

Abstract
The myristoylated, alanine-rich protein kinase C substrate (MARCKS) is a cytoskeletal protein implicated in the regulation of cell spreading, stress fiber formation, and focal adhesion assembly in nonmuscle cells. However, its precise role in cardiomyocyte growth, and its PKC-dependent regulation have not been fully explored. In this report, we show that MARCKS is expressed and phosphorylated under basal conditions in cultured neonatal and adult rat ventricular myocytes (NRVM and ARVM, respectively). The PKC activators phenylephrine, angiotensin II, and endothelin-1 (ET) further increased MARCKS phosphorylation, with ET inducing the greatest response. To determine which PKC isoenzyme was responsible for agonist-induced MARCKS phosphorylation, NRVM and ARVM were infected with replication-defective adenoviruses (Adv) encoding wildtype (wt) and constitutively active (ca) mutants of PKCε, PKCδ, and PKCα. Only PKCε increased phosphorylated MARCKS (pMARCKS). In contrast, Adv-mediated overexpression of a dominant-negative (dn) mutant of PKCε reduced basal and ET-stimulated pMARCKS. dnPKCε overexpression also prevented ET-induced, apparent co-localization of pMARCKS with f-actin staining structures. Adv-mediated overexpression of GFP-tagged, wtMARCKS (wtMARCKS-GFP) increased phosphorylation of focal adhesion kinase (FAK) and also increased NRVM surface area. In constrast, overexpression of a GFP-tagged, non-phosphorylatable (np) MARCKS mutant (npMARCKS-GFP) decreased basal and ET-induced endogenous MARCKS and FAK phosphorylation, and blocked the ET-induced increase in NRVM surface area. We conclude that MARCKS is expressed in cardiomyocytes, is phosphorylated by PKCε, and participates in the regulation of FAK phosphorylation and cell spreading.
Keywords: adenovirus, hypertrophy, signal transduction, Focal Adhesion Kinase, PKC substrate
INTRODUCTION
The myristoylated, alanine-rich protein kinase C substrate (MARCKS) is a major cytoskeletal protein and protein kinase C (PKC) substrate implicated in the regulation of actin filament dynamics. MARCKS is ubiquitously expressed in nonmuslce cells, where it binds to and cross-links actin filaments, and thus participates in diverse cellular processes including cell spreading, adhesion, and focal adhesion formation (1–4). Under basal conditions, MARCKS is unphosphorylated and bound to the cell membrane through an amino-terminal myristoylation domain and an internal serine phosphorylation site domain (PSD), which is also a site for high affinity calcium/calmodulin binding (4). Upon PKC phosphorylation of at least 3 serine residues in the PSD, MARCKS dissociates from the plasma membrane and translocates to the cytosol where it cross-links actin filaments (4). A number of elegant studies have addressed the role of MARCKS during integrin-mediated actin dynamics and cell spreading in skeletal muscle myoblasts (2, 5, 6). In the heart, however, the precise role of MARCKS and its PKC-dependent regulation have not been fully explored.
PKC activation has been shown to play regulatory roles in a number of physiological and pathological events in the heart, including hypertrophic and apoptotic signaling processes (7–12). Both peptide growth factors and adhesion to the extracellular matrix (ECM) have been shown to activate various isoforms of PKC, but only two isoforms, PKCε and PKCδ, are activated in response to endothelin-1 (ET) (13, 14). We have previously shown that ET-induced PKCε activation in neonatal rat ventricular myocytes (NRVM) results in cell spreading, increased focal adhesion formation, and focal adhesion kinase (FAK) activation (11). FAK is an important constituent of focal adhesions, which are sites that link integrins to the ECM and the actin cytoskeleton. FAK undergoes activation and tyrosine autophosphorylation at residue Y397 after integrin engagement and stimulation by growth factors in cultured NRVM (15–17). FAK and other focal adhesion proteins thus provide both structural and functional roles in integrin-dependent, bi-directional signal transduction in the heart.
In the present study, we investigated agonist- and isoform-specific PKC-induced MARCKS phosphorylation in both cultured NRVM and freshly isolated and cultured adult rat ventricular myocytes (ARVM). We used replication-defective adenoviruses encoding wild-type and mutant forms of PKCε PKCδ and PKCα to examine which isoenzyme is involved in MARCKS phosphorylation. Additionally, we used adenoviruses encoding wildtype and mutant forms of MARCKS to determine whether MARCKS is involved in FAK phosphorylation and cell spreading.
MATERIALS AND METHODS
Reagents.
PC-1 tissue culture medium was purchased from BioWhittaker (Walkersville, MD). Medium 199 (M199), Ca2+-free and Mg2+-free Hanks’ balanced salt solution (HBSS), acid-soluble calf skin collagen, and antibiotic/antimycotic solution were obtained from Sigma Chemical (St. Louis, MO). DMEM was obtained from GIBCO-BRL (Grand Island, NY), type II collagenase from Worthington (Lakewood, NJ), and penicillin-streptomycin (P/S) from Fisher Scientific/MediaTech (Itasca, IL). Phospho-specific MARCKS (N-19) rabbit polyclonal antibody (pAb) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and anti-GAPDH mouse monoclonal antibody (mAb) was purchased from Novus Biologicals (Littleton, CO). mAbs to PKCε, PKCδ and PKCα were obtained from Signal Transduction Laboratories (Lexington, KY). Anti-FAK rabbit pAb was obtained from Upstate (Lake Placid, NY). Phospho-specific Y397 rabbit anti-FAK pAb was purchased from Biosource (Camarillo, CA). FITC-phalloidin, goat-anti-rabbit and goat-anti-mouse secondary Abs were obtained from Molecular Probes (Eugene, OR). All other reagents were of the highest grade commercially available and were obtained from Sigma and Baxter S/P (McGaw Park, IL).
Cell culture.
Animals used in these experiments were handled in accordance with the Guiding Principles in the Care and Use of Animals, approved by the Council of the American Physiological Society. NRVM were isolated from the hearts of 2-day old Sprague-Dawley rats by collagenase digestion, as previously described (18). Myocytes were pre-plated for 1h in serum-free PC-1 medium to reduce nonmyocyte contamination. The nonadherent NRVM were then plated at a density of 1600 cells per mm2 onto collagen-coated chamber slides or 35mm dishes, and left undisturbed in a 5% CO2 incubator for 14–18h. Unattached cells were removed by aspiration, washed twice in HBSS, and the attached cells were maintained in a solution of DMEM/Medium 199 (4:1) containing antibiotic/antimycotic solution. Cardiomyocytes were either treated with drugs or infected (60 min, 25°C with gentle agitation) with replication-defective adenoviruses diluted in DMEM/Medium 199. Medium was then replaced with virus-free DMEM/Medium 199, and the cells cultured for an additional 24–48h. ARVM were isolated according to Westfall et al. (19), with modification as described previously (12). Myocytes were plated in DMEM-P/S onto laminin-coated (15 μg/ml) 35 mm dishes for 60 min in a 5% CO2 incubator. Unattached myocytes were removed and attached myocytes were maintained in DMEM/F12-PC-1 (2:1) containing P/S. Myocytes were either infected with adenoviruses for 24–48h or treated with drugs and immediately utilized for Western blotting.
Adenoviral constructs.
Replication-defective adenoviruses encoding caPKCε (Adv-caPKCε), caPKCδ (Adv-caPKCδ), wtPKCε (Adv-wtPKCε), wtPKCδ (Adv-wtPKCδ) and wtPKCα (Adv-wtPKCα) were constructed as previously described (10, 20, 21). Dominant-negative (dn) PKCε adenovirus was kindly provided by Dr. Peipei Ping, University of California–Los Angeles and constructed as previously described (22). The protein-coding region of MARCKS was amplified from neonatal rat brain RNA by RT-PCR. Total RNA (1.0 μg) was reverse transcribed and amplified using ThermoScript reverse transcriptase and Platinum PCR SuperMix (Invitrogen), with addition of PCRx Enhancer (2x, Invitrogen) to overcome secondary structure due to the GC-rich sequence of MARCKS. First strand cDNA was primed with random hexanucleotides, and PCR primers corresponded to the rat MARCKS cDNA sequence (GenBank accession No. XM215403; upper primer, 5’-CGTCGTTACACCAACCCAAGGCTC-3’; lower primer, 5’-AGCTTACTCGGCCACCGGTGCGGGG-3”). The lower primer sequence was modified to incorporate an AgeI restriction site. PCR conditions were 94°, 60 sec; 65°, 90 sec for 30 cycles.
To generate a MARCKS-GFP fusion protein, the 1003 base pair PCR product was cloned into pCR2.1 cloning vector using the TA Cloning Kit (Invitrogen). A HindIII-AgeI fragment was cloned into pEGFP-N1 (Clontech) to make an in-frame, MARCKS-GFP fusion protein. The MARCKS-GFP coding region was then subcloned into the adenovirus shuttle vector pShuttle (Clontech) using NheI-XbaI restriction sites. The rat MARCKS protein contains 5 potential phosphorylatable serine residues at amino acids 152, 156, 160, 163 and 167. Serine to alanine mutations were made sequentially at residues 156, 160, and 163 using the QuikChange Mutagenesis kit (Stratagene). The mutations and in-frame GFP fusion were confirmed by DNA sequencing. Transfection of HEK-293 cells with wildtype and mutant MARCKS plasmids resulted in GFP-fluorescence. Western blots probed with anti-MARCKS antibody (Santa Cruz) were used to confirm expression of transfected MARCKS protein. Phospho-MARCKS (pMARCKS) antibody (Santa Cruz) was used to confirm that the mutated MARCKS-GFP construct was not phosphorylated after phorbol myristate acetate (PMA) treatment (10 min, 200 nM PMA), in contrast to both endogenous and wildtype transfected MARCKS. Adenoviral vectors containing the GFP-tagged, wt and nonphosphorylatable (np) MARCKS coding regions were constructed in the AdEasy vector system (kindly provided by Dr. B. Vogelstein; (23)). Shuttle plasmids were transfected into BJ5183-Ad1 cells (Stratagene), which contain the AdEasy1 adenoviral backbone vector, to allow recombination of shuttle and vector plasmids. Adenoviral recombinants were isolated and transfected into HEK-293 cells to generate wtMARCKS-GFP and npMARCKS-GFP adenoviruses. Replication-defective adenoviruses encoding either cytoplasmic (cyto) or nuclear-encoded (ne) β-galactosidase (βgal); were used to control for nonspecific effects of adenoviral infection (11). The multiplicity of viral infection (moi) for each virus was determined by dilution assay in HEK-293 cells.
Immunolocalization.
NRVM grown on chamberslides were fixed and permeabilized as previously described (24). Cells were stained with a polyclonal pMARCKS antibody and FITC-conjugated phalloidin to visualize actin filaments. Fluorescently-labeled cells were then viewed using a Zeiss LSM 510 laser scanning confocal microscope.
Western blotting.
NRVM were washed once in ice-cold PBS and homogenized in lysis buffer containing 1% Triton X-100 and 0.1% SDS (25). Equal amounts of extracted cellular proteins were separated on 7.5% SDS-polyacrylamide gels with 4% stacking gels. Proteins were transferred to PVDF membrane using the recommended transfer buffer. Western blots were probed with antibodies specific for the phosphorylated form of MARCKS at ser160/163, the phosphorylated form of FAK at Y397, or total FAK. Due to the fact that the antibody for total (phosphorylated and unphosphorylated) MARCKS (Santa Cruz) appeared to have a higher affinity for the phosphorylated forms of MARCKS, we used an antibody specific for GAPDH as a protein-loading control. Primary antibody binding was detected with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies and visualized by enhanced chemiluminescence (Amersham, Arlington Heights, IL). Band intensity was quantified using laser densitometry.
Cell size and cellular composition.
For determination of cell surface area, NRVM were first loaded with 2’7’-bis (2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF)-AM [2 μM in a modified Krebs medium composed of (in mM) 135 NaCl, 5.9 KCl, 1.5 CaCl2, 1.2 MgCl2, 11.5 glucose, and 11.6 HEPES, pH 7.3, supplemented with 0.1% BSA and 0.2% Pluronic F-127 detergent] for 1 h, followed by 1 h incubation in BCECF-free Krebs buffer. Cells were viewed with a laser scanning confocal microscope. Optical sections through the base of the cells (~20 cells/field) were stored as digital images and analyzed with Image-1 software (Universal Imaging, West Chester, PA). A binary mask was created by setting the threshold brightness which distinguished the fluorescent cells from the black background. Cell area was determined as an exact count of the number of pixels that made up the objects binary mask, multiplied by the area of a unit pixel (26). Total cellular protein and DNA content were measured as described previously (26).
Data analysis.
Results were expressed as means ± SEM. Normality was assessed using the Kolmogorov-Smirnov test, and homogeneity of variance was assessed using Levene’s test. Data were compared by 1-way blocked ANOVA, followed by the Student-Newman-Keuls test. Differences among means were considered significant at P<0.05. Data were analyzed using the SigmaStat Statistical Software Package, Ver. 1.0 (Jandel Scientific, San Rafael, CA).
RESULTS
ET treatment increases MARCKS phosphorylation in NRVM and ARVM.
Initial experiments were performed to investigate whether hypertrophic agonists that activate PKCs induce the phosphorylation of MARCKS. As seen in Figure 1A, MARCKS was phosphorylated under basal conditions in NRVM. Stimulation with angiotensin II (AngII; 100nM) and phenylephrine (PE; 50μM) for either 5 or 10 min resulted in a modest increase in MARCKS phosphorylation. However, treatment with endothelin-1 (ET; 10nM), had a much greater effect. A time course for ET-stimulation in NRVM (Fig 1B) revealed that MARCKS phosphorylation increased by 2 min, peaked at 5-fold of control at 10 min, and remained elevated throughout the 60 min period. Likewise in freshly isolated ARVM, ET-treatment resulted in peak (3-fold) MARCKS phosphorylation at 10 min (Fig 1C). The results of 5–6 experiments are summarized in Figure 1D and 1E.
Figure 1. Hypertrophic agonists increase MARCKS phosphorylation in NRVM and ARVM.
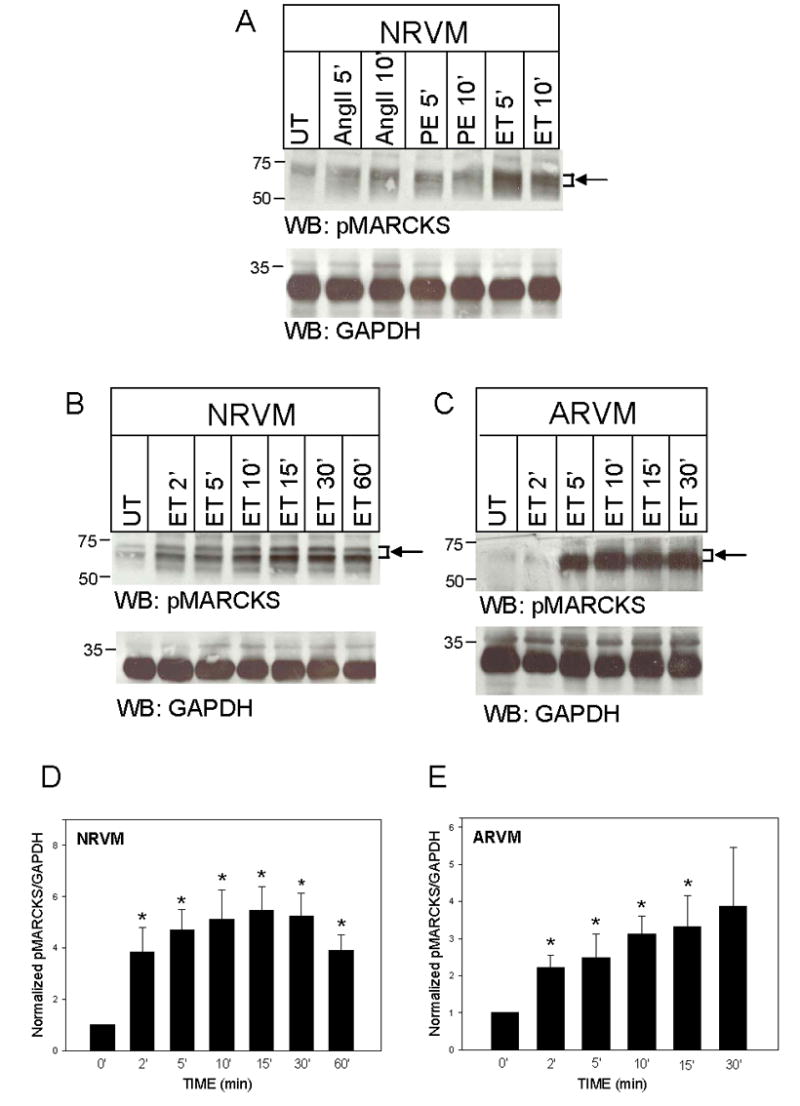
NRVM were maintained in control medium and (A) treated with angiotensin II (AngII; 100nM), phenylephrine (PE; 50μM), or endothelin-1 (ET; 10nM) for 5–10 minutes (‘), or (B) treated with ET (10nM) for 0–60’. In panel (C), freshly isolated ARVM were treated with ET (10nM, 2–30’). Western blots (50μg of extracted protein) were probed with either an antibody specific for MARCKS phosphorylated at Serine 160/163, or an antibody that recognizes GAPDH. The positions of molecular weight standards are indicated to the left of each blot. Arrow and bracket sets indicate band area of pMARCKS used for quantitation. Panels D and E depict the quantitative analysis of 5–6 Western blotting experiments. Levels of phosphorylated MARCKS were normalized to GAPDH at each time point, and then normalized to respective levels of untreated cells (UT). Data are means ± SEM; P<0.05 vs. 0 time point.
caPKCε overexpression increases MARCKS phosphorylation in both NRVM and ARVM.
To determine which PKC isoenzymes expressed in cardiomyocytes were responsible for agonist-induced MARCKS phosphorylation, NRVM and ARVM were infected with replication-defective Adv encoding caPKCε, caPKCδ, wtPKCε, wtPKCδ, or wtPKCα. Adv-neβgal was used to control for nonspecific effects of Adv infection. Of the adenoviruses tested, Adv-caPKCε resulted in the greatest degree of MARCKS phosphorylation, with a 3.5-fold increase in NRVM and a 6.5-fold increase in ARVM (Fig 2A and B; left panels). Adv-wtPKCε had a similar effect (data not shown). In contrast, wtPKCα overexpression actually reduced MARCKS phosphorylation, which was sustained following acute ET treatment (Fig 2A and B, right panels). Our ability to ascertain the effect of PKCδ on MARCKS phosphorylation was partially hampered by cross-reactivity of the phosphoMARCKS antibody with the overexpressed PKCδ protein, and their limited separation by SDS-PAGE. Figure 2C depicts MARCKS phosphorylation following caPKCδ (top) and wtPKCδ (bottom) overexpression. Note the large, nonspecific PKCδ band just above MARCKS in PKCδ-overexpressing myocytes. Nevertheless, overexpression of PKCδ did not appear to alter MARCKS phosphorylation. Levels of overexpressed PKCε, PKCα, and PKCδ are also shown, and are consistent with the levels of PKC overexpression obtained in previous studies from our laboratory (21, 27). Data summarizing the results of 4–5 experiments following caPKCε expression in NRVM and ARVM are depicted in Fig 2D.
Figure 2. caPKCε overexpression increases MARCKS phosphorylation in NRVM and ARVM.
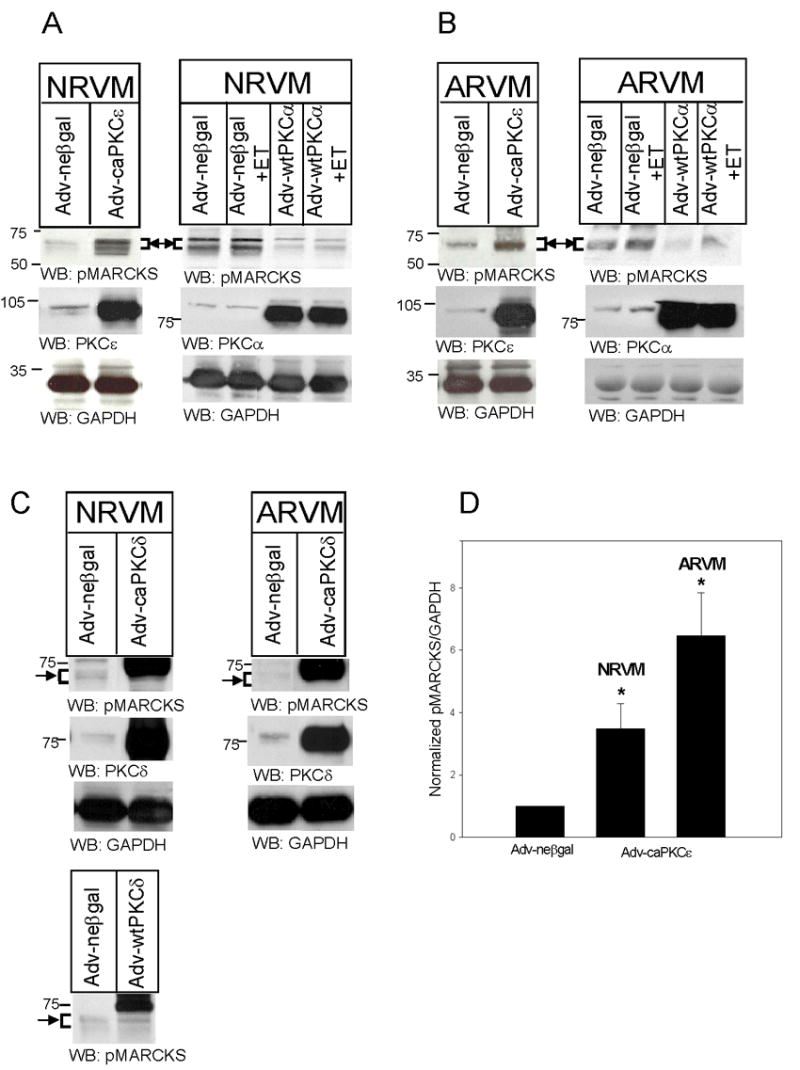
NRVM (A) and ARVM (B) were infected with Adv-neβgal and Adv-caPKCε, or Adv-neβgal and Adv-wtPKCα (25MOI, 24h), and then treated with ET (10nM, 10 min). In Panel C, NRVM and ARVM were infected with Adv-neβgal, Adv-caPKCδ, or Adv-wtPKCδ (25MOI, 24h). Western blots (50μg of extracted protein) were probed with either an antibody specific for MARCKS phosphorylated at Serine 160/163, antibodies specific for PKC-ε, -δ, or -α, or an antibody that recognizes GAPDH. Western blots depicted in Panel C were run for a longer period of time in order to maximize the separation of bands. The positions of molecular weight standards are indicated to the left of each blot. Arrow and bracket sets indicate band area of pMARCKS. Panel D depicts the quantitative analysis of 4–5 Western blotting experiments. Levels of phosphorylated MARCKS were first normalized to their respective GAPDH, and then normalized to levels in neβgal-expressing cells. Data are means ± SEM; P<0.05 vs. Adv-neβgal.
dnPKCε overexpression reduces basal and ET-induced MARCKS phosphorylation.
To determine whether PKCε was necessary for basal and agonist-induced MARCKS phosphorylation, we infected NRVM and ARVM with a replication-defective Adv encoding dnPKCε and analyzed MARCKS phosphorylation by Western blotting. Adv-cytoβgal was used to control for nonspecific effects of Adv infection. In NRVM, Adv-dnPKCε decreased both basal and ET-induced MARCKS phosphorylation (Fig 3A). The results of 5–7 experiments are quantitatively assessed in Figure 3C and demonstrate that dnPKCε overexpression significantly reduced basal levels of phosphorylated MARCKS to 0.5±0.1-fold of Adv-cytoβgal infected cells. ET-induced MARCKS phosphorylation was also significantly decreased from 2.5±0.4-fold in Adv-cytoβgal infected NRVM, to 1.0±0.3-fold following dnPKCε overexpression. However, ET was still able to phosphorylate MARCKS when compared to levels in dnPKCε-expressing myocytes without ET stimulation (3.4±1.0-fold). In ARVM, both basal and ET-induced MARCKS phosphorylation also decreased somewhat following dnPKCε overexpression; however, these differences did not reach statistical significance (Figure 3B and D).
Figure 3. dnPKCε overexpression partially inhibits ET-induced MARCKS phosphorylation in both NRVM and ARVM.
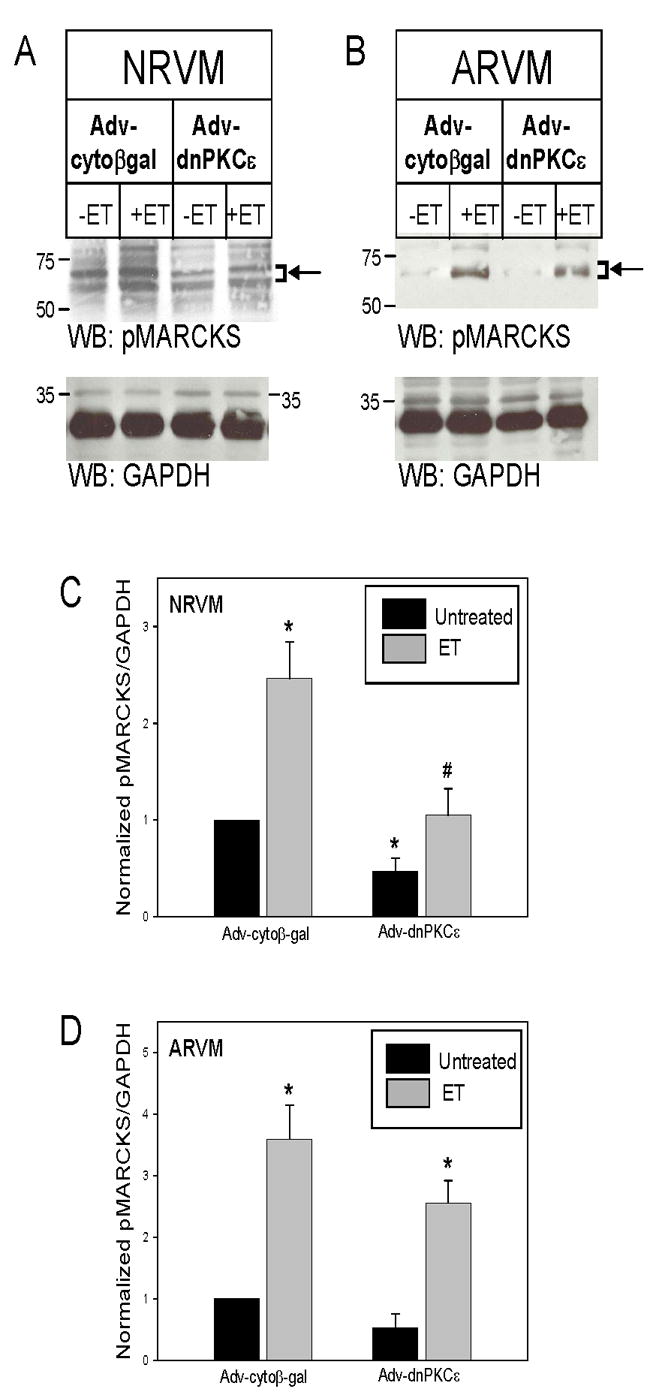
NRVM (A) and ARVM (B) were infected with Adv-cytoβgal or Adv-dnPKCε (750MOI, 48h), and then treated with ET (10nM, 10min). Western blots (50μg of extracted protein) were probed with either an antibody specific for MARCKS phosphorylated at erine 160/163, or an antibody that recognizes GAPDH. The positions of molecular weight standards are indicated to the left of each blot. Arrow and bracket sets indicate band area of pMARCKS used for quantitation. Panels C and D depict the quantitative analysis of 5–7 Western blotting experiments. Levels of phosphorylated MARCKS were normalized to their respective GAPDH, and then normalized to levels in cytoβgal-expressing cells. Data are means ± SEM; *P<0.05 vs. cytoβgal, #P<0.05 vs. cytoβgal +ET.
dnPKCε overexpression prevents ET-stimulated redistribution of endogenous, phosphorylated MARCKS to myofilaments.
NRVM and ARVM were infected with either Adv-cytoβgal or Adv-dnPKCε, and then acutely stimulated with ET. Following cell fixation, pMARCKS was detected with the phosphoMARCKS-specific antibody and rhodamine-conjugated secondary antibody, whereas FITC-conjugated phalloidin was used to visualize actin filaments. Optical sections (~1μm) were then obtained by confocal microscopy. Under basal conditions, phosphorylated MARCKS was detected in cytoplasmic regions of the myocytes in NRVM, and both plasma membrane and cytoplasmic regions in ARVM (Figure 4). The level of phosphoMARCKS staining appeared to increase in response to ET stimulation in Adv-cytoβgal infected cells, but not in cells infected with Adv-dnPKCε. Furthermore, ET increased the apparent co-localization of pMARCKS with f-actin staining structures in Adv-cytoβgal myocytes (i.e., increased yellow staining in the MERGED images) in both NRVM and ARVM. In contrast, expression of dnPKCε (especially in ARVM) decreased the amount of pMARCKS present in the membrane regions, and acute ET-stimulation failed to increase the apparent co-localization of pMARCKS with f-actin staining structures.
Figure 4. Expression of dnPKCε prevents ET-stimulated redistribution of endogenous phosphorylated MARCKS to the myofilaments.
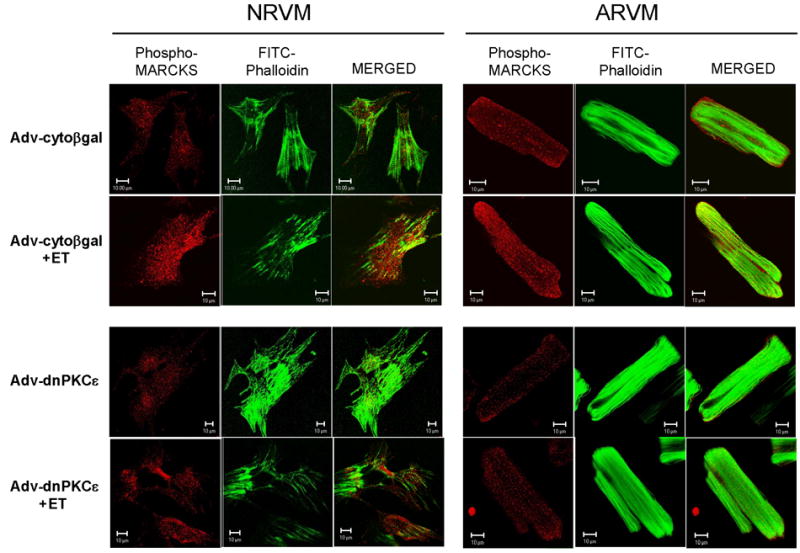
NRVM and ARVM were infected with Adv encoding either cytoβgal or dnPKCε (750 MOI, 24h), and then treated with ET (10nM, 10min). Cells were fixed and labeled with anti-pMARCKS pAb. Rhodamine-congugated rabbit anti-goat IgG (red) and FITC-conjugated phalloidin (green) were used for visualization by laser confocal microscopy (~1μm optical sections). Areas of apparent co-localization appear as yellow in the MERGED images.
Effects of wtMARCKS and npMARCKS overexpression on FAK phosphorylation.
Previously, we demonstrated that PKCε is involved in ET-induced activation of focal adhesion kinase (FAK) (14). Due to the fact that MARCKS phosphorlyation also appears dependent on PKCε, we examined the ability of MARCKS to regulate the activation of FAK. Adenoviruses encoding GFP-tagged, wtMARCKS and npMARCKS were generated and used to overexpress the wildtype and mutant proteins in NRVM and ARVM. As seen in Figure 5, overexpression of wtMARCKS-GFP in both NRVM and ARVM resulted in the accumulation of a phosphorylated MARCKS protein of approximately 95kDa by Western blotting, consistent with the expected migration of GFP-tagged wtMARCKS. The expressed wtMARCKS-GFP was itself highly phosphorylated (Fig 5A and B, left panels), and also resulted in a 4-fold increase in the phosphorylation of endogenous MARCKS, although the mechanism(s) responsible for this observation are not known. Acute ET stimulation of wtMARCKS-overexpressing NRVM and ARVM resulted in further phosphorylation of both exogenous and endogenous MARCKS. Interestingly, overexpression of wtMARCKS also significantly increased the phosphorylation of FAK at Y397 in both NRVM and ARVM. The results of 4–5 experiments are summarized in Figures 5C and 5d, respectively.
Figure 5. Expression of GFP-labeled wild-type (wt) MARCKS phosphorylates FAK, whereas expression of GFP-labeled non-phosphorylatable (np) MARCKS blocks both basal and ET-induced FAK phosphorylation.
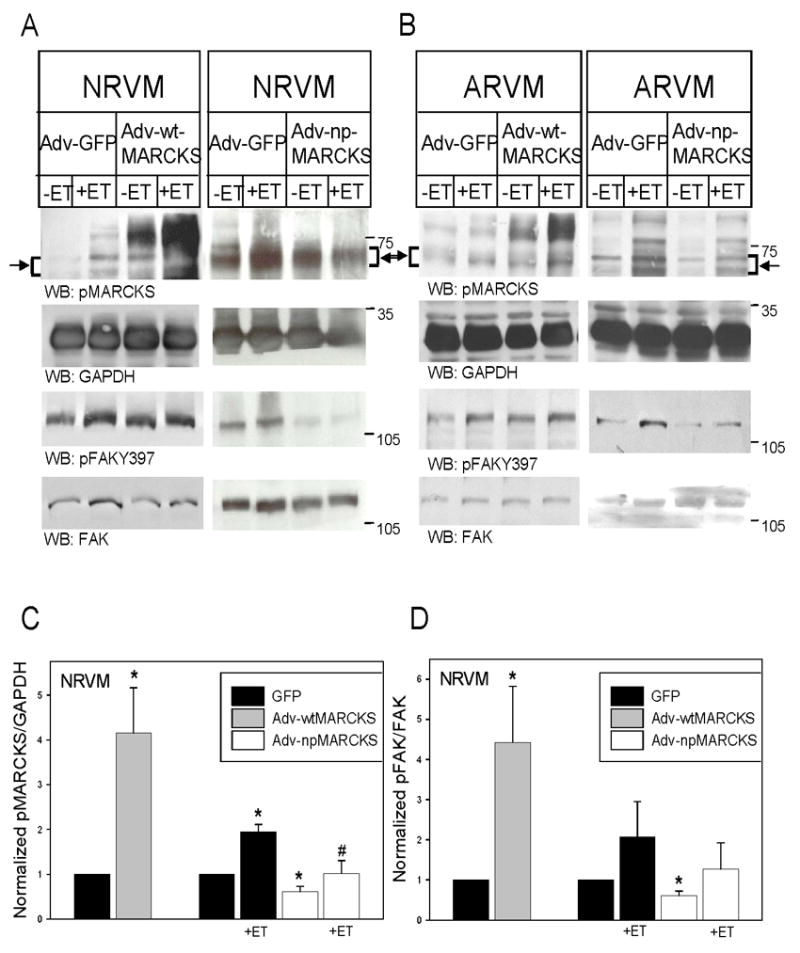
NRVM were infected with an adenovirus encoding either green fluorescent protein (GFP), (A) wtMARCKS-GFP (10moi, 24h), or (B) npMARCKS-GFP (25MOI, 48h). Cells expressing wtMARCKS and npMARCKS were then treated with ET (10nM, 10’). Western blots (50μg of extracted protein) were probed with an antibody specific for MARCKS phosphorylated at Serine 160/163, an antibody that recognizes GAPDH, an antibody that recognizes FAK phosphorylated at Y397, or an antibody that recognizes both phosphorylated and nonphosphorylated FAK. The position of molecular weight markers is indicated to the right of each blot. Arrow and bracket sets indicate band area of pMARCKS used for quantitation. Panels C and D depict the quantitative analysis of 3–5 Western blotting experiments. Levels of phosphorylated MARCKS were normalized to their respective GAPDH, and then normalized to levels in GFP-expressing cells. Phosphorylated FAK at Y397 was normalized to total FAK levels, and then normalized to levels in GFP-expressing cells. Data are means ± SEM; *P<0.05 vs. GFP, #P<0.05 vs. GFP+ET-treated.
Expression of npMARCKS in NRVM and ARVM acted as a dominant-negative inhibitor by reducing the phosphorylation of endogenous MARCKS and partially inhibiting ET-induced endogenous MARCKS phosphorylation (Fig 5A and B, right panels). Note that the expressed GFP-tagged npMARCKS protein is not detected due to the Ser → Ala mutations at 160/163. Basal FAK activation in NRVM and ARVM were reduced following npMARCKS expression, although only basal differences of phosphorylated FAK in NRVM reached statistical significance. In addition, ET-induced phosphorylation of FAK following npMARCKS expression was reduced somewhat when compared to ET-induced levels in GFP control cells. However, no difference was found when compared to basal levels in npMARCKS-expressing myocytes. The summarized experiments in NRVM are depicted in Figures 5C and 5D.
Effects of wtMARCKS and npMARCKS overexpression on cell spreading.
Since MARCKS has been shown to induce cell spreading in a number of other cell types, we examined the effects wtMARCKS and npMARCKS overexpression on NRVM surface area under basal conditions, and in response to prolonged stimulation with ET. NRVM were infected with Adv encoding GFP, wtMARCKS-GFP, or npMARCKS-GFP (24 or 48h), and then treated with ET (16 or 24h). Cells were then dye-loaded with BCECF and viewed under a confocal microscope in order to visualize the margins of the cell membrane. As seen in Figure 6, chronic ET treatment of Adv-GFP infected NRVM resulted in a near 2-fold increase in cell surface area. wtMARCKS overexpression alone increased cell spreading as compared to GFP-expressing cells, and ET did not further increase this response. Conversely, the average surface area of npMARCKS-overexpressing cells was similar to controls cells, but npMARCKS overexpression prevented the ET-induced increase in surface area. In order to relate alterations in cell surface area to that of cellular hypertrophy, protein/DNA ratios were measured in npMARCKS-GFP expressing NRVM treated chronically with PMA (200nM, 24h). Overexpression of npMARCKS-GFP lowered basal protein/DNA ratio, and prevented the PMA-induced increase in protein/DNA ratio (GFP=22.0±0.9; GFP+PMA=25.5±0.9; npMARCKS-GFP=20.2±0.7; npMARCKS-GFP+PMA=20.1±0.3 μg/μg; n=2 experiments).
Figure 6. Effects of wtMARCKS-GFP and npMARCKS-GFP on NRVM surface area.
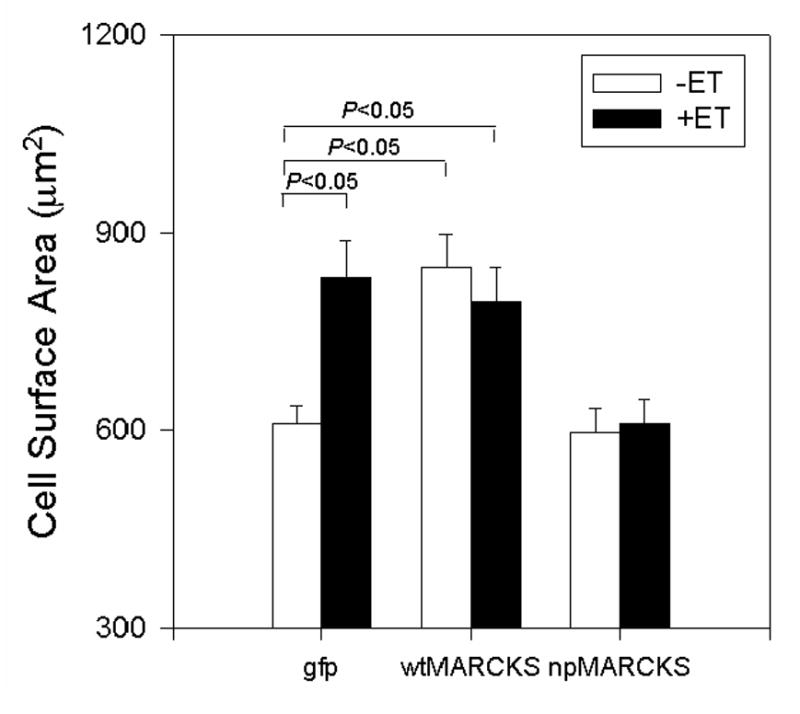
NRVM were infected with an adenovirus encoding either green fluorescent protein (GFP), wtMARCKS-GFP (10moi, 24h), or dnMARCKS-GFP (25moi, 48h), then treated with ET for either 16 or 24h. Cells were dye-loaded with BCECF and viewed by confocal microscopy in order to visualize the margins of the cell membrane.
DISCUSSION
Despite its initial description in 1982 (28), very little is known about the function and regulation of MARCKS phosphorylation in cardiomyocytes. McGill and Brooks (29) investigated the presence of MARCKS in whole rat ventricles and cardiomyocytes from developing rat hearts and determined that MARCKS expression changed dramatically during postnatal cardiac development. MARCKS protein was elevated at 5 days, decreased during 6 and 8 days, and then increased again at 11 days after birth. Furthermore, the reduction in MARCKS expression correlated with a peak in PKC activity at 8 days. Interestingly, although the MARCKS gene was expressed in both neonatal (2 day) and adult (42 day) hearts, MARCKS protein was not detected in adult cardiomyocytes in this study (29). In contrast, here we report the presence of phosphorylated MARCKS in both cultured neonatal myocytes, as well as both freshly isolated and cultured adult ventricular myocytes under basal conditions. The levels of phosphorylated MARCKS in adult cells were, however, approximately 5-fold less than those observed in neonatal cardiomyocytes (data not shown). These reduced levels in adult myocytes may help to explain the previous difficulty in detecting MARCKS protein in the adult heart.
A number of agonists which lead to phosphoinositide turnover such as PE, AngII and ET result in PKC activation through the generation of membrane diacylglycerols (30). Agonist-induced MARCKS phosphorylation has previously been demonstrated in NRVM in response to PE (14). PE-induced MARCKS phosphorylation increased 2-fold within 1 min and slowly returned to baseline within 15 min. In addition to PE, in the present study we investigated the ability of AngII and ET to elevate MARCKS phosphorylation in both NRVM and ARVM. We found that although all of the agonists tested resulted in an increase in the phosphorylation state of MARCKS within 5 min, ET induced the greatest elevation. This robust ET-induced MARCKS phosphorylation is not surprising, since ET substantially activates the novel PKC isoenzymes PKCε and PKCδ, but not the calcium-dependent PKC isoenzyme PKCα in both NRVM (13, 14) and ARVM (14). Furthermore, ET induced a more sustained activation of PKCε as compared to PKCδ (13) whereas PE-induced activation of PKCδ was barely detectable. The fact that PE increased MARCKS phosphorylation 2-fold (as demonstrated earlier (14) as well as in the present study), but only weakly activates PKCδ, is further support for the selective involvement of PKCε in MARCKS phosphorylation.
Our present results also support a previous study which examined the effect of selective down-regulation of PKC isoenzymes on MARCKS phosphorylation (14). PMA-induced activation and subsequent downregulation of PKCα, PKCε, and PKCδ were found to occur at distinctively different rates. Following a 4-hr treatment of NRVM with PMA, the immunoreactivity of PKCα and PKCδ were reduced to less than 20% and 10% of their basal values, whereas PKCε immunoreactivity remained elevated. It was deduced that because the MARCKS protein also remained heavily phosphorylated after a 4-hr PMA treatment, PKCε must be the isoenzyme responsible for phosphorylating MARCKS. The combination of the indirect evidence for PKCε phosphorylating MARCKS in this early study, and the more direct effect of PKCε overexpression presented here, makes a strong case for isoenzyme-selective PKC regulation of MARCKS in cardiomyocytes.
Although PKCε overexpression was sufficient to induce MARCKS phosphorylation, we found that overexpression of dnPKCε only partially reduced basal and ET-induced pMARCKS. There are a number of factors that may account for this observation. It is likely that additional pathways are involved in basal and ET-induced MARCKS phosphorylation which do not require PKCε. However, it seems unlikely that an incomplete inhibition of PKCε was responsible. Our group has previously shown that the same dnPKCε construct completely prevented ET-induced membrane translocation of endogenous PKCε activity, as assessed with [32P]ATP and a PKCε-specific substrate peptide (26). Nevertheless, a potentially confounding factor is the physical interaction of receptor for activated C kinase (RACK1) with PKC isoenzymes. Although RACK1 preferentially binds to Ca2+-dependent PKCs such as PKCα, Pass et al. (31) have shown that RACK1 can also bind to PKCε when overexpressed in mouse cardiomyocytes. We have also reported cross-talk between PKCα and PKCε. Overexpression of dnPKCε, even at moderate levels, reduced endogenous PKCα levels in NRVM (21). Conversely, overexpression of dnPKCα decreased endogenous PKCε levels. Despite these nonspecific effects, the likelihood that PKCα is involved in MARCKS phosphorylation seems remote, considering that overexpression of wtPKCα actually reduced MARCKS phosphorylation in the present study.
We have previously shown that caPKCε overexpression results in increased FAK autophosphorylation at Y397, whereas overexpression of caPKCδ reduced Y397 FAK phosphorylation. Conversely, dnPKCε overexpression reduced both basal and ET-induced FAK phosphorylation. Due to the fact that MARCKS is an actin filament cross-linking protein and has been implicated in cell spreading, adhesion, and focal adhesion formation in response to PKC activation in noncardiac cells (1–4), it seemed reasonable to expect that MARCKS is involved in PKCε-induced FAK phosphorylation in cardiomyocytes. Indeed, we now show that overexpression of wtMARCKS results in increased FAK phosphorylation at Y397 in both NRVM and ARVM. Conversely, overexpression of npMARCKS partially inhibited basal and ET-induced FAK phosphorylation. These data highlight the importance of MARCKS not only as an intermediate in ET-induced alterations in cell signaling through focal adhesions, but also suggests that maintenance of FAK function is partially dependent on the basal phosphorylation state of MARCKS.
We also report here that ET stimulation results in the redistribution of pMARCKS to portions of the cardiomyocyte actin cytoskeleton. Furthermore, dnPKCε overexpression partially prevented this ET-induced redistribution. Previous studies have shown that PKC-mediated phosphorylation of serines in the PSD domain decreases MARCKS’s affinity for the plasma membrane and leads to its translocation to actin filaments in the cytosol of nonmuscle cells (32, 33). The movement of signaling intermediates to specific actin-based adhesion sites can thus be be viewed as an integral step in the transmission of mechanochemical signaling.
As demonstrated in Figures 5 and 6, overexpression of wtMARCKS increased FAK phosphorylation and stimulated cell spreading in the absence of agonist stimulation. Conversely, a nonphosphorylatable MARCKS mutant reduced basal and ET-induced FAK phosphorylation, and also prevented ET-induced cell spreading and cellular hypertrophy. It is conceivable that the inability of the mutated MARCKS protein to undergo phosphorylation within its PSD domain rendered the molecule incapable of translocation to appropriate sites within the cytoskeleton, thereby regulating downstream functions related to focal adhesion formation and actin filament assembly. Of note, the importance of MARCKS phosphorylation and localization in cell spreading has been explored in α5 integrin-expressing myoblasts plated on fibronectin (2, 6). These investigators demonstrated that MARCKS expression and phosphorylation were both necessary and sufficient to induce myoblast FAK phosphorylation and cell spreading onto a fibronectin-coated substratum. Similarly, we found that expression of npMARCKS prevented both the ET-induced increase in cardiomyoycte cell spreading, as well as PMA-induced increases in protein/DNA. Nevertheless, future studies using the GFP-tagged, wt and npMARCKS in living cells will be necessary to better define the role of MARCKS translocation and phosphorylation in response to agonists that induce PKCε activation and cardiomyocyte hypertrophy.
In summary, we conclude that MARCKS is expressed in neonatal and adult cardiomyocytes, is phosphorylated by PKCε, and participates in the regulation of FAK phosphorylation and cell spreading.
Acknowledgments
These studies were supported by NIH RO1 grants HL34328 and HL63711, and a grant to the Cardiovascular Institute from the Ralph and Marian Falk Trust for Medical Research. Dr. Heidkamp was a recipient of NIH National Research Service Award HL68476 during the time these studies were performed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Manenti S, Malecaze F, Darbon JM. The major myristoylated PKC substrate (MARCKS) is involved in cell spreading, tyrosine phosphorylation of paxillin, and focal contact formation. FEBS Lett. 1997;419:95–98. doi: 10.1016/s0014-5793(97)01438-5. [DOI] [PubMed] [Google Scholar]
- 2.Disatnik MH, Boutet SC, Lee CH, Mochly-Rosen D, Rando TA. Sequential activation of individual PKC isozymes in integrin-mediated muscle cell spreading: a role for MARCKS in an integrin signaling pathway. J Cell Sci. 2002;115:2151–2163. doi: 10.1242/jcs.115.10.2151. [DOI] [PubMed] [Google Scholar]
- 3.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–622. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 4.Aderem A. The MARCKS family of protein kinase-C substrates. Biochem Soc Trans. 1995;23:587–591. doi: 10.1042/bst0230587. [DOI] [PubMed] [Google Scholar]
- 5.Disatnik MH, Rando TA. Integrin-mediated muscle cell spreading. The role of protein kinase c in outside-in and inside-out signaling and evidence of integrin cross-talk. J Biol Chem. 1999;274:32486–32492. doi: 10.1074/jbc.274.45.32486. [DOI] [PubMed] [Google Scholar]
- 6.Disatnik MH, Boutet SC, Pacio W, Chan AY, Ross LB, Lee CH, Rando TA. The bidirectional translocation of MARCKS between membrane and cytosol regulates integrin-mediated muscle cell spreading. J Cell Sci. 2004;117:4469–4479. doi: 10.1242/jcs.01309. [DOI] [PubMed] [Google Scholar]
- 7.Molkentin JD, Dorn IG., 2nd Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 8.Sugden PH, Clerk A. Cellular mechanisms of cardiac hypertrophy. J Mol Med. 1998;76:725–746. doi: 10.1007/s001090050275. [DOI] [PubMed] [Google Scholar]
- 9.Zhu W, Zou Y, Aikawa R, Harada K, Kudoh S, Uozumi H, Hayashi D, Gu Y, Yamazaki T, Nagai R, Yazaki Y, Komuro I. MAPK superfamily plays an important role in daunomycin-induced apoptosis of cardiac myocytes. Circulation. 1999;100:2100–2107. doi: 10.1161/01.cir.100.20.2100. [DOI] [PubMed] [Google Scholar]
- 10.Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential activation of mitogen-activated protein kinase cascades and apoptosis by protein kinase C epsilon and delta in neonatal rat ventricular myocytes. Circ Res. 2001;89:882–890. doi: 10.1161/hh2201.099434. [DOI] [PubMed] [Google Scholar]
- 11.Heidkamp MC, Bayer AL, Scully BT, Eble DM, Samarel AM. Activation of focal adhesion kinase by protein kinase C epsilon in neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2003;285:H1684–1696. doi: 10.1152/ajpheart.00016.2003. [DOI] [PubMed] [Google Scholar]
- 12.Bayer AL, Ferguson AG, Lucchesi PA, Samarel AM. Pyk2 expression and phosphorylation in neonatal and adult cardiomyocytes. J Mol Cell Cardiol. 2001;33:1017–1030. doi: 10.1006/jmcc.2001.1369. [DOI] [PubMed] [Google Scholar]
- 13.Clerk A, Bogoyevitch MA, Anderson MB, Sugden PH. Differential activation of protein kinase C isoforms by endothelin-1 and phenylephrine and subsequent stimulation of p42 and p44 mitogen- activated protein kinases in ventricular myocytes cultured from neonatal rat hearts. J Biol Chem. 1994;269:32848–32857. [PubMed] [Google Scholar]
- 14.Puceat M, Hilal-Dandan R, Strulovici B, Brunton LL, Brown JH. Differential regulation of protein kinase C isoforms in isolated neonatal and adult rat cardiomyocytes. J Biol Chem. 1994;269:16938–16944. [PubMed] [Google Scholar]
- 15.Eble DM, Strait JB, Govindarajan G, Lou J, Byron KL, Samarel AM. Endothelin-induced cardiac myocyte hypertrophy: role for focal adhesion kinase. Am J Physiol Heart Circ Physiol. 2000;278:H1695–1707. doi: 10.1152/ajpheart.2000.278.5.H1695. [DOI] [PubMed] [Google Scholar]
- 16.Pham CG, Harpf AE, Keller RS, Vu HT, Shai SY, Loftus JC, Ross RS. Striated muscle-specific beta(1D)-integrin and FAK are involved in cardiac myocyte hypertrophic response pathway. Am J Physiol Heart Circ Physiol. 2000;279:H2916–2926. doi: 10.1152/ajpheart.2000.279.6.H2916. [DOI] [PubMed] [Google Scholar]
- 17.Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275:19250–19257. doi: 10.1074/jbc.M909099199. [DOI] [PubMed] [Google Scholar]
- 18.Samarel AM, Engelmann GL. Contractile activity modulates myosin heavy chain-beta expression in neonatal rat heart cells. Am J Physiol. 1991;261:H1067–1077. doi: 10.1152/ajpheart.1991.261.4.H1067. [DOI] [PubMed] [Google Scholar]
- 19.Westfall MV, Rust EM, Albayya F, Metzger JM. Adenovirus-mediated myofilament gene transfer into adult cardiac myocytes. Methods Cell Biol. 1997;52:307–322. [PubMed] [Google Scholar]
- 20.Strait JB, Samarel AM. Isoenzyme-specific protein kinase C and c-Jun N-terminal kinase activation by electrically stimulated contraction of neonatal rat ventricular myocytes. J Mol Cell Cardiol. 2000;32:1553–1566. doi: 10.1006/jmcc.2000.1191. [DOI] [PubMed] [Google Scholar]
- 21.Porter MJ, Heidkamp MC, Scully BT, Patel N, Martin JL, Samarel AM. Isoenzyme-selective regulation of SERCA2 gene expression by protein kinase C in neonatal rat ventricular myocytes. Am J Physiol Cell Physiol. 2003;285:C39–47. doi: 10.1152/ajpcell.00461.2002. [DOI] [PubMed] [Google Scholar]
- 22.Ping P, Zhang J, Cao X, Li RC, Kong D, Tang XL, Qiu Y, Manchikalapudi S, Auchampach JA, Black RG, Bolli R. PKC-dependent activation of p44/p42 MAPKs during myocardial ischemia-reperfusion in conscious rabbits. Am J Physiol. 1999;276:H1468–1481. doi: 10.1152/ajpheart.1999.276.5.H1468. [DOI] [PubMed] [Google Scholar]
- 23.He X, Goldsmith CM, Marmary Y, Wellner RB, Parlow AF, Nieman LK, Baum BJ. Systemic action of human growth hormone following adenovirus-mediated gene transfer to rat submandibular glands. Gene Ther. 1998;5:537–541. doi: 10.1038/sj.gt.3300622. [DOI] [PubMed] [Google Scholar]
- 24.Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM. GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ Res. 2002;90:1282–1289. doi: 10.1161/01.res.0000023201.41774.ea. [DOI] [PubMed] [Google Scholar]
- 25.Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strait JB, 3rd, Martin JL, Bayer A, Mestril R, Eble DM, Samarel AM. Role of protein kinase C-epsilon in hypertrophy of cultured neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2001;280:H756–766. doi: 10.1152/ajpheart.2001.280.2.H756. [DOI] [PubMed] [Google Scholar]
- 27.Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential activation of mitogen-activated protein kinase cascades and apoptosis by protein kinase C epsilon and delta in neonatal rat ventricular myocytes. Circ Res. 2001;89:882–890. doi: 10.1161/hh2201.099434. [DOI] [PubMed] [Google Scholar]
- 28.Wu WC, Walaas SI, Nairn AC, Greengard P. Calcium/phospholipid regulates phosphorylation of a Mr “87k” substrate protein in brain synaptosomes. Proc Natl Acad Sci U S A. 1982;79:5249–5253. doi: 10.1073/pnas.79.17.5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGill CJ, Brooks G. Expression and regulation of 80K/MARCKS, a major substrate of protein kinase C, in the developing rat heart. Cardiovasc Res. 1997;34:368–376. doi: 10.1016/s0008-6363(97)00041-2. [DOI] [PubMed] [Google Scholar]
- 30.Sugden PH, Clerk A. Endothelin signalling in the cardiac myocyte and its pathophysiological relevance. Curr Vasc Pharmacol. 2005;3:343–351. doi: 10.2174/157016105774329390. [DOI] [PubMed] [Google Scholar]
- 31.Pass JM, Zheng Y, Wead WB, Zhang J, Li RC, Bolli R, Ping P. PKCepsilon activation induces dichotomous cardiac phenotypes and modulates PKCepsilon-RACK interactions and RACK expression. Am J Physiol Heart Circ Physiol. 2001;280:H946–955. doi: 10.1152/ajpheart.2001.280.3.H946. [DOI] [PubMed] [Google Scholar]
- 32.Thelen M, Rosen A, Nairn AC, Aderem A. Regulation by phosphorylation of reversible association of a myristoylated protein kinase C substrate with the plasma membrane. Nature. 1991;351:320–322. doi: 10.1038/351320a0. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Gambhir A, Hangyas-Mihalyne G, Murray D, Golebiewska U, McLaughlin S. Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J Biol Chem. 2002;277:34401–34412. doi: 10.1074/jbc.M203954200. [DOI] [PubMed] [Google Scholar]