

Abstract
Incomplete intrinsic penetrance is the failure of some genetically susceptible individuals (e.g., monozygotic twins of those who have a trait) to exhibit that trait. For the first time, we examine penetrance of susceptibility genes for multiple MHC gene-determined traits in the same subjects. Serum levels of IgA, IgD, IgG3, but not IgG4, in 50 pairs of monozygotic twins discordant for type 1 diabetes (T1D) correlated more closely in the twins than in random paired controls. The frequencies of subjects deficient in IgA (6%), IgD (33%) and IgG4 (12%), but not in IgG3, were higher in the twins than in controls. We postulate that this was because the MHC haplotypes (and possible non-MHC genes) that predispose to T1D also carry susceptibility genes for certain immunoglobulin deficiencies. Immunoglobulin deficiencies were not associated with T1D. Pairwise concordance for the deficiencies in the twins was 50% for IgA, 57% for IgD and 50% for IgG4. There were no significant associations among the specific immunoglobulin deficiencies except that all IgA-deficient subjects had IgD deficiency. Thus, intrinsic penetrance is a random process independently affecting different MHC susceptibility genes. Because multiple different external triggers would be required to explain the results, differential environmental determinants appear unlikely.
Keywords: Immunoglobulin deficiency, Type 1 diabetes, Incomplete penetrance, Major histocompatibility complex
Introduction
Incomplete penetrance of susceptibility genes is characteristic of many complex diseases, particularly those with susceptibility genes within the major histocompatibility complex (MHC) [1]. The best-studied example of incomplete penetrance is type 1 diabetes (T1D) [2]. About 6% of sibs of a patient are concordant for T1D. Because about 16% of MHC-identical sibs of a patient also have T1D, this is strong evidence for a susceptibility gene within the MHC. On the other hand, since 33 to 42% of monozygotic twins of a T1D patient also have the disease [3] (probandwise concordance), it is clear that non-MHC susceptibility genes are also required for susceptibility. We refer to the probandwise concordance rate in (genetically identical) monozygotic twins as intrinsic penetrance, whereas we refer to penetrance observed in other relatives of a patient or among unrelated persons as apparent penetrance [1]. Apparent penetrance varies with the number of all susceptibility genes shared with a patient.
Incomplete penetrance of susceptibility genes for gluten-sensitive enteropathy (GSE), another MHC allele-associated autoimmune disorder, has also been found in monozygotic twins and MHC-identical siblings of patients with the disease [4]. We previously found incomplete penetrance for certain MHC-determined immunoglobulin deficiencies (Igds) [5–7]. Thus, incomplete penetrance could be a general intrinsic property of some MHC genes. Studies of Igds in monozygotic twins discordant for T1D would provide critical information regarding the interrelationship of penetrance rates.
Another important question that can be answered by studies in monozygotic twins is whether a quantitative trait such as the level of immunoglobulin in serum is genetically controlled. Previous studies of twins have indicated genetic control of serum concentrations of IgE [8], IgM [9] and total IgG [9]. If the monozygotic twins are discordant for T1D, one can also obtain information about the influence of the disease on Ig levels in persons with identical genes.
In the present study, we examined serum Ig levels in 50 pairs of monozygotic twins discordant for T1D, since T1D patients are enriched in conserved extended haplotypes (CEHs) that confer susceptibility to the Igds of interest. The questions asked were: (a) Are IgA, IgD, IgG3 and IgG4 serum concentrations more similar in twins than in random unrelated pairs of individuals? (b) Are rates of IgAd, IgDd, IgG3d or IgG4d in persons with T1D and/or fully genetically susceptible to it but healthy different from the rates in normal controls? (c) Are the rates of IgAd, IgDd, IgG3d or IgG4d in patients with T1D different from those in their healthy monozygotic twins? (d) What are the rates of co-occurrence for IgAd, IgDd, IgG3d and IgG4d and for T1D among the discordant monozygotic twins? (e) What are the rates of concordance/discordance for IgAd, IgDd, IgG3d and IgG4d among monozygotic twins discordant for T1D?
Materials and methods
Twins discordant for T1D and controls
Identical twin pairs were selected from the British Diabetic Twin Study [3]. Twins from the registry are ascertained by referral through their physicians. Monozygosity was established in all twin pairs, using both clinical data and at least 22 blood groups, as described previously [3]. This establishes identity with a less than 3% chance of error [10]. Type 1 diabetes was defined according to the National Diabetes Data Group criteria [11] and diabetes was excluded in the non-diabetic co-twins by a 75 g oral glucose tolerance test and random whole blood glucose at testing of less than 7.0 mmol/L. Blood glucose was estimated on venous whole blood (YSI, Yellow Springs, OH). All diabetic twins had been treated from the time of diagnosis with insulin and were taking highly purified human insulin at least twice daily. None of the twins had a clinical history of susceptibility to infections. Mean ± Standard Deviation (SD) duration of diabetes was 17 ± 11 years in the diabetic twin.
Of 451 twin pairs we selected 50 identical pairs discordant for T1D, which were eligible according to the following criteria: 1) European origin, 2) Affected twins had T1D, 3) Both twins of each pair were available for study, 4) Neither twin was receiving drugs other than human insulin in the index case, 5) The non-diabetic twins were all tested for the T1D-associated autoantibodies to glutamic acid decarboxylase (GAD) and insulinoma-associated antigen-2 (IA-2) using established methodology [12]. Briefly, radioimmunoprecipitation assays for anti-GAD65 and anti-IA-2 (aa 603–979) utilized transcription and translation systems (Promega, Madison, WI). For both antibody assays 0.8–1.0 mg DNA was transcribed and translated with SP6 (IA-2ic) and T7 (GAD65) RNA polymerase in a TNT-coupled reticulocyte lysate system (Promega) in the presence of 35S methionine (0.8 μCi) (Amersham, UK). Fifty μL aliquots of 35S methionine- (50–75,000 cpm) labeled antigen were incubated overnight with 2 μL serum (final dilution 1:25) in trisbuffered saline. The immune complexes were isolated by adding 1 mg protein A-Sepharose and were counted in a multiwell Wallac counter. All samples were tested in duplicate, including positive and negative control standard sera. Each assay for IA-2 and GAD65 antibodies included serially diluted sera from a prediabetic individual standardized to the WHO DASP antibody workshop [13] standard serum sample to evaluate the cutoff level for positivity for anti-GAD and anti-IA-2. Values > control + 3 SD were taken as positive. Twins were selected to be negative for these tests and to have normal glucose tolerance. In the DASP antibody workshop, our assays for anti-GAD had a sensitivity of 74% and a specificity of 98% and for anti-IA-2 a sensitivity of 62% and a specificity of 100% [13]. This gave negative subjects less than a 2% risk of developing T1D. All subjects gave informed consent and the study was approved by the ethics committee of the Royal Hospital Trusts and the Institutional Review Board of The CBR Institute for Biomedical Research.
Serum immunoglobulin concentrations
Serum levels of IgA, IgG3, IgG4 and IgD were determined by ELISA [14] or radial immunodiffusion [15] using specific polyclonal goat antisera (Incstar, Clearwater, MN) or mouse monoclonal antibodies (Pierce, Rockford, IL) or kits (The Binding Site, San Diego, CA). We used Ig class and subclass levels in 187 unrelated healthy Caucasian individuals, as reported earlier [7], to define Igds as the geometric means − 2 SD (IgD < 0.14 mg/dL, IgG3 < 20.1 mg/dL, IgG4 < 3.8 mg/dL). IgAd was taken as < 5 mg/dL [16].
Statistical analyses
In general, the log-transformed levels in all populations of subjects were more normally distributed than direct values. Therefore, the log-transformed serum concentrations were used to calculate the mean - 2 SD, using the JMP (SAS Institute, Cary, NC) statistical program. The frequencies in each population of subjects of individual Igds (values < geometric mean in normal control subjects − 2 SD) were compared for statistical significance using 2 × 2 contingency tables and χ2 analysis or Fisher's exact test, as appropriate. The comparison of observed and expected co-occurrences of different Igds was evaluated for statistical significance by χ2 analysis where χ2 = (observed – expected)2/expected. Apparent penetrance was analyzed as described previously [1,2]. To determine whether levels in the twin pairs correlated more closely than in pairs of random unrelated persons, the concentrations of each of the Ig classes and subclasses were measured in serum from 134 unrelated healthy individuals assigned randomly to pairs. IgA, IgD, IgG3 and IgG4 serum levels were compared in these paired samples as were those from the monozygotic twin pairs. Two measures of co-variation were used: (a) the paired t-test, in which the null hypothesis was that the mean difference between Ig levels in the twin pairs was zero (the t-statistic has a t-distribution with n − 1 degrees of freedom, with n being equal to the number of twin pairs) and (b) the significance of the differences between the correlation coefficients (r) in the monozygotic twins compared with those in the random pairs for each Ig. In situations where the t-test gave borderline p values, we re-tested the significance of the difference of mean values using a sensitive F-test that is expressed as variances of two populations. The null hypothesis is that if the two variances are the same, then F = 1. The F-test is sensitive to the effect of the disease on Ig levels.
Results
Serum IgA, IgD, IgG3 and IgG4 levels in monozygotic twin pairs and in randomly paired unrelated individuals
Table 1 shows the results of analysis for correlation between serum Ig levels in the monozygotic twin pairs (Table 1a) compared with levels in serum from unrelated randomly paired controls (Table 1b). It is clear that the levels of IgA, IgDd and IgG3 (but not of IgG4) were significantly correlated in the monozygotic twins but not in the random subjects.
Table 1.
Correlation analysis of serum immunoglobulin levels
| (a) in monozygotic twin pairs | ||||
| Statistic | IgA | IgD | IgG3 | IgG4 |
| t value (paired t-test ) | 3.24 | −1.89 | 2.77 | 0.23 |
| r | 0.674 | 0.609 | 0.619 | 0.150 |
| pa | < 0.0001 | < 0.0001 | < 0.0001 | 0.298 |
| (b) in randomly paired unrelated control subjects | ||||
| Statistic | IgA | IgD | IgG3 | IgG4 |
| t value (paired t-test ) | 1.35 | 0.44 | 0.11 | 0.03 |
| r | −0.092 | 0.023 | −0.015 | −0.203 |
| pa | 0.527 | 0.851 | 0.862 | 0.094 |
p represents probability value of t-test.
Statistical significance is p < 0.05 (correlation probability).
The frequencies of IgAd, IgDd, IgG3d and IgG4d in the twins compared with normal controls
As is seen in Table 2, the frequencies of Igds among the T1D patients and their healthy twins of this study were higher for IgAd, IgDd and IgG4d than those in normal individuals. The observed excesses ranged from approximately 43-fold for IgAd to approximately 2 to 5-fold for IgG4d and IgDd. There were no significant differences in Igd frequencies in the members of the twin pairs who had T1D compared with those who did not (not shown).
Table 2.
Immunoglobulin deficiency frequencies in T1D twins and normal subjects
| Population | IgAd | IgDd | IgG3d | IgG4d |
|---|---|---|---|---|
| T1D twins | 0.060 | 0.330 | 0.040 | 0.121 |
| Normals | 0.0014a | 0.067 | 0.045 | 0.063 |
| Ratio T1D/Normal | 43 | 4.9 | 0.67 | 1.9 |
The frequency of IgAd in normal Caucasians is taken from the literature [16].
Surprisingly, although we earlier observed a marked increase in frequency of IgG3d among homozygotes and, to a lesser extent, heterozygotes for the [HLA-B8, SC01, DR3] CEH, and the frequency of this CEH is increased among T1D patients, the rate of IgG3d among the twins (0.040) with T1D was no higher than that in our normal controls (0.045).
Mean immunoglobulin levels in patients with T1D, their healthy monozygotic twins and normal controls
Table 3 shows mean serum Ig concentrations in the monozygotic twins with T1D compared with those who were healthy as well as in 53 normal controls. The mean levels of IgA were significantly higher (p = 0.002) and of IgG3 were significantly lower (p < 0.02) in the twins with T1D than in those who were healthy. IgA levels in the healthy twins were higher (p = 0.0015) and IgD levels were lower (p = 0.0022) than controls. Thus, the mean levels of IgA and IgD in the healthy twins were intermediate between those of T1D patients and controls. The mean serum IgA, IgD, IgG3 and IgG4 levels were all significantly different in all of the twins compared with normal controls. The levels of IgG4 in T1D and control groups showed a weak difference.
Table 3.
Mean immunoglobulin levels in patients with T1D, their healthy monozygotic twins and normal controlsa,b
| Immunoglobulin | T1D patient Mean level mg/dL | Healthy twin Mean level mg/dL | Controls Mean level mg/dL |
|---|---|---|---|
| IgA | 413 ± 44 | 309 ± 28 | 187 ± 13 |
| IgD | 1.02 ± 0.19 | 1.36 ± 0.21 | 2.70 ± 0.30 |
| IgG3 | 63 ± 5 | 72 ± 5 | 42 ± 2 |
| IgG4 | 29 ± 4 | 22 ± 3 | 39 ± 3 |
Unpaired t test with unequal variance. Mean levels are given + 1 SEM.
IgA levels in T1D patients vs control t probability p < 0.0001, IgA levels in healthy twins vs control t probability p = 0.0002, IgD levels in T1D patients vs control t probability p < 0.001, IgD levels in healthy twins vs control t probability p = 0.0004, IgG3 levels in T1D patients vs control t probability p = 0.0003, IgG3 levels in healthy twins vs control t probability p < 0.0001, IgG4 levels in T1D patients vs control t probability p = 0.02, (Fprob = 0.69), IgG4 levels in healthy twins vs control t probability p < 0.0001.
Serum Ig levels in the monozygotic twins with and without T1D and in normal controls are shown in Fig. 1. It is striking that, for IgA, IgG3 and IgG4, the deficient subjects had levels quite apart from those of other subjects. In contrast, IgD levels formed a continuum in the twins (with or without T1D) that is far less striking in the normal subjects.
Fig. 1.
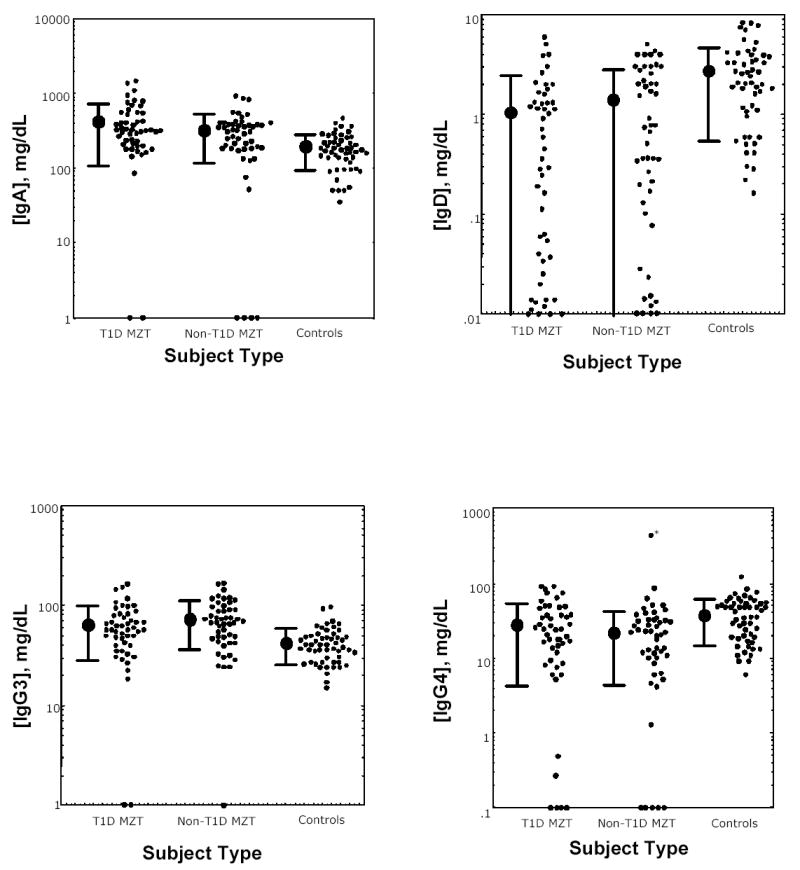
IgA (upper left), IgD (upper right), IgG3 (lower left) and IgG4 (lower right) serum concentrations in monozygotic twins with (left) and without (middle) T1D and healthy controls (right) for each immunoglobulin. Individual subject serum levels and group geometric mean levels ± 1 SD are shown. An outlier (*) was omitted from the calculation of the mean in the lower right panel.
The co-occurrence of immunoglobulin deficiencies
Fifty-eight (58%) of the 100 individuals of the monozygotic twin pairs had 1 or more Igds. Of these, 43 were single Igds (28 IgDd alone, 13 IgG4d alone and 2 IgG3d alone). Of 13 with 2 Igds, 7 were IgDd and IgG4d, 4 were IgAd and IgDd, and 1 each was IgDd and IgG3d and IgG3d and IgG4d. An important question was whether the frequency of co-occurrence of individual Igds in single individuals differed from chance. If co-occurrence is by chance, the frequency of co-occurrence (observed) should be equal to the product of the individual frequencies (expected). Of the various double-Igd combinations, only IgAd and IgDd co-occurred with significantly increased frequency, as seen in Table 4 and as we have observed in other IgAd patients without T1D (data not shown). There was no disproportional co-occurrence of any Igd with T1D among the twins.
Table 4.
Co-occurrence of immunoglobulin deficiencies and T1D in individual members of T1D-discordant monozygotic twin pairs
| Co-occurrence | Observed | Expected | p |
|---|---|---|---|
| IgAd-IgDd | 6 | 2.0 | < 0.005 |
| IgAd-IgG3d | 0 | 0.2 | n.s.a |
| IgAd-IgG4d | 2 | 0.7 | n.s. |
| IgDd-IgG3d | 1 | 1.0 | n.s. |
| IgDd-IgG4d | 4 | 4.0 | n.s. |
| IgG3d-IgG4d | 1 | 0.4 | n.s. |
| IgAd-T1D | 2 | 3.0 | n.s. |
| IgDd-T1D | 19 | 17.0 | n.s. |
| IgG3d-T1D | 1 | 1.5 | n.s. |
| IgG4d-T1D | 6 | 6.0 | n.s. |
n.s. = not significant.
Concordance for IgAd, IgDd, IgG3d and IgG4d in the monozygotic twin pairs discordant for T1D
Pairwise concordances for the Igds studied are shown in Table 5. The observed concordance rate (intrinsic penetrance) for IgAd was 50% and the rates for IgDd and IgG4d were 57% and 50%, respectively. Since only 4 subjects had IgG3d, it was not possible to interpret the observed lack of concordance.
Table 5.
Concordance for immunoglobulin deficiencies in T1D-discordant twins
| Pairwise concordance | IgAd | IgDd | IgG3d | IgG4d |
|---|---|---|---|---|
| Concordant no./no. of affected twin pairs | 2/4 | 12/21 | 0/4 | 4/8 |
| Concordant frequency | 0.50 | 0.57 | 0 | 0.50 |
Discussion
For the first time, the relationships among the rates of concordance (intrinsic penetrance) for more than one MHC-influenced trait have been determined in a group of monozygotic twins (who were also discordant for T1D). The effort was helped considerably by the remarkably increased frequencies of a number of Igds among patients with T1D. This is most likely the result of the carrying of increased frequencies of susceptibility genes for Igds by many of those haplotypes that confer susceptibility to T1D. This is certainly true of MHC susceptibility genes but should also hold for non-MHC susceptibility genes for both kinds of traits, one definitely polygenic (T1D), the other possibly polygenic (Igds). A previous report from Italy [17] found IgAd in 7 of 191 T1D patients for a rate of 3.7% compared with 0.2% in Italian controls.
Why would two supposedly genetically identical individuals differ in the expression of T1D? The explanation offered almost 80 years ago [18] and still considered most likely [19] is that one monozygotic twin was exposed to an environmental trigger that the other was not. Attempts to identify such an environmental trigger (a favorite has been a viral infection) have not been successful [20]. Moreover, concordance in dizygotic twins was identical to the rate in sibs in general in a large-scale study [21], suggesting no detectable difference in the frequency of environmental triggers between the sibs with very similar and less similar environments.
We had earlier explored the nature and extent of apparent penetrance of Igds through the study of unrelated individuals who were homozygous, heterozygous or non-carrying for CEHs [5–7] known or suspected to carry susceptibility genes for MHC-determined Igds such as IgAd [16], IgDd [22], IgG3d and IgG4d. CEHs are ≥ 1 Mb stretches of fixed genomic DNA defined by their HLA-B, complotype, and HLA-DR alleles [23]. It is CEHs that provide almost all of the individual MHC markers for HLA-associated diseases [2,24]. Among normal Caucasian MHC haplotypes, CEHs account for at least 30% [23] and up to 50% or more [25] of MHC haplotypes. The CEH [HLA-B8, SC01, DR3], where SC01 represents the complotype BF*S, C2*C, C4A*Q0, C4B*1, is the most common CEH of northern European Caucasians (8% in our Boston database) and is particularly common in the British Isles. It is elevated in frequency in patients with a variety of autoimmune diseases, including T1D [24], GSE [26], myasthenia gravis [27] and systemic lupus erythematosus [28]. It is also associated with IgAd [16], IgDd [22], IgG3d [5] and IgG4d [5].
Given the fixity of DNA on CEHs, if a susceptibility gene occurs on a CEH, as evidenced by its elevated frequency among patients compared with ethnically-matched controls, it will occur on virtually all independent examples of that CEH in the population. Based on this concept, we developed the prospective method for analyzing the mode of inheritance and estimating apparent penetrance of genes on CEHs [1,5]. If homozygotes but few, if any, heterozygotes or non-carriers for a CEH exhibit an associated trait, the susceptibility gene carried by the CEH is expressed recessively. On the other hand, if both homozygotes and heterozygotes but few, if any, non-carriers express the trait, the susceptibility gene is dominantly expressed.
In our earlier work on Igds, homozygotes, but not heterozygotes, for the CEH [HLA-B8, SC01, DR3] had markedly increased rates of IgAd and IgG4d compared with heterozygotes and controls, suggesting recessive inheritance [5]. In contrast, heterozygotes, as well as homozygotes, had increased rates of IgDd and IgG3d, consistent with dominant inheritance. Rates of IgDd and IgG3d in homozygotes were higher than in heterozygotes. Because the Igds occurred, in general, independently of each other and because similar Ig studies of another DR3-carrying CEH [HLA-B18, F1C30, DR3] [6] revealed only IgDd (but not IgAd, IgG3d nor IgG4d), it was clear that the susceptibility genes for each of the Igds were distinct from each other. Since the IgDd frequencies in [HLA-B18, F1C30, DR3] homozygotes (37%) and heterozygotes (19%) [6] were very similar to those in [HLA-B8, SC01, DR3] homozygotes and heterozygotes, (37% and 20%, respectively) [5], it may be that the susceptibility gene for IgDd is the same on both haplotypes.
Penetrance, if it were the result of an external triggering event, would be expected to affect the whole organism. For a dominant trait, it should be irrelevant for trait expression whether the host has one or two susceptibility genes [1]. On the other hand, if penetrance were a property of the susceptibility gene on each chromosome, homozygotes with two susceptibility genes would be expected to have a higher rate than heterozygotes with only one. Since the rates of IgG3d and IgDd in [HLA-B8, SC01, DR3] and of IgDd in [HLA-B18, F1C30, DR3] homozygotes were higher than those in heterozygotes, this suggested a stochastic basis for penetrance affecting homologous genes on each chromosome. In addition, it appeared that penetrance was a manifestation of the MHC susceptibility genes, since the only difference between the subjects was in their MHC haplotypes. If MHC genes are recessively expressed, penetrance will be the same for the external trigger and intrinsic switch mechanisms, providing no information, although there is no reason to believe that the intrinsic switch isn’t also operative in recessive as well as dominant MHC-determined trait expression.
There is almost no literature on Igds in monozygotic twins. We are aware of only a single report of a pair of monozygotic twins discordant for IgAd [29]. The rate of pairwise concordance among the IgAd twins in our report was 50%, among the IgDd twins was 57% and among those with IgG4d was 50%. These rates are considerably higher than the reported pairwise monozygotic twin rate for T1D of 27% [21]. The fact that, for the most part, twin pair concordances and co-occurrence for the Igds in the monozygotic twin pairs were independent of each other (Table 3) and of concordance for and co-occurrence with T1D is consistent with intrinsic penetrance being a stochastic process [1,6]. If there were a specific environmental trigger for T1D, then there would have to be separate and different triggers for each of the Igds.
The one exception to independence of occurrence of Igds is the finding that all 6 of the IgAd patients also had IgDd, although most cases of IgDd occurred without IgAd. Of 13 IgAd patients (without T1D) that we have studied, 10 or 77%, had IgDd (unpublished findings), far in excess of the considerably less than the 2.6% expected if co-occurrence were random. The association is thus independent of susceptibility to T1D. It is unclear why having IgAd predisposes to IgDd (or perhaps vice versa).
Certainly, an important question is whether any of the Igds occurring at high frequencies among patients with T1D contributes to the known high incidence of infections among them [30]. It is unlikely that IgDd contributes since there is no clear function identified for serum IgD. IgAd is associated with increased frequency of sinopulmonary infections [31] in some patients and IgG4d has been reported to be associated with increased susceptibility to bacterial infections as well [32,33]. A previous study of Ig levels in T1D patients revealed no strong link between low levels and infections in T1D patients [34]. Moreover, none of the patients in the current study had known increased frequency of infections.
Neither the MHC susceptibility loci nor the molecular mechanisms of IgAd (or any other Igd) or T1D are known, rendering purely speculative any conjecture regarding the mechanism of incomplete penetrance for these MHC-associated disorders. Genetic expression of the five genetic traits (T1D and the four Igds) were unrelated to one another. The stochastic nature of the intrinsic penetrance for these traits suggests to us the likelihood of intrinsic epigenetic regulation operating independently on different MHC susceptibility genes [1,2]. The likelihood of an external environmental trigger for these independent traits, for which there is no clear evidence, is reduced because each trait would require a separate differential trigger.
There are some interesting parallels between IgAd and autoimmunity. An important issue for some IgAd patients is the presence of anti-IgA in their serum that can cause anaphylactic reactions upon transfusion with IgA-containing blood. The frequency of anti-IgA antibodies varies from 20–40%, with a United Kingdom study reporting that 1 of 3 IgAd patients had antibodies to IgA [35]. Although the relationship of IgA deficiency to the presence of anti-IgA antibodies is not clearly established, this autoimmune phenomenon may be related to the fact that the inability to produce IgA in IgA deficiency is due to the increased destruction of a subset of B cells in these patients [36].
Acknowledgments
This work was supported by a grant from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL29583; CAA, ZH, CEL, DPD, RS, CD, AB), The British Diabetic Twin Research Trust (RDL), and the Juvenile Diabetes Research Foundation International (RDL). We thank Pam Sawtell for handling samples. We thank Ms. Louise Viehmann for outstanding secretarial services.
References
- 1.Alper CA, Awdeh Z. Incomplete penetrance of MHC susceptibility genes: prospective analysis of polygenic MHC-determined traits. Tissue Antigens. 2000;56:199–206. doi: 10.1034/j.1399-0039.2000.560301.x. [DOI] [PubMed] [Google Scholar]
- 2.Larsen CE, Alper CA. The genetics of HLA-associated disease. Curr Opin Immunol. 2004;16:660–7. doi: 10.1016/j.coi.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, et al. Heterogeneity of type 1 diabetes. Analysis of monozygotic twins in Great Britain and the United States. Diabetologia. 2001;44:354–62. doi: 10.1007/s001250051626. [DOI] [PubMed] [Google Scholar]
- 4.Greco L, Romino R, Coto I, Di Cosmo N, Percopo S, Maglio M, et al. The first large population based twin study of coeliac disease. Gut. 2002;50:624–8. doi: 10.1136/gut.50.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alper CA, Marcus-Bagley D, Awdeh Z, Kruskall MS, Eisenbarth GS, Brink SJ, et al. Prospective analysis suggests susceptibility genes for deficiencies of IgA and several other immunoglobulins on the [HLA-B8, SC01, DR3] conserved extended haplotype. Tissue Antigens. 2000;56:207–16. doi: 10.1034/j.1399-0039.2000.560302.x. [DOI] [PubMed] [Google Scholar]
- 6.Calvo B, Castaño L, Marcus-Bagley D, Fici DA, Awdeh Z, Alper CA. The [HLA-B18, F1C30, DR3] conserved extended haplotype carries a susceptibility gene for IgD deficiency. J Clin Immunol. 2000;20:216–20. doi: 10.1023/a:1006693614974. [DOI] [PubMed] [Google Scholar]
- 7.Alper CA, Xu J, Cosmopoulos K, Dolinski B, Stein R, Uko G, et al. Immunoglobulin deficiencies and susceptibility to infection among homozygotes and heterozygotes for C2 deficiency. J Clin Immunol. 2003;23:297–305. doi: 10.1023/a:1024540917593. [DOI] [PubMed] [Google Scholar]
- 8.Strachan DP, Wong HJ, Spector TD. Concordance and interrelationship of atopic disease and markers of allergic sensitization among adult female twins. J Allergy Clin Immunol. 2001;108:901–7. doi: 10.1067/mai.2001.119408. [DOI] [PubMed] [Google Scholar]
- 9.Anton-Guirgis H, Culver BD, Kurosaki T, Elston R. A study of multiple biological markers in twins. Acta Genet Med Gemellol. 1985;34:153–65. doi: 10.1017/s0001566000004670. [DOI] [PubMed] [Google Scholar]
- 10.Barnett AH, Eff C, Leslie RD, Pyke DA. Diabetes in identiclal twins. A study of 200 pairs. Diabetologia. 1981;20:87–93. doi: 10.1007/BF00262007. [DOI] [PubMed] [Google Scholar]
- 11.National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes. 1980;28:1039–57. doi: 10.2337/diab.28.12.1039. [DOI] [PubMed] [Google Scholar]
- 12.Hawa MI, Fava D, Medici F, Deng YJ, Notkins AL, De Mattia G, et al. Antibodies to IA-2 and GAD65 in type 1 and type 2 diabetes: isotype restriction and polyclonality. Diabetes Care. 2000;23:228–33. doi: 10.2337/diacare.23.2.228. [DOI] [PubMed] [Google Scholar]
- 13.Bingley PJ, Bonifacio E, Mueller PW Participating Laboratories. Diabetes antibody standardization program: first assay proficiency evaluation. Diabetes. 2003;52:1128–36. doi: 10.2337/diabetes.52.5.1128. [DOI] [PubMed] [Google Scholar]
- 14.Engvall E, Perlmann P. Enzyme-linked immunosorbent assay, ELISA. III. Quantitation of specific antibodies by enzyme-labeled anti-immunoglobulin in antigen-coated tubes. J Immunol. 1972;109:129–35. [PubMed] [Google Scholar]
- 15.Mancini G, Carbonara AO, Heremans JF. Immunochemical quantitation of antigens by single radial immunodiffusion. Immunochemistry. 1965;2:235–54. doi: 10.1016/0019-2791(65)90004-2. [DOI] [PubMed] [Google Scholar]
- 16.Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clin Exp Immunol. 2000;120:225–31. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerutti F, Urbino A, Sacchetti C, Palomba E, Zoppo M, Tovo PA. Selective IgA deficiency in juvenile-onset insulin-dependent diabetes mellitus (in Italian) Pediatr Med Chir. 1988;10:197–201. [PubMed] [Google Scholar]
- 18.Gunderson E. Is diabetes of infectious origin? J Infect Dis. 1927;41:197–202. [Google Scholar]
- 19.Leslie RDG, Elliott RB. Early environmental events as a cause of IDDM. Diabetes. 1994;43:843–50. doi: 10.2337/diab.43.7.843. [DOI] [PubMed] [Google Scholar]
- 20.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–9. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 21.Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs. Diabetes. 2003;52:1052–5. doi: 10.2337/diabetes.52.4.1052. [DOI] [PubMed] [Google Scholar]
- 22.Fraser P, Schur P. Hypoimmunoglobulinemia D: frequency, family studies, and association with HLA. Clin Immunol Immunopathol. 1981;19:67–74. doi: 10.1016/0090-1229(81)90048-9. [DOI] [PubMed] [Google Scholar]
- 23.Awdeh ZL, Raum D, Yunis EJ, Alper CA. Extended HLA/complement allele haplotypes: evidence for T/t-like complex in man. Proc Natl Acad Sci USA. 1983;80:259–63. doi: 10.1073/pnas.80.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raum D, Awdeh Z, Yunis EJ, Alper CA, Gabbay KH. Extended major histo-compatibility complex haplotypes in type I diabetes mellitus. J Clin Invest. 1984;74:449–54. doi: 10.1172/JCI111441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alper CA, Larsen CE, Dubey DP, Awdeh ZL, Fici DA, Yunis EJ. The haplotype structure of the human major histocompatibility complex. Hum Immunol. 2006;67:73–84. doi: 10.1016/j.humimm.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Alper CA, Fleischnick E, Awdeh Z, Katz AJ, Yunis EJ. Extended major histocompatibility complex haplotypes in patients with gluten-sensitive enteropathy. J Clin Invest. 1987;79:251–6. doi: 10.1172/JCI112791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Degli-Esposti MA, Andreas A, Christiansen FT, Schalke B, Albert E, Dawkins RL. An approach to the localization of the susceptibility gene for generalized myasthenia gravis by mapping recombinant ancestral haplotypes. Immunogenetics. 1992;35:355–64. doi: 10.1007/BF00179791. [DOI] [PubMed] [Google Scholar]
- 28.Schur PH, Marcus-Bagley D, Awdeh Z, Yunis EJ, Alper CA. The effect of ethnicity on major histocompatibility complex complement allotypes and extended haplotypes in patients with systemic lupus erythematosus. Arthritis Rheum. 1990;33:985–92. doi: 10.1002/art.1780330710. [DOI] [PubMed] [Google Scholar]
- 29.Lewkonia RM, Gairdner D, Doe WF. IgA deficiency in one of identical twins. Br Med J. 1976;1:311–3. doi: 10.1136/bmj.1.6005.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pozzilli P, Leslie RDG. Infections and diabetes: mechanisms and prospects for prevention. Diabet Med. 1994;11:935–41. doi: 10.1111/j.1464-5491.1994.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 31.Edwards E, Razvi S, Cunningham-Rundles C. IgA deficiency: clinical correlates and responses to pneumococcal vaccine. Clin Immunol. 2004;111:93–7. doi: 10.1016/j.clim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Umetsu DT, Ambrosino DM, Quinti I, Siber GR, Geha RS. Recurrent sinopulmonary infection and impaired antibody response to bacterial capsular polysaccharide antigen in children with selective IgG-subclass deficiency. N Engl J Med. 1985;313:1247–51. doi: 10.1056/NEJM198511143132002. [DOI] [PubMed] [Google Scholar]
- 33.Moss RB, Carmack MA, Esrig S. Deficiency of IgG4 in children: association of isolated IgG4 deficiency with recurrent respiratory infections. J Pediatr. 1992;120:16–21. doi: 10.1016/s0022-3476(05)80590-6. [DOI] [PubMed] [Google Scholar]
- 34.Liberatore RDR, Jr, Barbosa SFC, Alkimin MDG, Bellinati-Pires R, Florido MPC, Isaac L, et al. Is immunity in diabetic patients influencing the susceptibility to infections? Immunoglobulins, complement and phagocytic function in children and adolescents with type 1 diabetes mellitus. Pediatr Diabetes. 2005;6:206–12. doi: 10.1111/j.1399-543X.2005.00136.x. [DOI] [PubMed] [Google Scholar]
- 35.Munks R, Booth JR, Sokol RJ. A comprhensive IgA service provided by a blood transfusion center. Immunohematol. 1998;14:155–60. [PubMed] [Google Scholar]
- 36.Husain Z, Holodick N, Day C, Szymanski I, Alper CA. Increased apoptosis of CD20+ IgA+ B cells is the basis for IgA deficiency: the molecular mechanism for correction in vitro by IL-10 and CD40L. J Clin Immunol. 2006;26:113–25. doi: 10.1007/s10875-006-9001-y. [DOI] [PubMed] [Google Scholar]