

Abstract
The binding of phosphoenolpyruvate (PEP) to the single allosteric site on phosphofructokinase (EC 2.7.1.11) from Bacillus stearothermophilus (BsPFK) diminishes the ability of the enzyme to bind the substrate fructose 6-phosphate (Fru-6-P). Comparisons of crystal structures with either Fru-6-P or phosphoglycolate, an analog of PEP, bound have shown that Arg-162 interacts with the negatively charged Fru-6-P. Upon the binding of phosphoglycolate, Arg-162 is virtually replaced by Glu-161, which introduces a potential coulombic repulsion between enzyme and substrate [Schirmer, T. & Evans, P. R. (1990) Nature (London) 343, 140–145]. It has previously been proposed that this structural transition explains the allosteric inhibition in BsPFK, and this explanation has appeared in textbooks to illustrate how an allosteric ligand can influence substrate binding at a distance. Site-directed mutagenesis has been employed to create three mutants of BsPFK that substitute an alanine residue for Glu-161, Arg-162, or both. The E161A mutation does not affect the inhibition of BsPFK by PEP at 25°C, and while the R162A mutation decreases BsPFK's affinity for Fru-6-P by approximately 30-fold, R162A diminishes the effectiveness of PEP inhibition by only 1/3. Combining E161A and R162A produces behavior comparable to R162A alone. These and other data suggest that the movement of Glu-161 and Arg-162 does not play the central role in producing the allosteric inhibition by PEP as originally envisioned in the Schirmer and Evans mechanism.
Phosphofructokinase (EC 2.7.1.11) has been studied extensively both structurally and functionally. It is often looked upon as establishing a paradigm for how regulatory enzymes come to be imbued with allosteric properties. Phosphoenolpyruvate (PEP) allosterically inhibits phosphofructokinase from Bacillus stearothermophilus (BsPFK) by diminishing the affinity the enzyme displays for its substrate, fructose 6-phosphate (Fru-6-P). In 1990 Schirmer and Evans (1) proposed a mechanism to explain this inhibition based on analyses of x-ray crystal structures of BsPFK. This proposal has become generally accepted as a quintessential example of how allosteric ligands achieve their effects as evidenced by its inclusion in popular biochemistry textbooks (2–4).
A comparison of the structure of the substrate-bound form of BsPFK (5) to that of BsPFK with an inhibitor analog of PEP, phosphoglycolate, bound in the allosteric site (6)† reveals a different conformation in the active site of these two forms. It was proposed that this conformational change is responsible for the ability of phosphoglycolate, and by extension PEP,‡ to diminish the affinity of BsPFK for Fru-6-P. The conformational change involves a substantial movement of residues Glu-161 and Arg-162, depending on whether substrate or inhibitor is bound. When Fru-6-P is bound to the active site of BsPFK, the positively charged side chain of Arg-162 protrudes into the active site and favorably interacts with the negatively charged phosphate group of Fru-6-P. Glu-161 is well removed from the binding site of this structure. When phosphoglycolate is bound to the allosteric site of BsPFK with the active site empty, the Arg-162 residue is positioned away from the active site, and its former location is occupied by the negatively charged side chain of Glu-161. This virtual switch in the position of these two oppositely charged residues introduces a negative charge into the binding site for Fru-6-P and causes a putative electrostatic repulsion between the side chain of Glu-161 and an incoming Fru-6-P molecule, presumably resulting in a decrease in the affinity for Fru-6-P.
We were prompted to reevaluate this mechanism for the following reasons: First, the mechanism does not address the issue of reciprocity that must occur between the binding of a ligand to the allosteric site and the binding of a substrate in the active site. Because PEP increases the K1/2 for Fru-6-P approximately 500-fold at 25°C (7), thermodynamic linkage requires that Fru-6-P inhibit the binding of PEP to the same extent. It is not clear how the movement of Glu-161 and Arg-162, both located in the Fru-6-P binding site, might influence the binding of PEP to the allosteric site some 20 Å away. Indeed, only a relatively small repositioning of residues lining the allosteric site is evident, with no obvious consequences, in the Schirmer and Evans structures (1).
Second, the two crystal structures that form the basis of the proposal do not by themselves provide enough information. The actions of a K-type allosteric inhibitor, such as PEP or phosphoglycolate, are quantitatively described, both in nature and magnitude, by a coupling free energy, ΔGay (7, 8). ΔGay in turn is the standard free energy for the following disproportionation equilibrium:
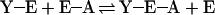 |
1 |
where E–A and Y–E represent the binary complexes of either substrate or allosteric ligand bound to the enzyme, respectively (9–11). E represents native enzyme with no ligands bound and Y–E–A represents the tertiary complex formed when both substrate and allosteric ligand are bound. In the present case, A represents Fru-6-P and Y represents PEP or phosphoglycolate. One cannot speculate about the poise of the equilibrium depicted in Eq. 1 without making explicit or implicit assumptions about the chemical potential of the unexamined enzyme forms. E–A and Y–E may be very different, as are the Fru-6-P-bound and phosphoglycolate-bound forms of BsPFK according to the x-ray structures, but that difference alone does not speak directly to the reason why Eq. 1 attains a particular equilibrium value because both of those enzyme forms appear on the same side of that equilibrium. No adequate structural information is currently available pertaining to the E and Y–E–A enzyme forms.
Finally, the proposed mechanism might be interpreted, particularly by readers of general biochemistry texts, as suggesting that simple coulombic interactions between Glu-161 or Arg-162 and Fru-6-P are solely responsible for the different binding affinities displayed by Fru-6-P in the absence and presence of the inhibitor. If this were the case, then heat should be released and absorbed in these two cases, respectively. However, van't Hoff analyses for wild-type BsPFK have indicated that the enthalpy change introduced by PEP actually favors activation, and entropy, not enthalpy, is driving the observed inhibition by PEP at 25°C (7, 8). A simple mechanism dominated by coulombic interactions does not easily explain this observation.
In 1995 Auzat et al. (12) constructed mutants in BsPFK in which the active site Glu-161 residue was mutated to either an alanine or a glutamine residue. While they concluded that Glu-161 was not a crucial residue for inhibition by PEP, they did not characterize the extent to which removing residue 161 affected the overall coupling free energy between Fru-6-P and PEP nor how Glu-161 affects the enthalpic and entropic contributions to the overall coupling free energy.
In an attempt to better characterize the role of both active site residues Glu-161 and Arg-162 in the ability of PEP to inhibit BsPFK, we have used site-directed mutagenesis to construct three mutants of BsPFK in which the active site residues Glu-161 and Arg-162 were first individually, and then simultaneously, changed to alanine. By exposing these mutants to the same rigorous thermodynamic characterizations as has previously been conducted on wild-type BsPFK (7), a more complete understanding of the extent of participation of these residues in establishing the coupling free energy between Fru-6-P and PEP has been obtained.
Materials and Methods
Materials.
All chemical reagents used for protein purification, kinetic assays, and fluorescence measurements were of analytical grade and purchased from Aldrich, Fisher, or Sigma. The Matrix Blue A-agarose used for purification of BsPFK was purchased from Amicon. The coupling enzymes aldolase, triosephosphate isomerase, and glycerol-3-phosphate dehydrogenase were purchased as ammonium sulfate suspensions from Boehringer Mannheim. Before use in kinetic assays, the coupling enzymes were dialyzed extensively against 50 mM 3-(N-morpholino)propanesulfonic acid–KOH (Mops–KOH), pH 7.0/100 mM KCl/14 mM MgCl2/0.1 mM EDTA. Creatine kinase, creatine phosphate, NADH, and sodium salts of Fru-6-P, ATP, and PEP were purchased from Sigma. Bicinchoninic acid reagents used in determining protein concentration were purchased from Pierce. Site-directed mutagenesis was carried out by using the Promega Altered Sites In Vitro Mutagenesis System, which included the pALTER vector and pALTER control vector, T4 DNA polymerase, T4 DNA ligase, T4 polynucleotide kinase, and the oligonucleotides used to confer ampicillin resistance. All other oligonucleotides were synthesized by using an Applied Biosystems 392 DNA/RNA synthesizer at the Gene Technologies Laboratory at the Institute of Developmental and Molecular Biology at Texas A&M University. Deionized distilled water was used throughout.
Site-Directed Mutagenesis.
The plasmid pBR322/Bspfk (13), which contains the gene for BsPFK, was received as a generous gift from Simon H. Chang (Louisiana State University). The plasmid was further modified to place the gene for BsPFK in pALTER behind a lac promoter as described elsewhere (M. L. Riley-Lovingshimer and G.D.R., unpublished results).
Mutagenesis was performed according to the protocol for the Promega Altered Sites In Vitro Mutagenesis System, using single-stranded DNA derived from the pMR/Bspfk plasmid containing the gene for BsPFK in pALTER under the lac promoter. Three mutagenesis reactions were conducted; one to construct each of the single mutations E161A and R162A and one to construct the double mutation E161A/R162A. Selected ampicillin-resistant mutants were sequenced at the Gene Technologies Laboratory by the Sanger dideoxynucleotide method, using an Applied Biosystems sequencer and dye-labeled terminators to verify the sequence of the mutant DNA. The resultant recombinant expression vectors were transformed into the DF1020 Escherichia coli host strain by using the calcium chloride method (14). DF1020 was the generous gift of Robert Kemp (Chicago Medical School) and is a strain that produces no wild-type PFK-1 (15, 16).
Protein Purification.
Purifications of active site mutants of BsPFK were performed according to Valdez et al. (17) with the following minor modifications: DF1020 cells containing the appropriate mutant plasmid were grown to stationary phase in Luria–Bertani (LB) broth (Bacto tryptone 10 g/liter, Bacto yeast extract 5 g/liter, and NaCl 10 g/liter) containing 100 μg/ml ampicillin at 37°C. Matrix blue A-agarose (Amicon) was used rather than Cibacron blue 3GA-agarose type 3000-CL-L (Sigma). After elution from the column, BsPFK was desalted by dialysis into storage buffer [50 mM N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid) (EPPS)/100 mM KCl/14 mM MgCl2/0.1 mM EDTA, pH 8.0], concentrated with an Amicon concentrator apparatus equipped with a 30,000 molecular weight cut-off filter, and then redialyzed into storage buffer before being stored at 4°C. Purity of final enzyme preparation was estimated by using SDS/polyacrylamide gel electrophoresis (18) and staining with Coomassie blue. Protein concentration was determined by using the bicinchoninic acid protein assay (19).
Enzymatic Activity Assays.
Activities of BsPFK were measured by coupling the reaction catalyzed by BsPFK to the oxidation of NADH and monitoring the corresponding decrease in the absorbance at 340 nm. Assays were carried out in 1.0-ml reaction volumes containing storage buffer, 0.2 mM NADH, 2 mM DTT, 250 μg of aldolase, 50 μg of glycerol-3-phosphate dehydrogenase, and 5 μg of triosephosphate isomerase adjusted to pH 8.0 at the appropriate temperature. Creatine kinase and creatine phosphate were added to regenerate MgATP from MgADP to alleviate the effects of MgADP on the enzyme. The concentration of MgATP was kept constant and equal to 3 mM, and the concentrations of Fru-6-P and the inhibitor PEP were as indicated. Assays were initiated by the addition of 10 μl of BsPFK that had been appropriately diluted so as not to exceed a change of 0.1 absorbance unit at 340 nm per min. One unit of activity is defined as the amount of enzyme needed to produce 1 μmol of fructose 1,6-bisphosphate per min. Activity measurements were conducted on Beckman Series 600 spectrophotometers, using a linear regression calculation to convert change in absorbance at 340 nm to enzyme activity.
Steady-State Fluorescence.
Steady-state fluorescence intensity was measured by using an SLM-4800 fluorometer upgraded with ISS photon-counting and data acquisition electronics. Samples were excited at 300 nm with a xenon arc lamp and fluorescence was detected either through a monochromator or through a 2-mm-thick WG 335-nm cut-on filter. Fluorescence experiments were carried out in 2-ml reaction volumes containing storage buffer at pH 8.0 with protein subunit concentration varying from 1 to 5 μM. Fru-6-P or PEP were titrated into the sample and the emission intensity was monitored as a function of ligand concentration. All emission measurements were corrected for blank contributions and dilution.
Data Analysis.
All data analysis was performed on a Power Macintosh 7100/80AV using Kaleidagraph 3.08 (Synergy Software) to fit to the following equations. The concentration of Fru-6-P that resulted in half-maximal activity, K1/2, was determined by using the Hill equation:
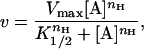 |
2 |
where v equals the steady-state rate of turnover, Vmax represents the maximal specific activity, and nH is the Hill coefficient. The variation in K1/2 as a function of PEP concentration was fit to the following equation:
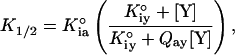 |
3 |
where K1/2 is the concentration of Fru-6-P resulting in half-maximal activity obtained from Eq. 2; K°ia is the dissociation constant for the substrate Fru-6-P in the absence of any allosteric ligand; K°iy is the dissociation constant for the allosteric ligand in the absence of substrate Fru-6-P; and Qay is the coupling parameter describing the extent to which the binding of the allosteric ligand affects the binding of substrate and vice versa as defined by the following equation:
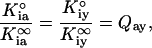 |
4 |
where Kia∞ and Kiy∞ represent the dissociation constants for A and Y, respectively, in the saturating presence of the other ligand. To be consistent with previously adopted conventions (8, 20, 21), A represents the substrate Fru-6-P and Y represents the allosteric inhibitor PEP. By resolving both the terms K°iy and Qay, Eq. 3 allows separate quantification of both effector binding affinity and the action of the effector once it is bound, respectively.
The coupling parameter, Qay, describes both the nature and magnitude of the effect that allosteric ligands have on the binding of substrate. If Qay < 1 the allosteric ligand is an inhibitor, and if Qay > 1 the allosteric ligand is an activator. If Qay = 1, then the allosteric ligand has no effect on the binding of substrate. In the case of the inhibitor PEP, the smaller the value of Qay, the greater the extent of inhibition of substrate binding. Qay is related to the standard coupling free energy between Fru-6-P and PEP, ΔGay, which in turn is related the standard coupling enthalpy and entropy, ΔHay and ΔSay respectively, by the following relationships:§
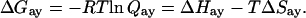 |
5 |
Note that ΔGay will be positive for an inhibitor and negative for an activator.
Results
A comparison of the maximal specific activities, affinities for the substrate Fru-6-P, and cooperativity of substrate binding of the E161A, R162A, and E161A/R162A mutants of BsPFK with the corresponding values previously obtained for wild-type BsPFK is presented in Fig. 1 and Table 1. Fig. 1 depicts the dependence of activity as a function of substrate concentration for wild-type BsPFK and the three active site mutants. Table 1 lists the kinetic parameters obtained from these data. The only parameter that differs between wild type and the mutants to a significant degree is the approximately 30-fold increase in K1/2 for Fru-6-P when Arg-162 is mutated to Ala. Thus both the R162A and the E161A/R162A mutants exhibit a high K1/2 for Fru-6-P, whereas the K1/2 exhibited by E161A is very similar to that for wild type.
Figure 1.

Fru-6-P saturation profiles for wild type (●) and E161A (○), R162A (■), and E161A/R162A (□) mutants of BsPFK. MgATP concentration was 3 mM. Other conditions were as described in the text.
Table 1.
Steady-state kinetic parameters for wild type and active site mutants of BsPFK
| Enzyme | Vmax, units/mg | K1/2, mM | Hill number nH |
|---|---|---|---|
| Wild type | 111 ± 1 | 0.034 ± 0.001 | 1.6 ± 0.1 |
| E161A | 99 ± 1 | 0.035 ± 0.001 | 1.2 ± 0.1 |
| R162A | 122 ± 2 | 1.10 ± 0.06 | 1.0 ± 0.1 |
| E161A/R162A | 108 ± 1 | 1.21 ± 0.05 | 1.1 ± 0.1 |
To determine whether the residues Glu-161 and Arg-162 influence the binding of PEP or its action in diminishing PFK's affinity for Fru-6-P, respectively, one or both of the active site residues were mutated to an alanine. Fig. 2 shows the dependence of K1/2 for Fru-6-P as a function of PEP concentration determined from steady-state kinetics for wild-type BsPFK and for the three active site mutants. It is clear from these data that PEP inhibits all three mutants, as well as wild type, by increasing K1/2. Because the rapid-equilibrium assumption is valid for BsPFK (7), the maximum extent of this increase determines the value of Qay as indicated by Eq. 4. Qay is virtually identical for wild type and the E161A mutant. The curves pertaining to the mutants containing R162A are both displaced upward, consistent with a diminished affinity for Fru-6-P at all concentrations of PEP. PEP causes a further increase in K1/2 in these mutants, although not quite to the same extent as wild type. Table 2 lists the parameters obtained from these analyses. Note that, although the mutations also perturb the binding affinity for PEP to various degrees as indicated by K°iy in Table 2, the coupling free energy, ΔGay, retains at least two-thirds of its wild-type value in all mutants.
Figure 2.

Dependence of the apparent K1/2 for Fru-6-P on increasing concentrations of the inhibitor PEP for wild type (●) and E161A (○), R162A (■), and E161A/R162A (□) mutants of BsPFK. K1/2 values were obtained from substrate saturation profiles such as those shown in Fig. 1. Curves correspond to the best fit of these data to Eq. 3 described in the text.
Table 2.
Thermodynamic coupling parameters for wild type and all three active site mutants of BsPFK at 25°C
| Enzyme | Kia○, mM | Kiy○, mM | Qay × 103 | ΔGay, kcal/mol |
|---|---|---|---|---|
| Wild type | 0.028 ± 0.002 | 0.040 ± 0.004 | 1.4 ± 0.1 | 3.89 ± 0.04 |
| E161A | 0.025 ± 0.002 | 0.106 ± 0.011 | 2.5 ± 0.7 | 3.55 ± 0.17 |
| R162A | 1.16 ± 0.06 | 1.01 ± 0.09 | 23 ± 4 | 2.23 ± 0.10 |
| E161A/R162A | 0.76 ± 0.06 | 0.166 ± 0.019 | 14 ± 1 | 2.53 ± 0.04 |
Parameters were determined from steady-state kinetics with MgATP = 3 mM.
Often ligand binding can also be followed by monitoring the changes the ligands induce in intrinsic protein fluorescence. For E. coli PFK, the interactions between substrates and effectors have been studied extensively in this manner (20–22). However, in the case of wild-type BsPFK, the single tryptophan is for the most part unresponsive to conformational changes induced by ligand binding (23). The only change in the fluorescence properties of wild-type BsPFK is a 10% decrease in the intensity upon the binding of the inhibitor PEP to the allosteric site (23). This change has proven to be sufficient, in wild type and in the E161A/R162A mutant, to assess the coupling interactions between Fru-6-P and PEP by monitoring the change in intensity upon PEP binding as a function of Fru-6-P concentration. As indicated in Eq. 4, the binding antagonism between Fru-6-P and PEP should be equally manifest in these experiments. The resulting values for the apparent dissociation constants for PEP, Kiy, varied with Fru-6-P concentration as shown in Fig. 3 These data were fit to an equation analogous to Eq. 3 in which the K°ia and K°iy terms are switched—i.e., A is the independent variable and Kiy is the dependent variable. The results are presented in Table 3. The discrepancy between the values presented in Tables 2 and 3 is likely because of the influence of MgATP, which is present at a saturating concentration in the kinetics experiments but absent from the fluorescence experiments. Substantial antagonism between MgATP and Fru-6-P binding in E. coli PFK has been described previously (24). Despite these differences, the value of ΔGay for the mutant is still roughly two-thirds the value for wild type, in agreement with the behavior observed in the kinetics experiments.
Figure 3.

Dependence of the apparent dissociation constant for PEP, Kiy, on the concentration of Fru-6-P for wild type (●) and the E161A/R162A mutant (□) of BsPFK. Kiy was determined from changes in intrinsic fluorescence after additions of PEP. Error bars represent ±SE. Curves represent the best fit of the data to an equation analogous to Eq. 3 as described in the text.
Table 3.
Thermodynamic coupling parameters for wild type and the E161A/R162A mutant of BsPFK at 25°C
| Enzyme | Kia○, μM | Kiy○, μM | Qay × 103 | ΔGay, kcal/mol |
|---|---|---|---|---|
| Wild type | 0.7 ± 0.2 | 17.9 ± 2.7 | 2.9 ± 0.5 | +3.47 ± 0.10 |
| E161A/R162A | 14.4 ± 4.7 | 33.9 ± 6.2 | 23 ± 4 | +2.24 ± 0.10 |
Parameters were determined from titrations of intrinsic fluorescence with [MgATP] = 0.
By using van't Hoff analyses, the inhibition of wild-type BsPFK by PEP was previously determined to be entropically dominated (7, 8). Fig. 4 shows a comparison of van't Hoff plots obtained for wild-type and E161A BsPFK. Table 4 lists the entropic and enthalpic energy contributions for the PEP–Fru-6-P coupling obtained from these data according to Eq. 5. As is true for wild type, the inhibition of E161A BsPFK by PEP is also entropically dominated, and the value of ΔHay has a negative sign as indicated by the positive slope in each case. Note that the temperature at which the two lines cross is very close to 25°C (1/K = 0.00335).
Figure 4.

van't Hoff analyses of the coupling between Fru-6-P and PEP for wild type (●) and the E161A mutant (○) of BsPFK. Wild-type data are from ref. 7.
Table 4.
Summary of thermodynamic parameters at 25°C obtained from van't Hoff analyses of the coupling parameters for wild type and the E161A mutant of BsPFK
| Enzyme | ΔGay, kcal/mol | ΔHay, kcal/mol | TΔSay, kcal/mol |
|---|---|---|---|
| Wild type | 3.61 ± 0.81 | −4.31 ± 0.81 | −7.93 ± 0.01 |
| E161A | 3.58 ± 0.76 | −12.77 ± 0.76 | −16.35 ± 0.05 |
Discussion
It is clear that the roles played by Glu-161 and Arg-162 in the allosteric inhibition by PEP of BsPFK are greatly overstated by the mechanism proposed by Schirmer and Evans (1). If this explanation of PEP inhibition were correct, then removal of the negatively charged residue Glu-161 should greatly reduce PEP's ability to decrease PFK's affinity for Fru-6-P. However, the present study shows that removal of this residue causes only a barely significant decrease in the ability of PEP to inhibit Fru-6-P binding at 25°C (Fig. 2, Table 2). Also, changing Arg-162 to Ala causes only a 10-fold decrease in Qay (Fig. 2, Table 2). The same result was obtained when a fluorescence-based thermodynamic assay was used to determine the Fru-6-P–PEP coupling in the double mutant E161A/R162A. (Fig. 3, Table 3). These data indicate that residues Glu-161 and Arg-162 play relatively minor roles in the mechanism of PEP inhibition at 25°C, since removal of both residues still allows PEP to inhibit with a coupling free energy equal to two-thirds the wild-type value. However, even this conclusion suggests an over-simplified picture of how these residues contribute, or fail to contribute, to the structural basis for the allosteric behavior of BsPFK.
Structural data, such as crystal structure coordinates, are often useful in establishing reaction mechanisms of enzyme-catalyzed reactions. However, discrepancies can arise when structural data are exclusively utilized to deduce thermodynamic parameters associated with protein–ligand interactions. The proposed mechanism of inhibition of BsPFK by PEP is one such example. The essential issue when considering the basis for the actions of a K-type allosteric ligand is: How does the binding equilibrium for substrate come to be altered when an allosteric ligand is bound at a remote site? To understand why a shift occurs in a binding equilibrium, one must consider the changes in the chemical potential of both reactants and products of that equilibrium that are introduced by the prior binding of the allosteric ligand. Each of the two x-ray structures upon which the Schirmer and Evans mechanism is based represents, respectively, only one participant of each of these two equilibria. The Fru-6-P-bound form of the enzyme represents the product of the substrate binding reaction in the absence of allosteric ligand, and the phosphoglycolate-bound form represents the reactant of the second substrate-binding equilibrium that occurs while phosphoglycolate remains bound. Thus the structural attributes of neither of the two equilibria to be compared are completely established. This mechanistic proposal therefore must presume that when Fru-6-P binds with phosphoglycolate bound, Glu-161 remains in place, creating a relatively destabilized product complex for the second equilibrium, and that Arg-162 is predisposed to position itself in the active site of free enzyme without Fru-6-P being present. No information is known about the structure of either the free enzyme form or the ternary complex enzyme form (i.e., with both Fru-6-P and phosphoglycolate bound) to support either of these assumptions.¶
Our initial skepticism was based in part on the expectation that a simple coulombic mechanism implies that the basis for the allosteric effects is primarily enthalpic. This implication seemed to disagree with our observation of entropy-dominated couplings in BsPFK (7). The data presented in Fig. 4 indicate that factors other than a pure coulombic repulsion between Glu-161 and the substrate must be responsible for the inhibition. Despite the fact that the binding free energy for Fru-6-P in the E161A mutant is virtually identical to wild-type, the change in ΔH for binding indeed becomes more negative in the absence of that negatively charged glutamate, as might be predicted if coulombic repulsion were involved (Table 4). However, this effect is completely compensated by an opposing change in ΔS, so the effect on phosphoglycolate inhibition, given by ΔGay, is negligible at 25°C. An entropy “problem” continues to diminish the propensity for Fru-6-P to bind after PEP, whether or not Glu-161 is present, and is actually larger when Glu-161 is absent.
The importance of a full thermodynamic reconciliation is evident when one further considers the influence of the E161A mutation at other temperatures as revealed in Fig. 4. At low temperatures, replacing the glutamate does make the enzyme less susceptible to PEP inhibition because Qay is greater for the mutant than for wild type. This is a direct consequence of the more negative value of ΔHay, which favors activation by PEP and which therefore acts to decrease the inhibition when low temperature diminishes the contribution that ΔSay makes to ΔGay. However, at high temperatures, replacing the glutamate actually causes PEP to become a better inhibitor. The adverse coupling entropy, which is larger in absolute value in the mutant, becomes weighted more heavily in its contribution to ΔGay at high temperatures. Thus, Glu-161 is involved in the allosteric communication as evidenced by the thermodynamic perturbations resulting from its removal, but this involvement is quite different and apparently more indirect than that envisioned by the Schirmer and Evans mechanism.
Finally, the currently available structural information fails to address the reciprocity requirement of allosteric behavior. The data in Fig. 3 demonstrate directly that Fru-6-P binding must also inhibit PEP binding to an identical extent. Changes in the binding sites themselves would seem unlikely to provide a sufficient explanation for allosteric behavior in general because the effect of each ligand on the other must be equal in nature and magnitude. There must therefore be a region of mutual influence to serve as the “conduit” for this information transfer from one site to another. Although the location or even the nature of this conduit cannot be predicted a priori, it must extend continuously from one site to another. Moreover, the energetic perturbations that occur throughout the conduit will contribute to the overall energetics, and hence the observed phenomenology, of the allosteric effect. It is very unlikely that structural changes localized to only one site can provide a complete explanation for the mechanism underlying an allosteric effect. In the case of BsPFK, clearly more investigation will be required before a convincing mechanism can be proposed.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM33216) and the Robert A. Welch Foundation (A1368). J.L.K. was supported in part by a Chemistry–Biology Interface training grant from the National Institutes of Health (T32 GM08523).
Abbreviations
- PFK
phosphofructokinase
- BsPFK
PFK from Bacillus stearothermophilus
- PEP
phosphoenolpyruvate
- Fru-6-P
fructose 6-phosphate
Footnotes
A higher-resolution structure was deposited in the Brookhaven Protein Data Bank (www.rcsb.org) in 1998 as file pdb6pfk.ent.
A comparison of the functional similarities and differences between the inhibition by phosphoglycolate and PEP has been presented previously (8).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050588097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050588097
The superscript usually included to denote standard state (°) has been omitted to avoid confusion with the notation used in the previous equations.
A structure has been determined from crystals produced by incubating BsPFK with Fru-6-P and Pi during crystallization in which the Fru-6-P was removed by subsequent washing. However, two equivalents of Pi remain bound in these crystals, with one occupying the same position as the phosphate moiety of Fru-6-P when the latter is bound. This position is approximately 3 Å from Arg-162 and therefore mimics the electrostatic situation with Fru-6-P bound.
References
- 1.Schirmer T, Evans P R. Nature (London) 1990;343:140–145. doi: 10.1038/343140a0. [DOI] [PubMed] [Google Scholar]
- 2.Zubay G. Biochemistry. 3rd. Ed. Dubuque, IA: Brown; 1993. pp. 263–266. [Google Scholar]
- 3.Voet D, Voet J G. Biochemistry. 2nd Ed. New York: Wiley; 1995. pp. 472–474. [Google Scholar]
- 4.Voet D, Voet J G, Pratt C W. Fundamentals of Biochemistry. New York: Wiley; 1999. pp. 408–409. [Google Scholar]
- 5.Evans P R, Hudson P J. Nature (London) 1979;279:500–504. doi: 10.1038/279500a0. [DOI] [PubMed] [Google Scholar]
- 6.Evans P R, Farrants G W, Lawrence M C. J Mol Biol. 1986;191:713–720. doi: 10.1016/0022-2836(86)90455-9. [DOI] [PubMed] [Google Scholar]
- 7.Tlapak-Simmons V L, Reinhart G D. Biophys J. 1998;75:1010–1015. doi: 10.1016/S0006-3495(98)77589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tlapak-Simmons V L, Reinhart G D. Arch Biochem Biophys. 1994;308:226–230. doi: 10.1006/abbi.1994.1032. [DOI] [PubMed] [Google Scholar]
- 9.Weber G. Biochemistry. 1972;11:864–878. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- 10.Weber G. Adv Protein Chem. 1975;29:1–83. doi: 10.1016/s0065-3233(08)60410-6. [DOI] [PubMed] [Google Scholar]
- 11.Reinhart G D, Hartleip S B, Symcox M M. Proc Natl Acad Sci USA. 1989;86:4032–4036. doi: 10.1073/pnas.86.11.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auzat I, Byrnes W M, Garel J-R, Chang S H. Biochemistry. 1995;34:7062–7068. doi: 10.1021/bi00021a018. [DOI] [PubMed] [Google Scholar]
- 13.French B A, Valdez B C, Younathan E S, Chang S H. Gene. 1987;59:279–283. doi: 10.1016/0378-1119(87)90335-0. [DOI] [PubMed] [Google Scholar]
- 14.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 15.Daldal F. J Mol Biol. 1983;168:285–305. doi: 10.1016/s0022-2836(83)80019-9. [DOI] [PubMed] [Google Scholar]
- 16.Hellinga H W, Evans P R. Eur J Biochem. 1985;149:363–373. doi: 10.1111/j.1432-1033.1985.tb08934.x. [DOI] [PubMed] [Google Scholar]
- 17.Valdez B C, French B A, Younathan E S, Chang S H. J Biol Chem. 1989;264:131–135. [PubMed] [Google Scholar]
- 18.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Smith D K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk B C. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 20.Johnson J L, Reinhart G D. Biochemistry. 1994;33:2635–2643. doi: 10.1021/bi00175a036. [DOI] [PubMed] [Google Scholar]
- 21.Johnson J L, Reinhart G D. Biochemistry. 1997;36:12814–12822. doi: 10.1021/bi970942p. [DOI] [PubMed] [Google Scholar]
- 22.Johnson J L, Reinhart G D. Biochemistry. 1994;33:2644–2650. doi: 10.1021/bi00175a037. [DOI] [PubMed] [Google Scholar]
- 23.Kim S-J, Chowdhury F N, Stryjewski W, Younathan E S, Russo P S, Barkley M D. Biophys J. 1993;65:215–226. doi: 10.1016/S0006-3495(93)81070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson J L, Reinhart G D. Biochemistry. 1992;31:11510–11518. doi: 10.1021/bi00161a032. [DOI] [PubMed] [Google Scholar]