unchanged over the time course of these conditions.
We examined whether these misfolded proteins colocalized with autophagosome markers Atg7, Atg8, and LAMP-1. By immunofluorescence microscopy, WT opsin reached the cell surface and did not colocalize with these three markers (Figure 7A). In contrast, P23H opsin formed intracellular aggregates (Figure 7B). Upon 6 hours of starvation-enhanced autophagy, P23H opsin was found to colocalize with Atg7 and Atg 8 (Figure 7B), indicating that the misfolded P23H opsin incorporates into AVs. LAMP-1, which labels late autophagosomes and autolysosomes, colocalized with the P23H opsin aggregates in the absence of autophagy induction, suggesting a role of the lysosomal pathway in degradation of these aggregates. Similarly, immunofluorescence studies with CFTR revealed that WT CFTR reached the cell surface and did not colocalize with the three markers (Figure 8A). ΔF508 CFTR was retained within the cell but did not form any visible aggregates (Figure 8B). Like P23H opsin, ΔF508 CFTR colocalized with Atg7 and Atg 8 under starvation, suggesting sequestration of misfolded proteins in AVs.
FIGURE 7.

Misfolded aggregates of P23H opsin colocalized with Atg proteins. Immunofluorescence localization of opsin (shown in green) and autophagy markers Atg7, Atg8, and lysosomal associated membrane protein 1 (LAMP-1) (shown in red) was performed on HEK-293 cells expressing WT opsin (A) and P23H opsin (B) under fed and starved conditions for 6 hours after tetracycline removal. P23H opsin forms intracellular aggregates, whereas WT opsin was visualized at the cell surface. The Atg proteins and LAMP-1 were found to colocalize with the P23H opsin aggregates upon enhancing autophagy by starvation.
FIGURE 8.

Colocalization of ΔF508 CFTR with Atg proteins. Immuno-fluorescence localization of HA-tagged CFTR (shown in green) and autophagy markers Atg7, Atg8 and lysosomal associated membrane protein 1 (LAMP-1. (shown in red) was performed on BHK cells expressing WT CFTR (A) and ΔF508 CFTR (B) under fed and starved conditions for 6 hours. ΔF508 CFTR does not form aggregates in cells, but is retained within the cell, while the WT CFTR is present at the cell membrane and within the cytosol. Under starvation conditions, ΔF508 CFTR colocalized with Atg7 and Atg8.
To better visualize these autophagic responses, AV structure was examined by electron microscopy and quantified the autophagic responses by morphometrics. AVs were observed in P23H opsin-expressing cells, under fed and starved conditions (Figure 9, A and B). A 60% increase of AVs was observed in fed cells expressing P23H opsin compared to cells expressing WT opsin (Figure 9A). This suggests an increase in the basal level of autophagy in P23H cells, presumably due to the presence of the misfolded protein. The fractional volume of AVs was significantly increased when the P23H opsin-expressing cells were starved for amino acids and serum or treated with rapamycin (Figure 9B). Morphometric analysis showed a twofold increase in the fractional volume of AVs in P23H opsin-expressing cells under both autophagic conditions. Interestingly, AVs were observed in between or next to the aggregates. We have shown that P23H opsin localizes with Atg7, Atg8, and LAMP-1. To further substantiate that this protein is in AVs, the presence of P23H opsin in AVs was examined by immunogold methods. These studies revealed that some of the AVs contained P23H opsin (Figure 9C) as observed by the presence of gold particles within the vacuoles, thus providing convincing evidence for sequestration and degradation of P23H opsin by autophagy.
FIGURE 9.

Enhanced formation of AVs containing P23H opsin in starved cells as shown by ultrastructure and immunogold. HEK 293 cells stably expressing the WT opsin or P23H opsin were either fed or starved for amino acids and serum or treated with 50 nM rapamycin and the presence of autophagosomes (AVi, arrowheads) and acid phosphatase positive autolysosomes (AVd, arrows) determined by their characteristic ultrastructure (A, B). Morphometric quantification of AVs (acid phosphatase negative and positive autophagic vacuoles) was done and presented as the fractional volume (n=20). Samples with significant t test P values (<.05) relative to fed controls are marked with asterisks. In panel A, cells expressing P23H opsin showed an enhanced basal level of autophagy compared to cells expressing WT opsin. In panel B, a significant increase in AVs was observed when cells were starved for amino acids or treated with rapamycin. Next, tetracycline-treated HEK 293 cells expressing P23H opsin were starved for amino acids and serum for 6 hours and the P23H opsin localized by immunogold cytochemistry (C). Autophagic vacuoles with (arrow) and without opsin could be visualized.
DISCUSSION
Under the conditions of our experiments, rapamycin can significantly reduce the cellular amounts of P23H opsin while autophagy is clearly upregulated. Also, this effect shows selectivity in its ability to preferentially reduce the amounts of misfolded polytopic integral membrane protein, which form aggregates like P23H opsin, and ΔF508 CFTR, which does not, while minimally affecting the wild-type proteins. Under conditions whereby autophagy is enhanced (Figure 6B), it was demonstrated that P23H opsin and ΔF508 CFTR, but not WT forms, are preferentially and rapidly reduced in the cell (Figures 1 and 2). This effect is suppressed in the presence of 3MA, an inhibitor of autophagy, but not when cells are treated with the proteasome inhibitor, MG132. Furthermore, it was demonstrated that these mutant proteins are sequestered within AVs. It was shown that P23H opsin can be found within AVs (Figure 6C) and that the distinct punctate staining of autophagic vacuole (AV) markers, Atg7 and Atg8, colocalizes with P23H and ΔF508 CFTR proteins (Figures 4 and 5). Atg7 is a key autophagic gene encoding a protein resembling E1 ubiquitin-activating enzyme required for formation of AVs.42 Atg7 promotes the conjugation of Atg8, a microtubule-associated protein light chain 3, to the lipids that form the sequestering membranes of the AVs.43,44
The preferential loss of misfolded polytopic integral membrane proteins in the presence of rapamycin appears to be a conserved eukaryotic pathway because this was observed in hamster and human cell lines. This selectivity may be attributed to the ability of the cell to recognize protein aggregates and certain epitopes exposed specifically on the misfolded proteins, thereby stimulating autophagy. Indeed, we have observed an increase in basal autophagy in cells expressing P23H opsin (Figure 6A). An alternative possibility for preferential degradation of mutant proteins may be the differential cellular localizations of mutant and WT proteins. That is, membrane proteins at the cell surface may be inaccessible to sequestration by AVs. Further studies need to be done to better understand the autophagic response to protein aggregation and its surveillance mechanisms to preferentially degrade these mutant proteins.
Is autophagy relevant to the photoreceptor cells? The seminal, beautiful work of Remé’s group demonstrated that by changing the lighting conditions for rats, rod photoreceptor inner segments regulate the amount of WT rhodopsin by autophagic degradation.45 This clearly suggests that the molecular machinery required for autophagy is present in the rod cell and participates in controlling steady-state levels of rhodopsin. Although not directly demonstrated, Remé’s work also implies that any disease process that might affect the amount of folded rhodopsin produced by the rod cell would be monitored by the autophagy pathway. We are studying this hypothesis in transgenic mice.
Drugs like rapamycin that induce autophagy may be an important therapeutic strategy for autosomal dominant toxic gain-of-function diseases, specifically reducing the amounts of the misfolded mutant protein while leaving the wild-type protein untouched. An increasing number of inherited retinopathies appear to be protein conformational disorders, such as forms of retinitis pigmentosa associated with genes other than rod opsin, myocilin glaucoma, Malattia Leventinese, or age-related macular degeneration.46–49 These retinal degenerative disorders may also prove amenable to treatment with molecules, like rapamycin, that target the mTOR signaling network. Our data suggests that autophagy may participate in the removal of misfolded proteins but does not exclude the possibility that other pathways affected by rapamycin in the cell may also contribute to the time-dependent loss of the misfolded protein. Collectively, these effects may allow the cell to establish a normal physiological state. On the basis of the results presented, we have initiated transgenic mice studies and also have screened and identified novel compounds that have activities like rapamycin. Our ability to study the molecular pathways that participate in the fate mutant opsin will help to delineate the pathogenesis of retinitis pigmentosa and simultaneously permit the development of therapeutics that are so badly needed for this disease
ACKNOWLEDGMENTS
Ritu Malhotra, MSc, Mark Krebs, PhD, and William Dunn Jr, PhD, helped with the experiments and ideas presented in this thesis.
REFERENCES
- 1.Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 2.Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 3.Sung CH, Schneider BG, Agarwal N, et al. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;19:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- 5.Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- 6.Mickle JE, Cutting GR. Genotype-phenotype relationships in cystic fibrosis. Med Clin North Am. 2000;84:597–607. doi: 10.1016/s0025-7125(05)70243-1. [DOI] [PubMed] [Google Scholar]
- 7.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 8.Heda GD, Tanwani M, Marino CR. The Delta F508 mutation shortens the biochemical half-life of plasma membrane CFTR in polarized epithelial cell. Am J Physiol Cell Physiol. 2001;280:C166–C174. doi: 10.1152/ajpcell.2001.280.1.C166. [DOI] [PubMed] [Google Scholar]
- 9.Cheng SH, Gregory R , Marshall J, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 10.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 11.Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;95:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 12.Galy A, Roux MJ, Sahel JA, et al. Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for RhodopsinPro23His human retinitis pigmentosa. Hum Mol Genet. 2005;14:2547–2557. doi: 10.1093/hmg/ddi258. [DOI] [PubMed] [Google Scholar]
- 13.Noorwez SM, Kuksa V, Imanishi Y, et al. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J Biol Chem. 2003;278:14442–14450. doi: 10.1074/jbc.M300087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noorwez SM, Malhotra R, McDowell JH, et al. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- 15.Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci U S A. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petters RM, Alexander CA, Wells KD, et al. Genetically engineered large animal model for studying cone photoreceptor survival and degeneration in retinitis pigmentosa. Nat Biotechnol. 1997;10:965–970. doi: 10.1038/nbt1097-965. [DOI] [PubMed] [Google Scholar]
- 17.Sung CH, Makino C, Baylor D, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;10:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deretic D, Schmerl S, Hargrave PA, et al. Regulation of sorting and post-Golgi trafficking of rhodopsin by its C-terminal sequence QVS(A)PA. Proc Natl Acad Sci U S A. 1998;95:10620–10625. doi: 10.1073/pnas.95.18.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deretic D, Williams AH, Ransom N, et al. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Natl Acad Sci U S A. 2005;102:3301–3306. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuang JZ, Vega C, Jun W, Sung CH. Structural and functional impairment of endocytic pathways by retinitis pigmentosa mutant rhodopsin-arrestin complexes. J Clin Invest. 2004;114:131–140. doi: 10.1172/JCI21136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- 22.Portera-Cailliau C, Sung CH, Nathans J, Adler R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci U S A. 1994;91:974–978. doi: 10.1073/pnas.91.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travis GH. Mechanisms of cell death in the inherited retinal degenerations. Am J Hum Genet. 1998;62:503–508. doi: 10.1086/301772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li T, Franson WK, Gordon JW, et al. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci U S A. 1995;92:3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrell RW. Cell toxicity and conformational disease. Trends Cell Biol. 2005;15:574–580. doi: 10.1016/j.tcb.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Oh KT, Weleber RG, Lotery A, et al. Description of a new mutation in rhodopsin, Pro23Ala, and comparison with electroretinographic and clinical characteristics of the Pro23His mutation. Arch Ophthalmol. 2000;118:1269–1276. doi: 10.1001/archopht.118.9.1269. [DOI] [PubMed] [Google Scholar]
- 27.Veenhuis M, Douma A, Harder W, Osumi M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch Microbiol. 1983;134:193–203. doi: 10.1007/BF00407757. [DOI] [PubMed] [Google Scholar]
- 28.Yokota S. Formation of autophagosomes during degradation of excess peroxisomes induced by administration of dioctyl phthalate. Eur J Cell Biol. 1993;61:67–80. [PubMed] [Google Scholar]
- 29.Bergamini E, Detata V, Cubeddu TL, et al. Increased degradation in rat liver induced by antilipolytic agents: a model for studying autophagy and protein degradation in liver? Exp Molec Pathol. 1987;46:114–122. doi: 10.1016/0014-4800(87)90035-9. [DOI] [PubMed] [Google Scholar]
- 30.Luiken JJFP, Vandenberg M , Heikoop JC, Meijer AJ. Autophagic degradation of peroxisomes in isolated rat hepatocytes. FEBS Letters. 1992;304:93–97. doi: 10.1016/0014-5793(92)80596-9. [DOI] [PubMed] [Google Scholar]
- 31.Heikoop JC, Vandenberg M, Strijland A, et al. Turnover of peroxisomal vesicles by autophagic proteolysis in cultured fibroblasts from Zellweger patients. Eur J Cell Biol. 1992;57:165–171. [PubMed] [Google Scholar]
- 32.Kondo K, Makita T. Inhibition of peroxisomal degradation by 3-methyladenine (3MA) in primary cultures of rat hepatocytes. Anat Rec. 1997;247:449–454. doi: 10.1002/(SICI)1097-0185(199704)247:4<449::AID-AR2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 33.Serafini B, Stefanini S, Ceru MP, Sartori C. Lysosomal involvement in the removal of clofibrate-induced rat liver peroxisomes. A biochemical and morphological analysis. Biol Cell. 1998;90:229–237. doi: 10.1016/s0248-4900(98)80019-1. [DOI] [PubMed] [Google Scholar]
- 34.Masaki R, Yamamoto A, Tashiro Y. Cytochrome P-450 and NADPH-cytochrome P-450 reductase are degraded in the autolysosomes in rat liver. J Cell Biol. 1987;104:1207–1215. doi: 10.1083/jcb.104.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanner AJ, Dice JF. Batten disease and mitochondrial pathways of proteolysis. Biochem Mol Med. 1996;57:1–9. doi: 10.1006/bmme.1996.0001. [DOI] [PubMed] [Google Scholar]
- 36.Tolkovsky AM, Xue LZ, Fletcher GC, Borutaite V. Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie. 2002;84:233–240. doi: 10.1016/s0300-9084(02)01371-8. [DOI] [PubMed] [Google Scholar]
- 37.Fortun J, Dunn WA, Joy S, et al. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 39.Lu M, Echeverri F, Moyer BD. Endoplasmic reticulum retention, degradation, and aggregation of olfactory G-protein coupled receptors. Traffic. 2003;4:416–433. doi: 10.1034/j.1600-0854.2003.00097.x. [DOI] [PubMed] [Google Scholar]
- 40.Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 41.Tanida I , Tanida-Miyake E, Ueno T, Kominami E. The C-terminal region of an Apg7p/Cvt2p is required for homodimerization and is essential for its E1 activity and E1-E2 complex formation. J Biol Chem. 2001;276:1701–1706. doi: 10.1074/jbc.M007737200. [DOI] [PubMed] [Google Scholar]
- 42.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nature Rev Molec Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 43.Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. J Cell Sci. 2004;117:2805–2812. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunn WA. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reme CE, Wolfrum U, Imsand C, et al. Photoreceptor autophagy: effects of light history on number and opsin content of degradative vacuoles. Invest Ophthalmol Vis Sci. 1999;10:2398–2404. [PubMed] [Google Scholar]
- 46.Aherne A, Kennan A, Kenna PF, et al. On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa. Hum Mol Genet. 2004;13:641–650. doi: 10.1093/hmg/ddh061. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Vollrath D. Reversal of mutant myocilin non-secretion and cell killing: implications for glaucoma. Hum Mol Genet. 2004;13:1193–1204. doi: 10.1093/hmg/ddh128. [DOI] [PubMed] [Google Scholar]
- 48.Marmorstein LY, Munier FL, Arsenijevic Y, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:13067–13072. doi: 10.1073/pnas.202491599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiszniewski W, Zaremba CM, Yatsenko AN, et al. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 2005;14:2769–2778. doi: 10.1093/hmg/ddi310. [DOI] [PubMed] [Google Scholar]