

Abstract
We seek to evaluate the clinical consequences of resistance to antihormonal therapy by studying analogous animal xenograft models. Two approaches were taken. 1) MCF-7 tumors were serially transplanted into selective estrogen receptor modulator (SERM) -treated immunocompromised mice to mimic five years of SERM treatment. The studies in vivo were designed to replicate the development of acquired resistance to SERMs over years of clinical exposure. 2) MCF-7 cells were cultured long-term under SERM-treated or estrogen withdrawn conditions (to mimic aromatase inhibitors), and then injected into mice to generate endocrine-resistant xenografts. These tumor models have allowed us to define Phase I and Phase II antihormonal resistance according to their responses to E2 and fulvestrant. Phase I SERM-resistant tumors were growth stimulated in response to estradiol (E2), but paradoxically, Phase II SERM and estrogen withdrawn-resistant tumors were growth inhibited by E2. Fulvestrant did not support growth of Phase I and II SERM-resistant tumors, but did allow growth of Phase II estrogen withdrawn-resistant tumors. Importantly, fulvestrant plus E2 in Phase II antihormone-resistant tumors reversed the E2-induced inhibition and instead resulted in growth stimulation. These data have important clinical implications. Based on these and prior laboratory findings, we propose a clinical strategy for optimal third-line therapy: patients who have responded to and then failed at least two antihormonal treatments may respond favorably to short-term low-dose estrogen due to E2-induced apoptosis, followed by treatment with fulvestrant plus an aromatase inhibitor to maintain low tumor burden and avoid a negative interaction between physiologic E2 and fulvestrant.
Keywords: Breast Cancer, Estradiol, Tamoxifen, Raloxifene, Fulvestrant
1. Introduction
The target for endocrine therapy is the estrogen receptor (ER), and the translation of laboratory findings on the control of estrogen-regulated tumor growth has established the current treatment strategies which have been validated in clinical trials [1–7]. Tamoxifen (TAM), the prototype selective estrogen receptor modulator (SERM), is a current standard adjuvant treatment used for 5 years in all stages of ER-positive breast cancer [8–12]. However, aromatase inhibitors (AIs) are becoming the leading choice for antihormonal treatment of ER-positive breast cancer in postmenopausal patients. Still, there is a need to study the long-term therapeutic consequences of TAM because of its use in premenopausal ER-positive breast cancer [9, 10], and as a chemopreventive agent to reduce the risk of breast cancer in high-risk women [13]. There is also considerable interest in the use of raloxifene (RAL), a related SERM, as a chemopreventive agent [14, 15], since it has recently been shown in the STAR trial (Study of TAM and RAL) to exhibit equivalent efficacy as TAM in reducing the risk of breast cancer [16]. Additionally, RAL is used for the treatment and prevention of osteoporosis in postmenopausal women [17], has been noted to have endometrial safety [14, 15, 17], and reduces the risk of cardiovascular disease [18–20]. Since RAL may have to be given indefinitely to prevent osteoporosis, RAL-exposed breast cancer will almost certainly occur. Overall, there is a large and growing population of women at risk for developing endocrine therapy-resistant breast cancer.
It is important to emphasize that the successful treatment of patients with one endocrine agent and then failure, leads to exhaustive endocrine therapy with the succession of agents, each with decreasing efficacy. The failure of TAM as a first-line therapy forms the basis of the use of AIs or fulvestrant (ICI 182,780, Faslodex®) as second-line therapies for the treatment of breast cancer.
In postmenopausal women, the aromatase enzyme converts androgens to estrogens in peripheral tissues such as adipose tissue and in the breast cancer tissue itself [21, 22]. AIs block activity of this enzyme and fall into two classes, steroidal and non-steroidal [23–25]. Exemestane (Aromasin®) [26, 27], a steroidal AI, irreversibly binds aromatase at the catalytic site and inactivates the enzyme. Anastrozole (Arimidex®) [28, 29] and letrozole (Femara®) [30, 31], non-steroidal AIs, bind aromatase at a different site, a heme group, to reversibly inhibit the enzyme. AIs have been evaluated in advanced breast cancer and in the adjuvant setting [26–31]. In the largest adjuvant trial, the ATAC trial (Arimidex, TAM, Alone or in Combination), patients in the anastrozole arm versus the TAM arm showed significantly longer disease-free survival, reduced contralateral breast cancer, and reduced distant metastases [32, 33]. Indeed, AIs are now recommended and may replace TAM as the standard first-line antihormonal adjuvant therapy in postmenopausal ER-positive breast cancer patients. Further, due to the success of this and of other trials evaluating AIs for extended adjuvant therapy, AIs are also indicated after 5 years [34] and even 2 years of TAM [26, 29].
FUL is an analogue of E2 and the first in a new class of drugs that are complete antiestrogens, that is, they display no agonist activity via AF-1 or AF-2 of the ER [35, 36]. FUL also leads to potent downregulation of ER protein expression because FUL binding to ER induces an abnormal conformation that results in accelerated ubiquitylation and shuttling of the ER to the proteasome for degradation [35, 37]. Two large Phase III clinical trials have been conducted to evaluate FUL versus the AI anastrazole in postmenopausal advanced ER-positive breast cancer patients who have failed TAM. Both of these trials showed that FUL was equally effective as anastrazole in terms of time to progression and objective response rates [38, 39]. Hence, FUL has been approved as a second-line therapy. FUL is also currently being evaluated in combination with AIs [36].
Over the past two decades, we have developed unique MCF-7 breast cancer xenograft models of long-term SERM (TAM and RAL) treatment and models of long-term estrogen withdrawal that could reasonably mimic resistance to AIs. These tumor models were developed in vivo and in vitro. The in vivo tumor models were designed to mimic the selection process needed over years to develop acquired resistance in the clinic by serially implanting MCF-7 tumors into SERM-treated and ovariectomized immunodeficient mice also over a period of years [40–50]. The in vitro tumor models were developed by culturing MCF-7 cells in estrogen-free conditions, with or without SERM treatment if appropriate, for over 1 year to develop antihormone resistance, and then injecting these cells into ovariectomized athymic mice treated with the SERM, if appropriate, and allowing tumors to grow [51–53]. We now have in hand a panel of breast cancer xenograft and tissue culture models that have allowed us to define the evolution of resistance to antihormonal therapy into at least two phases, each of which exhibits distinct growth responses to E2 and FUL. We found that the growth of Phase I SERM-resistant tumors is stimulated by E2, while growth of Phase II SERM or estrogen withdrawn-resistant tumors is, paradoxically, inhibited by E2 treatment. Previous studies conducted by our group have shown that E2 not only inhibits growth of Phase II SERM and estrogen withdrawn-resistant tumors; it also induces apoptosis, leading to tumor regression. However, a fraction of these Phase II tumors eventually re-grow after E2-induced regression occurs, but these tumors are again re-sensitized to antihormonal therapy. We also found that while FUL does not support the growth of Phase I and II SERM-resistant tumors, it does allow growth of Phase II estrogen withdrawn-resistant tumors. Further, we found that while E2 blocked growth of Phase II antihormone-resistant tumors, the combination of E2 plus FUL resulted in robust growth. Phase II antihormonal resistance has not yet been widely recognized, but could be exploited by developing a novel third-line treatment plan based on short-term low-dose estrogen to debulk patients’ tumors who fail exhaustive endocrine therapy, followed by the combination of FUL plus an AI to maintain low tumor burden and avoid a negative interaction between physiologic E2 and FUL.
2. Materials and methods
2.1. Athymic mice, tumor inoculation, and tumor tracking
All procedures involving animals have been approved by the Fox Chase Cancer Center’s Internal Animal Care and Use Committee.
All animal studies employed female ovariectomized athymic BALB/c nude (nu/nu) mice (Taconic, Hudson, NY, USA) that were inoculated with tumor cells at 5–6 weeks of age. For experiments employing tumor models which were generated and serially propagated as xenografts (in vivo), 1 mm3 tumor sections were bilaterally transplanted using a trochar into the axillary mammary fat pads. For studies using tumor models which were generated and maintained in tissue culture (in vitro), cells were suspended in phosphate-buffered saline and bilaterally injected into axillary mammary fat pads at 107 cells per site.
Tumor growth was tracked by weekly measurements of tumor length (l) and width (w) using Vernier calipers, from which the tumor cross-sectional area was calculated using the equation: (l/2) × (w/2) × π. Tumor growth curves are expressed as the average cross-sectional tumor area per treatment group ± standard error (SE).
2.2. Drug treatments
Mice were treated with estrogen by implanting a 0.3 cm E2 silastic capsule subcutaneously into the intrascapular region on the back of the mouse at the time of tumor cell inoculation. The capsules were prepared by filling silicone tubing (0.078 inch inner diameter/ 0.125 inch outer diameter; Fisher) 0.3 cm in length with a 1:3 (w/w) mixture of E2 (Sigma-Aldrich, St. Louis, MO, USA) and silastic elastomer (Dow Corning, Midland, MI, USA), and then sealing the ends with silicone adhesive (Dow Corning) and sterilized by gamma irradiation. Athymic mice implanted with these capsules achieve mean serum levels of 83.8 pg/ml (308 pM) E2 [54], which approximates perimenopausal E2 levels in women. RAL and TAM were orally administered by gastric intubation at 1.5 mg/day 5 days per week. Evista tablets (Eli Lilly Pharmaceuticals, Indianapolis, IN, USA; purchased from the Fox Chase Cancer Center’s pharmacy), the clinically available form of RAL (60 mg/tablet), were initially dissolved in water, and then suspended at 10 mg/ml in 10% polyethylene glycol 400/Tween 80 (99.5% polyethylene glycol 400, 0.5% Tween 80) and 0.9% carboxymethyl cellulose. TAM (Sigma) was initially dissolved in ethanol (EtOH), and then suspended at 10 mg/ml in 10% polyethylene glycol 400/Tween 80 (99.5% polyethylene glycol 400, 0.5% Tween 80) and 0.9% carboxymethyl cellulose. FUL was administered by sc injection in the scruff of the neck at a total of 10 mg/week. For the experiment depicted in Figure 1, four different FUL formulations and dosing schedules were used: FUL was initially dissolved in 1) EtOH or 2) dimethylsulfoxide (DMSO), and then made into a suspension with peanut oil at 50 mg/ml and administered as a 5 mg injection twice per week; 3) FUL was dissolved in only DMSO at 50 mg/ml and administered as a 2 mg injection 5 days per week; or 4) the clinical faslodex preparation, a 50 mg/ml proprietary solution of FUL in primarily EtOH supplemented with castor oil as a release rate modifier, was administered as 2 mg injections 5 days per week. For all other experiments, only the clinical Faslodex preparation was used and administered as 2 mg injections 5 days per week. FUL powder was a kind gift of AstraZeneca (Macclesfield, United Kingdom), and the clinical Faslodex preparation was purchased from the Fox Chase Cancer Center’s pharmacy.
Fig. 1.

Growth inhibition of MCF-7/E2 tumors in response to different FUL formulations and dosing schedules. Thirty ovariectomized athymic nude mice were bitransplanted in the axillary mammary fat pads with MCF-7/E2 tumor pieces 1 mm3 in size. At the time of tumor implantation, the mice were separated into 6 treatment groups of 5 mice each, or 10 tumors per group. The treatment groups were Control (no treatment), 0.3 cm E2 silastic capsule implanted sc, and 4 groups of different formulations/dosing schedules of 10 mg total FUL per week plus the 0.3 cm E2 capsule sc. The 4 FUL formulations/dosing schedules corresponded to: 1) a 50 mg/ml suspension of FUL dissolved first in EtOH and then mixed with peanut oil, and administered 2 times per week as a 5 mg sc injection; 2) the clinically used Faslodex preparation consisting of a 50 mg/ml solution of FUL in EtOH and castor oil, and administered 5 times per week as a 2 mg sc injection; 3) a 50 mg/ml suspension of FUL dissolved first in DMSO and then mixed with peanut oil, and administered 2 times per week as a 5 mg sc injection; or 4) a 50 mg/ml solution of FUL in 100% DMSO, and administered daily 5 times per week as a 2 mg sc injection. Tumor growth was tracked by weekly measurements using Vernier calipers and calculating the tumor cross-sectional area according to the formula: (length/2 × width/2 × π ). The data shown represent the average tumor cross-sectional area (cm2) per group ± SE. The cross-sectional area of E2-treated tumors was statistically different from that of each of the four E2 + FUL groups (all P-values <0.0001). Also, the cross-sectional area of tumors in the E2 + 5 mg FUL (EtOH/Peanut Oil suspension given 2 days per week) was statistically different from those in the E2 + 5 mg FUL (DMSO/Peanut Oil suspension given 2 days per week) group (P = 0.0013). Likewise, the cross-sectional area of tumors in the E2 + 2 mg FUL (EtOH/Castor Oil solution given 5 days per week) group was statistically different from that of the E2 + 2 mg FUL (100% DMSO solution given 5 days per week) group (P = 0.0038).
2.3. Generation of MCF-7/E2 xenograft tumors
The MCF-7/E2 xenograft tumor model, representing the antihormonal-sensitive stage of breast cancer, was originally developed by bilateral injection of 107 MCF-7 cells, grown in tissue culture, into the axillary mammary fat pads of female ovariectomized athymic BALB/c nu/nu mice implanted with a 0.3 cm E2 capsule [40]. The resulting MCF-7/E2 tumors have been propagated in vivo by serial transplantation into likewise E2-treated ovariectomized athymic mice.
2.4. Generation of MCF-7/RAL1 xenograft tumors
The MCF-7/RAL1 (Phase I) SERM-resistant tumor model was derived by transplantation of MCF-7/E2 tumors into RAL-treated ovariectomized athymic mice. After extended RAL treatment, a small percentage of these tumors showed minimal but significant growth, and following re-transplantation into new RAL-treated ovariectomized athymic mice, these tumors then exhibited robust RAL-stimulated growth [50]. MCF-7/RAL1 tumors have been propagated in vivo for over 3 years by serial transplantation into RAL-treated ovariectomized athymic mice.
2.5. Generation of MCF-7/RAL2 xenograft tumors
The MCF-7/RAL2 (Phase II) SERM-resistant tumor model was developed in vitro by tissue culture of MCF-7 cells in estrogen-free medium supplemented with 1 μM RAL for over 1 year [51]. For every experiment involving MCF-7/RAL2 tumors, cells were grown in culture, and then bilaterally injected into the axillary mammary fat pads of ovariectomized athymic mice at 107 cells per site. MCF-7/RAL2 cells were maintained in culture in estrogen-free MEM plus 5% dextran-coated charcoal-treated calf serum (DCC-CS), 2 mM glutamine, 6 ng/ml bovine insulin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1× non-essential amino acids (all media components from Invitrogen, Carlsbad, CA, USA).
2.6. Generation of MCF-7/TAM2 xenograft tumors
The MCF-7/TAM2 (Phase II) SERM-resistant tumor model was developed in a similar manner as the MCF-7/RAL1 tumor model, by initial transplantation of MCF-7/E2 tumor pieces into TAM-treated ovariectomized athymic mice, and repeated transplantation of minimally growing tumors into new TAM-treated mice until robust TAM-stimulated growth occurred. These MCF-7/TAM tumors passed through different phases of SERM resistance; they were initially stimulated to grow by both TAM and E2 [41, 55], but have evolved over 5 years of serial propagation in vivo to a stage in which only TAM, but not E2, stimulates growth [47–49].
2.7 Generation of MCF-7 long-term estrogen-deprived cell culture models
MCF-7:ED cells were derived by maintaining a population of MCF-7 cells under estrogen-deprived conditions for >1 year to mimic AI treatment, and represent Phase I resistance to long-term estrogen withdrawal. MCF-7:ED cells were originally selected in phenol red-free MEM plus 5% dextran-coated charcoal-treated calf serum, but have more recently been maintained (this report) in phenol red-free RPMI, 10% dextran-coated charcoal-treated fetal bovine serum, 2 mM glutamine, 6 ng/ml bovine insulin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1× non-essential amino acids.
MCF-7/5C cells were clonally isolated from a population of MCF-7 cells grown under long-term estrogen-free conditions [56] and represent Phase II LTED resistance. MCF-7/5C cells were originally cultured in estrogen-free MEM plus 5% dextran-coated charcoal-treated calf serum, and under these conditions, MCF-7/5C cells exhibited estrogen and SERM independent growth [52, 56]. However, we have observed that when MCF-7/5C cells are switched to estrogen-free RPMI plus 10% dextran-coated charcoal-treated calf serum, the cells undergo rapid apoptosis when treated with 1 nM E2 [52, 53]. In all experiments described in this report, MCF-7/5C cells were maintained under the latter media conditions (estrogen-free RPMI plus 10% dextran-coated charcoal-treated calf serum, 2 mM glutamine, 6 ng/ml bovine insulin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1× non-essential amino acids).
2.8 Generation of MCF-7/5C estrogen-deprived xenograft tumors
The MCF-7/5C (Phase II) long-term estrogen withdrawn-resistant tumors were generated by bilateral injection of these cells grown in culture into the axillary mammary fat pads of ovariectomized athymic mice at 107 cells per site.
2.9 Cell proliferation assays
Wild-type MCF-7 cells were switched from phenol red-containing RPMI medium supplemented with 10% FBS to phenol red-free RPMI medium supplemented with 10% dextran-coated charcoal-treated fetal bovine serum for 4 days prior to beginning the proliferation assay. Since MCF-7/ED and MCF-7/5C cells are routinely maintained in estrogen-free media, no media switch was required. MCF-7, MCF-7/ED, and MCF-7/5C cells were seeded in estrogen-free RPMI containing 10% DCC-FBS at a density of 2x104 cells per well in 24-well plates. After 24 hours (Day 0), the medium was replaced with fresh estrogen-free RPMI medium and cells were treated with 0.1% ethanol vehicle (control), 1 nM E2, 10 nM FUL, or 1 nM E2 + 10 nM FUL. Cells were retreated with the drugs on days 2, 4, and 6 and the experiment was stopped on day 7. The DNA content of the cells, a measure of proliferation, was determined using a Fluorescent DNA Quantitation kit (Bio-Rad Laboratories, Hercules, CA). For each analysis, six replicate wells were used, and at least three independent experiments were performed. Proliferation of cell lines following 7 day of growth are shown as the mean DNA amount per well per treatment ± standard deviation (SD).
2.10 Statistical analyses
In tumor growth experiments in which treatments were started at the time of tumor innoculation, tumors were analyzed longitudinally with two-factor analysis of variance (ANOVA) to determine significant differences in cross-sectional areas between all tumors in each treatment group in a time-dependent manner (data in Figures 1 through 4, and 6 through 7). In the tumor growth experiments in which treatments were started after the tumors were established, one-factor ANOVA was used to determine significant differences in cross-sectional areas between all tumors in each treatment group on the last day of the experiment (data in Fig. 8, day 52). All statistical tests were two-sided and calculated using SAS (SAS Institute, Cary, NC, USA).
Fig. 4.
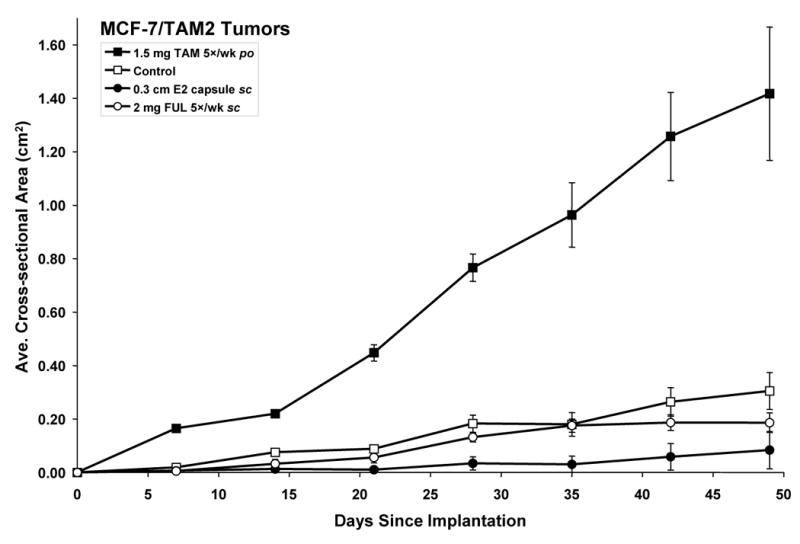
Growth inhibition of MCF-7/TAM2 tumors in response to E2 and FUL. Twenty ovariectomized athymic nude mice were implanted in the axillary mammary fat pads with 1 mm3 MCF-7/TAM2 tumor pieces and separated into 4 treatment groups of 5 mice each (10 tumors per group) corresponding to 1.5 mg/day TAM po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc, and Control (no treatment). The data shown represent the average tumor cross-sectional area (cm2) per group ± SE. The cross-sectional area of TAM-treated (P < 0.0001) and E2-treated MCF-7/TAM2 tumors (P = 0.0004) was significantly different from Control tumors (P < 0.0001).
Fig. 6.

Growth inhibition of MCF-7/5C tumors in response to E2 treatment, and resistance to FUL, and E2 plus FUL. Twenty ovariectomized athymic nude mice were bilaterally injected in the axillary mammary fat pads with 107 MCF-7/5C cells grown in culture and separated into 4 treatment groups of 5 mice each (10 tumors per group) corresponding to Control (no treatment), 0.3 cm E2 capsule sc, 2 mg/day FUL sc, and 0.3 cm E2 capsule sc + 2 mg/day FUL sc. The data are shown as a histogram on day 21 of the average tumor cross-sectional area (cm2) per group ± SE. The cross-sectional areas of Control-treated, FUL-treated, and E2 plus FUL-treated MCF-7/5C tumors were each significantly different from E2-treated (all P-values < 0.0001). However, the cross-sectional area of both FUL-treated and E2 plus FUL-treated MCF-7/5C tumors were not significantly different from that of control-treated MCF-7/5C tumors.
Fig. 7.

E2 plus FUL-stimulated growth of MCF-7/RAL2 tumors. Data are from Figure 3 on day 42 and shown as a histogram, but supplemented with the additional group of 5 ovariectomized athymic mice (10 tumors) treated with 0.3 cm E2 capsule sc plus 2 mg/day FUL sc. The cross-sectional area of E2 plus FUL-treated MCF-7/RAL2 tumors was significantly different from that of Control-treated, E2-treated, and FUL-treated MCF-7/RAL2 tumors (all P-values < 0.0001).
Fig. 8.

E2 plus FUL-stimulated growth of MCF-7/TAM2 tumors. Twenty-five ovariectomized athymic mice were implanted in the axillary mammary fat pads with 1 mm3 MCF-7/TAM2 tumor pieces, and then treated with 1.5 mg/day TAM po until the tumors were established at 0.24 cm2, then TAM was withdrawn for 1 week. Following 1 week of TAM withdrawal, the tumors reached an average cross-sectional area of 0.37 cm2 and the animals were separated into 5 treatment groups of 5 mice each (10 tumors per group) corresponding to 1.5 mg/day TAM po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc, 0.3 cm E2 capsule sc plus 2 mg/day FUL sc, and Control (no treatment). The data shown represent the average tumor cross-sectional area (cm2) per group ± SE. The cross-sectional areas of MCF-7/TAM2 tumors at day 52 were compared by 1-way ANOVA. The cross-sectional areas of TAM-treated (P = 0.0026), E2-treated (P = 0.0098), and E2 plus FUL-treated MCF-7/TAM2 tumors (P = 0.018) were significantly different from control-treated tumors.
3. Results
3.1 Growth of MCF-7/E2 tumors and responsiveness to FUL
MCF-7/E2 xenograft tumors are propagated in vivo by serial transplantation into 0.3 cm E2 capsule-implanted ovariectomized athymic mice. To explore the sensitivity of MCF-7/E2 tumors to FUL, MCF-7/E2 tumor cores were implanted into 30 ovariectomized athymic mice and separated into 6 groups of 5 mice each, or 10 tumors per group. The treatment groups were control (no treatment), 0.3 cm E2 capsule sc, or 0.3 cm E2 capsule sc plus 1 of 4 different formulations and dosing schedules of FUL totaling 10 mg/week sc (Fig. 1). Two of the FUL formulations were suspensions made with peanut oil, differing by whether FUL was initially dissolved in EtOH or in DMSO. These FUL (EtOH/peanut oil or DMSO/peanut oil) suspensions were administered as 5 mg sc injections given two days per week, totaling 10 mg/week. The third formulation was FUL dissolved in only DMSO, and was administered as a 2 mg sc injection given five days per week, totaling 10 mg/week. The fourth FUL formulation corresponded to the clinical Faslodex preparation, which is a proprietary solution of primarily EtOH and some castor oil as a slow release-rate modifier. The clinical Faslodex preparation was administered as a 2 mg sc injection given 5 times per week, totaling 10 mg/week.
The MCF-7/E2 tumors grew robustly when treated with the 0.3 cm E2 capsule, but did not grow in the control group (Fig. 1, E2 vs. Control, P < 0.0001), demonstrating that these tumors were dependent on E2. The implanted capsules produce E2 levels that are in the physiologic range observed in perimenopausal women. The cross-sectional areas of each of the four groups of MCF-7/E2 tumors treated with E2 plus FUL was significantly smaller than those tumors treated with E2 alone (Fig. 1, all P-values <0.0001). Therefore, FUL inhibited E2-stimulated growth of MCF-7/E2 tumors. However, the degree of growth inhibition varied depending upon the formulation. Comparing the FUL suspensions in peanut oil given 2 times per week, FUL initially dissolved in DMSO inhibited tumor growth significantly better than FUL initially dissolved in EtOH (Fig. 1, P = 0.001). Comparing the FUL formulations given 5 times per week, FUL dissolved in only DMSO inhibited tumor growth significantly better than the clinical Faslodex preparation (Fig. 1, P = 0.004). Although we did not measure circulating FUL levels, we hypothesize that circulating levels of FUL were higher when using DMSO-based formulations, leading to more potent inhibition of E2-stimulated tumor growth.
3.2 Growth of MCF-7/RAL1 tumors
MCF-7/RAL1 tumors are maintained in vivo by serial transplantation into 1.5 mg/day RAL-treated ovariectomized athymic mice. To illustrate the phase of SERM resistance the MCF-7/RAL1 tumor should be categorized into, MCF-7/RAL1 tumor cores were implanted into 20 ovariectomized athymic mice and separated into 4 treatment groups of 5 mice each (10 tumors/group) corresponding 1.5 mg/day RAL po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc (Faslodex preparation), and control (no treatment). The MCF-7/RAL1 tumors were significantly stimulated to grow by RAL treatment (P < 0.0001) and by E2 treatment (P < 0.0001) compared to control treatment (Fig. 2). However, a modest amount of growth was observed in the control-treated group, indicating that these tumors are not absolutely dependent upon an ER ligand with partial agonist activity. We have previously shown that primary cultures of MCF-7/RAL1 tumors exhibit equivalent levels of estrogen response element (ERE)-regulated reporter gene activity in the absence of E2 as did primary cultures of MCF-7/E2 tumors when treated with E2 [50]. Thus, the unliganded ER activity in MCF-7/RAL1 tumors is high and probably contributed to the modest growth of these tumors without the need of RAL or E2. FUL did not significantly effect the growth of MCF-7/RAL1 tumors (Fig. 2). Thus, either a SERM or E2, but not FUL, supports the growth of these MCF-7/RAL1 xenografts. Therefore, these tumors are categorized as Phase I SERM-resistant.
Fig. 2.

Growth stimulation of MCF-7/RAL1 tumors in response to E2, and inhibition by FUL. Twenty ovariectomized athymic nude mice were implanted in the axillary mammary fat pads with 1 mm3 MCF-7/RAL1 tumor pieces and separated into 4 treatment groups of 5 mice each (10 tumors per group) corresponding to 1.5 mg/day RAL po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc, and Control (no treatment). The data shown represent the average tumor cross-sectional area (cm2) per group ± SE. The cross-sectional area of RAL-treated (P < 0.0001) and E2-treated MCF-7/RAL1 tumors (P < 0.0001) was significantly different from control-treated tumors.
3.3 Growth of MCF-7/RAL2 tumors (Fig. 3)
Fig. 3.

Growth inhibition of MCF-7/RAL2 tumors in response to E2 and FUL. Twenty ovariectomized athymic nude mice were bilaterally injected in the axillary mammary fat pads with 107 MCF-7/RAL2 cells grown in culture and separated into 4 treatment groups of 5 mice each (10 tumors per group) corresponding to 1.5 mg/day RAL po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc, and Control (no treatment). The data shown represent the average tumor cross-sectional area (cm2) per group ± SE. The cross-sectional area of RAL-treated MCF-7/RAL2 tumors was significantly different from E2-treated, FUL-treated and Control-treated tumors (all P-values = < 0.0001).
MCF-7/RAL2 tumor cells are maintained in vitro by culture in media containing 1 μM RAL. To study the growth properties of MCF-7/RAL2 cells in vivo, the cells were grown in culture and injected into 20 ovariectomized athymic mice, which were separated into 4 groups of 5 (10 tumors/group) and treated with 1.5 mg/day RAL po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc (Faslodex preparation), or control (not treated). The MCF-7/RAL2 tumors only grew when treated with RAL (RAL vs. control, P<0.0001), and did not form any palpable tumors by day 42 when treated with E2, FUL or not treated (control) (Fig. 3). We have previously shown that when MCF-7/RAL2 tumors are allowed to grow by treating with TAM until they are established and then switching treatments to E2, E2 causes tumor regression by inducing apoptosis as measured by TUNEL staining [51]. Therefore, growth of the MCF-7/RAL2 tumors was dependent on RAL, but inhibited by E2 and FUL, which categorizes these tumors as Phase II SERM-resistant.
3.4 Growth of MCF-7/TAM2 tumors
MCF-7/TAM2 tumors are propagated in vivo by serial transplantation into 1.5 mg/day TAM-treated ovariectomized athymic mice. To characterize the growth properties of this tumor type, MCF-7/TAM2 tumor cores were implanted into 20 ovariectomized athymic mice, which were separated into 4 groups of 5 (10 tumors/group) and treated with 1.5 mg/day TAM po, 0.3 cm E2 capsule sc, 2 mg/day FUL (Faslodex) sc, or not treated (control). MCF-7/TAM2 tumors were stimulated to grow by TAM compared to the control group (Fig. 4, P < 0.0001). The control group did show a minimal amount of growth (Fig. 4), which is hypothesized to be due to substantial unliganded ER activity as in the MCF-7/RAL1 model. FUL did not significantly effect growth of the MCF-7/TAM2 tumors versus control treatment. Interestingly, E2 did significantly inhibit tumor growth compared to the control group (Fig. 4, P = 0.0004). As with the MCF-7/RAL2 tumors, we have previously demonstrated that E2 treatment leads to regression of MCF-7/TAM2 tumors [47, 49] by inducing apoptosis as detected by TUNEL staining [48]. Therefore, TAM stimulated growth, FUL did not support growth, and E2 inhibited growth of MCF-7/TAM2 tumors, defining this model as Phase II SERM-resistant.
3.5 Growth of long-term estrogen withdrawn-resistant models
Since having categorized each of the SERM-resistant tumor models as Phase I or II resistant, we characterized the growth properties of cells which have been cultured long-term under estrogen-free conditions to determine whether resistance to estrogen withdrawal (as a surrogate for AI resistance) also evolves through distinct stages. Initially, we compared the proliferation of parental MCF-7 cells with two cell lines resistant to long-term estrogen withdrawal, MCF-7/ED (estrogen-deprived) and MCF-7/5C cells. MCF-7/ED cells were originally selected by culture of parental MCF-7 in estrogen-free medium for > 1 year, but were not cloned as a subline, rather they remain a population of cells. In a similar manner, MCF-7/5C cells were also derived from parental MCF-7 cells following long-term estrogen withdrawal, but were cloned as a subline [56]. Notably, MCF-7/ED and MCF-7/5C cells were generated independently in different studies, that is, MCF-7/5C cells were not subcloned from the MCF-7/ED cells.
Growth of parental MCF-7, MCF-7/ED and MCF-7/5C cells was determined by measuring DNA amounts after 7 days in culture. Before beginning the experiment, parental MCF-7 cells were cultured for 4 days in estrogen-free media, since they had been maintained in fully-estrogenized medium. The experiment was started by seeding each of the cell lines in 24-well plates in estrogen-free medium. The cells were treated every 2 days with EtOH (vehicle control), 1 nM E2, 10 nM FUL, and 1 nM E2 plus 10 nM FUL. After 7 days, DNA quantities per well were determined using a fluorescence-based DNA assay. As expected in parental MCF-7 cells, E2 induced growth by 6.9-fold (E2 vs. control treatment), and this E2-stimulated proliferation was completely blocked by the addition of FUL (E2 + FUL vs. control) (Fig. 5). Hence, E2 stimulated proliferation of parental MCF-7 cells in an ER-dependent manner.
Fig. 5.
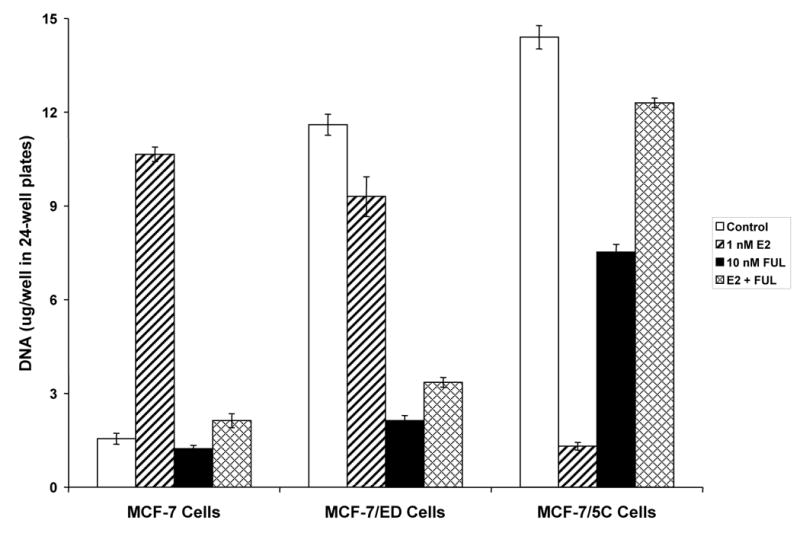
Differential proliferation of MCF-7 long-term estrogen withdrawn cell culture models in response to E2, FUL, and E2 plus FUL for 7 days. Cells were cultured under estrogen-free conditions for 4 days, and then seeded at 2 x 104 cells per well in a 24-well plate. Beginning 24 hours after seeding (day 0) and every 2 days thereafter up to 6 days (days 2, 4, and 6), the cells were treated with 1 nM E2, 10 nM FUL, 1 nM E2 + 10 nM FUL, or Control (0.1% EtOH)-treated. The experiment was stopped on day 7. As a measure of proliferation, the amount of DNA per well was determined using a fluorescence-based DNA quantitation assay. Data are shown as the mean of 6 replicate wells per group ± SD. The experiment was performed 3 times independently, and one representative experiment is shown.
Next, MCF-7/ED cells representing a population of cells resistant to estrogen withdrawal were characterized. MCF-7/ED cells grew maximally under estrogen-free conditions (control treatment, 100 % growth) and nearly maximally when treated with E2 (80% of control) (Fig. 5). However, FUL and E2 plus FUL treatment inhibited growth of MCF-7/ED cells (18% and 29%, respectively, of control) (Fig. 5). Thus, MCF-7/ED cell proliferation was largely unaffected by E2, but dependence on the ER was demonstrated by the sensitivity of the cells to FUL.
Finally, we evaluated MCF-7/5C cells, which were a clonal derivative of long-term estrogen-withdrawn cells. MCF-7/5C cells grew maximally under estrogen-free conditions (control treatment, 100% growth), but E2 treatment almost completely blocked proliferation (9% of control) (Fig. 5). Interestingly, FUL-treated MCF-7/5C cells exhibited significant growth (52% of control) (Fig. 5). Further, MCF-7/5C cells treated with E2 plus FUL showed still greater amounts of proliferation (85% of control) (Fig. 5). In prior studies, we have demonstrated that MCF-7/5C cells undergo apoptosis when treated with E2, and that co-treatment with FUL blocks this effect of E2 [53]. Hence, MCF-7/5C cells required ER to be unliganded for maximal proliferation and survival, whereas E2-bound ER led to cytostasis and apoptosis. Further, FUL reversed the apoptotic signal of E2 and promoted proliferation.
We next verified that the MCF-7/5C cells behaved similarly in vivo as a xenograft tumor as they did in vitro in cell culture. MCF-7/5C cells were grown in culture and injected into 20 ovariectomized athymic mice. The animals were separated into 4 treatment groups of 5 mice each (10 tumors/group), corresponding to control (not treated), 0.3 cm E2 capsule sc, 2 mg/day FUL sc (Faslodex), and 0.3 cm E2 capsule sc plus 2 mg/day FUL sc (Faslodex). MCF-7/5C cells rapidly formed substantial tumors at every injection site (10 out of 10) in control-treated mice by 21 days after inoculation, but only 1 palpable tumor formed out of 10 injection sites in mice treated with E2, resulting in a highly significant difference in the average tumor cross-sectional area between the two treatment groups (Fig. 6, P < 0.0001). In a prior report, we have shown that E2 induces tumor regression and apoptosis in established MCF-7/5C xenograft tumors [53]. Importantly, MCF-7/5C xenograft tumors showed robust growth in the presence of FUL or E2 plus FUL, which was not significantly different than growth of the control (no treatment) group, but was significantly greater than in the E2 treatment group (Fig. 6, FUL vs. E2, P < 0.0001; E2+FUL vs. E2, P < 0.0001). Hence, the MCF-7/5C xenograft tumor model was resistant to growth inhibition by FUL, and FUL treatment abrogated E2-mediated growth inhibition.
Considering these varied growth responses together, parental MCF-7 cells model the therapeutic stage of antihormonal therapy; MCF-7/ED cells represent Phase I resistance to estrogen withdrawal since they grew independent of E2 yet remained sensitive to FUL; and MCF-7/5C tumors/cells were classified as Phase II resistant to estrogen withdrawal since E2 inhibited their growth, but were resistant to growth inhibition by FUL or E2 plus FUL.
3.6 Response of Phase II SERM-resistant tumor models to E2 plus FUL (Fig. 7 and 8)
Since we observed that MCF-7/5C cells grew better when treated with E2 plus FUL than with E2 alone, we examined the effect of FUL in a background of physiologic E2 in the Phase II SERM-resistant tumor models. The data from the MCF-7/RAL2 experiment depicted in Figure 3 was re-evaluated with an additional group of 5 animals (10 tumors) treated with a 0.3 cm E2 capsule sc plus 2 mg/day FUL (Faslodex). MCF-7/RAL2 tumors treated with E2 plus FUL showed robust growth compared to no palpable tumors in the E2 alone (P < 0.0001), FUL alone (P < 0.0001), or control groups (P < 0.0001) (Fig. 7). Therefore, E2 plus FUL, when combined, negated the growth inhibitory effects of either compound by itself.
We then tested whether this interaction between physiologic E2 and FUL also occurred in the MCF-7/TAM2 tumor model of Phase II SERM resistance. However, this experiment was designed to evaluate effects of different treatments on tumors once they are established by allowing tumors to grow in the presence of TAM until they were palpable, and then randomized to different treatment groups. MCF-7/TAM2 tumor cores were implanted into 25 ovariectomized athymic mice. All animals were treated with 1.5 mg/day TAM po until tumors grew to an average cross-sectional area of 0.24 cm2, at which time TAM treatment was withdrawn for 1 week to allow time for this drug to be completely metabolized and clear the animals’ systems. Following the 1 week of TAM withdrawal, the average cross-sectional area of all tumors was 0.37 cm2, and the animals were randomized into 5 groups of 5 mice each (10 tumors/group) corresponding to continuing 1.5 mg/day TAM po, 0.3 cm E2 capsule sc, 2 mg/day FUL sc, 0.3 cm E2 capsule sc plus 2 mg/day FUL sc, and control (no treatment). As would be predicted from the MCF-7/TAM2 experiment depicted in Figure 4, TAM treatment significantly stimulated growth (P = 0.0026) and E2 significantly inhibited growth (P = 0.0098) compared to the control group on day 52 (Fig. 8). The size of FUL treated tumors was not significantly different than the control group. In contrast, we noted that tumors treated with the combination of E2 + FUL did exhibit significantly greater growth than the control group (Fig. 8, P = 0.018). Thus, in a second model of Phase II SERM resistance, growth inhibition by E2 alone was negated in the presence of FUL, leading to growth stimulation.
4. Discussion
We sought to discover unifying principles involved in the development of antihormone resistance by systematically studying the growth properties of a panel of antihormonally resistant MCF-7-based breast cancer xenograft tumor models. We have confirmed and extended prior observations [40–53] that have allowed the categorization of these tumor models as either Phase I or Phase II antihormone resistant. Phase I SERM resistance was characterized by growth stimulation in response to either a SERM or E2 (MCF-7/RAL1 tumors, Figure 2), while in Phase II SERM resistance, only the SERM stimulated growth and E2 inhibited growth (MCF-7/RAL2 and MCF-7/TAM2, Fig. 3 and 4, respectively). Phase I long-term estrogen withdrawn (AI) -resistant cells in culture grew independently of E2 (MCF-7/ED cells, Figure 5), but Phase II resistant tumors were growth inhibited by E2 (MCF-7/5C tumors, Figure 6). Hence, long-term blockade of ER activity by either SERMs or estrogen withdrawal can lead to selection of cells in which E2 signals no longer proliferation, but rather inhibition, of growth, and as we have previously reported, apoptosis [48, 51, 53].
We also found that growth of Phase II SERM resistant tumors was not supported by FUL, but Phase II estrogen withdrawn-resistant tumors were cross-resistant to FUL (MCF-7/5C tumors, Fig. 6). Further, FUL combined with physiologic E2 nullified the inhibitory effects of either compound alone and led to stimulation of growth in Phase II SERM-resistant tumors (MCF-7/RAL2 and MCF-7/TAM2 tumors, Figures 7 and 8, respectively), and supported growth in Phase II estrogen withdrawn-resistant tumors (MCF-7/5C tumors, Fig. 6).
Noteworthy, the concentrations of FUL used in the cell culture proliferation experiments was 10 nM. We chose this concentration of FUL to reflect the circulating levels that are achieved clinically. In the clinic, FUL is not administered orally because of low bioavailability; rather it is given intra muscularly (as a single 250 mg dose once per month) to achieve slow constant release of the drug. In two independent multi-national Phase III clinical efficacy trials evaluating FUL in advanced breast cancer patients, the steady state circulating concentrations of FUL were determined to be approximately 6 – 7 μg/L (9.9 – 11.5 nM) in the European trial and 9 μg/L (14.8 nM) in the North American trial [57]. Hence, we used FUL at 10 nM in cell culture, reflecting the circulating concentrations of FUL achieved in women, but this concentration was much lower than the 100 nM to 1 μM FUL concentrations used in most cell culture studies. We hypothesize that the low circulating concentrations of FUL in patients may contribute to the lower than expected response rates in the clinic as would be predicted by the effectiveness of FUL in cell culture. In support of this hypothesis, we have found that while MCF-7/5C cells proliferate in the presence of 10 nM FUL, they do not in 1 μM FUL (unpublished, JS Lewis-Wambi and VC Jordan).
The distinct growth responses of the tumor and cell culture models studied here illustrate that resistance to hormonal blockade therapy continually evolves but can be separated into at least two phases (Fig. 9). Antihormonal resistance develops from selection of specific cell types that survive and proliferate when the ER is bound by a partial antiestrogen (Phase I SERM resistance) or unliganded (Phase I estrogen withdrawn-resistance). Prolonged hormonal blockade therapy maintains selective pressure, such that Phase I resistant cells continue to evolve to a Phase II resistant phenotype, and likely undefined additional phases. However, the study of Phase II antihormonal resistance has revealed a new biology of E2 action involved in apoptosis that could be exploited to benefit breast cancer patients who have been exhaustively treated with SERMs and AIs. Moreover, the finding that FUL in a background of physiologic E2 stimulated growth of Phase II resistant cells has important clinical implications. This knowledge can be implemented to optimize the application of third-line antihormonal therapy (Fig. 10). We propose that patients who have responded and then failed two antihormone therapies may exhibit Phase II resistant characteristics, and therefore respond to low-dose short-term estrogen therapy. The estrogen therapy would lead to apoptosis in the Phase II resistant cells and thereby debulk the tumor. Prior laboratory studies indicate that cells which remain are re-sensitized to first-line or second-line antihormonal therapy [47, 49]. Hence, the low-dose short-term estrogen therapy would be followed by FUL plus an AI, to avoid the possible selection of cells that could grow in response to FUL plus physiologic E2.
Fig. 9.
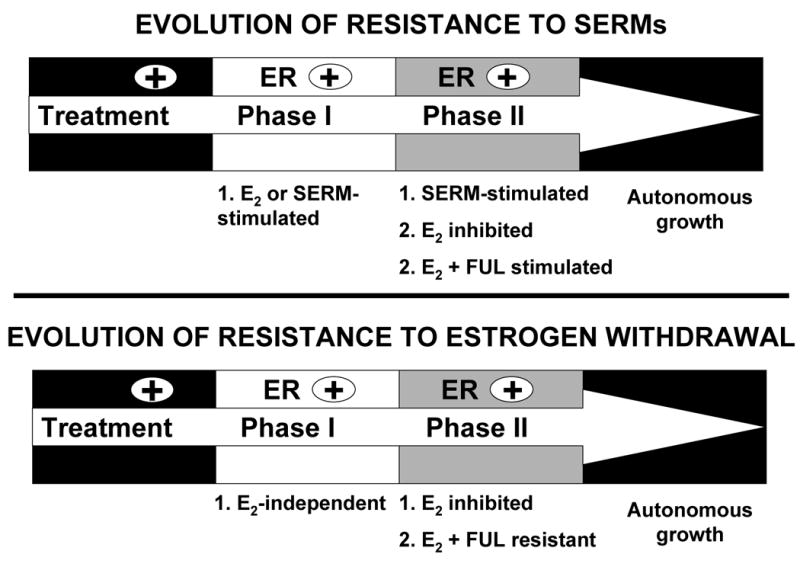
Evolution of antihormonal resistance in laboratory models of breast cancer. Long-term antihormonal therapy leads to selection of resistant cells that are stimulated to grow by a SERM or grow in an estrogen-depleted environment (AI resistance). Based on prior laboratory studies (4–10, 12, 13) and data presented here, the progression of antihormonal resistance can be separated into at least 2 phases defined by different growth responses to E2 and FUL. In Phase I resistant disease, tumor cells are either growth stimulated by E2 (as in SERM resistance) or grow independently of E2 (as in estrogen withdrawn/AI resistance). However, these Phase I resistant cells remain dependent on ER since they are sensitive to growth inhibition by FUL. Selection of tumor cells continues during exhaustive antihormonal therapy until Phase II resistance develops, which is characterized by a new biology of E2 action. Both Phase II SERM and estrogen withdrawn-resistant tumors respond to E2 with growth inhibition and apoptosis. FUL still inhibits growth of Phase II SERM-resistant tumors, but not of Phase II estrogen withdrawn-resistant tumors. Moreover, FUL interacts with E2 at physiologic concentrations to promote growth of both Phase II SERM and estrogen withdrawn-resistant disease. These emerging concepts on the evolution of antihormonal resistance based on laboratory studies have important implications for the utility of estrogen and FUL in the clinic.
Fig. 10.

A proposed clinical strategy for the optimal application of third-line antihormonal therapy. We propose that patients who have initially responded to, and then failed, two previous antihormonal therapies may exhibit Phase II antihormonal resistance, and would then benefit from short-term low-dose estrogen, which would induce apoptosis in the Phase II resistant cells and debulk the tumor. Prior laboratory evidence indicates that the small percentage of Phase II tumors which revert to an estrogen-stimulated stage after estrogen-induced regression, are also re-sensitized to antihormonal therapy [47, 49]. A low tumor burden would be maintained by FUL in an estrogen-depleted environment, i.e. FUL plus an aromatase inhibitor, to avoid the possible emergence of tumor growth through a negative interaction between FUL and physiologic estrogen.
Acknowledgments
This work is supported by the Department of Defense Breast Program under award number BC050277 Center of Excellence (V.C.J.) (Views and opinions of, and endorsements by the author(s) do not reflect those of the US Army or the Department of Defense), SPORE in Breast Cancer CA89018 (V.C.J.), R01 GM067156 (V.C.J.), the Avon Foundation (V.C.J.), the Weg Fund (Fox Chase Cancer Center), and NIH P30 CA006927 (Fox Chase Cancer Center).
Abbreviations
- AI
aromatase inhibitor
- ER
estrogen receptor
- E2
17β -estradiol
- FUL
fulvestrant
- RAL
raloxifene
- SERM
selective estrogen receptor modulator
Footnotes
Proceedings of the 17th International Symposium of the Journal of Steroid Biochemistry and Molecular Biology, ‘Recent Advances in Steroid Biochemistry and Molecular Biology’ (Seefeld, Tyrol, Austria, 31 May - 3 June, 2006).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jensen EV, Block GE, Smith S, Kyser K, DeSombre ER. Estrogen receptors and breast cancer response to adrenalectomy. Natl Cancer Inst Monogr. 1971;34:55–70. [PubMed] [Google Scholar]
- 2.McGuire WL, Carbone PP, Vollmer EP. Oestrogen receptors in human breast cancer: An overview. In: McGuire WL, Carbone PP, Vollmer EP, editors. Oestrogen receptors in human breast cancer. Vol. 6 Raven Press; New York: 1975. [Google Scholar]
- 3.Lerner LJ, Jordan VC. The development of antiestrogens for the treatment of breast cancer: Eighth cain memorial award lecture. Cancer Res. 1990;50:4177–4189. [PubMed] [Google Scholar]
- 4.Jordan VC. Fourteenth gaddum memorial lecture. A current view of tamoxifen for the treatment and prevention of breast cancer. Br J Pharmacol. 1993;110:507–517. doi: 10.1111/j.1476-5381.1993.tb13840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Early Breast Cancer Trialists' Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 6.Jensen EV, Jordan VC. The estrogen receptor: A model for molecular medicine. Clin Cancer Res. 2003;9:1980–1989. [PubMed] [Google Scholar]
- 7.Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6:181–202. [PubMed] [Google Scholar]
- 8.Strasser-Weippl K, Goss PE. Advances in adjuvant hormonal therapy for postmenopausal women. J Clin Oncol. 2005;23:1751–1759. doi: 10.1200/JCO.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 9.Early Breast Cancer Trialists' Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 10.Early Breast Cancer Trialists' Collaborative Group. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 11.Fisher B, Dignam J, Bryant J, Wolmark N. Five versus more than five years of tamoxifen for lymph node-negative breast cancer: Updated findings from the national surgical adjuvant breast and bowel project b-14 randomized trial. J Natl Cancer Inst. 2001;93:684–690. doi: 10.1093/jnci/93.9.684. [DOI] [PubMed] [Google Scholar]
- 12.Smith I. Breast cancer: Adjuvant treatment for early breast cancer. Ann Oncol. 2005;16:ii182–187. doi: 10.1093/annonc/mdi709. [DOI] [PubMed] [Google Scholar]
- 13.Cuzick J, Powles T, Veronesi U, Forbes J, Edwards R, Ashley S, Boyle P. Overview of the main outcomes in breast-cancer prevention trials. Lancet. 2003;361:296–300. doi: 10.1016/S0140-6736(03)12342-2. [DOI] [PubMed] [Google Scholar]
- 14.Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, Norton L, Nickelsen T, Bjarnason NH, Morrow M, Lippman ME, Black D, Glusman JE, Costa A, Jordan VC. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the more randomized trial. JAMA. 1999;281:2189–2197. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- 15.Martino S, Cauley JA, Barrett-Connor E, Powles TJ, Mershon J, Disch D, Secrest RJ, Cummings SR. Continuing outcomes relevant to evista: Breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst. 2004;96:1751–1761. doi: 10.1093/jnci/djh319. [DOI] [PubMed] [Google Scholar]
- 16.Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Jr, Wade JL, 3rd, Robidoux A, Margolese RG, James J, Lippman SM, Runowicz CD, Ganz PA, Reis SE, McCaskill-Stevens W, Ford LG, Jordan VC, Wolmark N. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: The NSABP study of tamoxifen and raloxifene (star) p-2 trial. JAMA. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 17.Cranney A, Tugwell P, Zytaruk N, Robinson V, Weaver B, Adachi J, Wells G, Shea B, Guyatt G., Iv Meta-analysis of raloxifene for the prevention and treatment of postmenopausal osteoporosis. Endocr Rev. 2002;23:524–528. doi: 10.1210/er.2001-4002. [DOI] [PubMed] [Google Scholar]
- 18.Barrett-Connor E, Cox DA, Anderson PW. The potential of serms for reducing the risk of coronary heart disease. Trends in Endocrinology and Metabolism. 1999;10:320–325. doi: 10.1016/s1043-2760(99)00182-4. [DOI] [PubMed] [Google Scholar]
- 19.Wenger NK, Barrett-Connor E, Collins P, Grady D, Kornitzer M, Mosca L, Sashegyi A, Baygani SK, Anderson PW, Moscarelli E. Baseline characteristics of participants in the raloxifene use for the heart (RUTH) trial. The American Journal of Cardiology. 2002;90:1204–1210. doi: 10.1016/s0002-9149(02)02835-7. [DOI] [PubMed] [Google Scholar]
- 20.Mosca L, Barrett-Connor E, Wenger NK, Collins P, Grady D, Kornitzer M, Moscarelli E, Paul S, Wright TJ, Helterbrand JD, Anderson PW. Design and methods of the raloxifene use for the heart (RUTH) study. The American Journal of Cardiology. 2001;88:392–395. doi: 10.1016/s0002-9149(01)01685-x. [DOI] [PubMed] [Google Scholar]
- 21.Evans CT, Ledesma DB, Schulz TZ, Simpson ER, Mendelson CR. Isolation and characterization of a complementary DNA specific for human aromatase-system cytochrome p-450 mRNA. Proc Natl Acad Sci U S A. 1986;83:6387–6391. doi: 10.1073/pnas.83.17.6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson LR, Bulun SE. Estrogen production and action. Journal of the American Academy of Dermatology. 2001;45:S116–S124. doi: 10.1067/mjd.2001.117432. [DOI] [PubMed] [Google Scholar]
- 23.Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. N Engl J Med. 2003;348:2431–2442. doi: 10.1056/NEJMra023246. [DOI] [PubMed] [Google Scholar]
- 24.Joensuu H, Ejlertsen B, Lonning PE, Rutqvist LE. Aromatase inhibitors in the treatment of early and advanced breast cancer. Acta Oncol. 2005;44:23–31. doi: 10.1080/02841860510007468. [DOI] [PubMed] [Google Scholar]
- 25.Geisler J, Lonning PE. Aromatase inhibition: Translation into a successful therapeutic approach. Clin Cancer Res. 2005;11:2809–2821. doi: 10.1158/1078-0432.CCR-04-2187. [DOI] [PubMed] [Google Scholar]
- 26.Coombes RC, Hall E, Gibson LJ, Paridaens R, Jassem J, Delozier T, Jones SE, Alvarez I, Bertelli G, Ortmann O, Coates AS, Bajetta E, Dodwell D, Coleman RE, Fallowfield LJ, Mickiewicz E, Andersen J, Lonning PE, Cocconi G, Stewart A, Stuart N, Snowdon CF, Carpentieri M, Massimini G, Bliss JM the Intergroup Exemestane Study. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med. 2004;350:1081–1092. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 27.Mouridsen HT. Exemestane following tamoxifen in postmenopausal women with primary breast cancer. J Clin Oncol. 2004;22:3833–3834. doi: 10.1200/JCO.2004.99.115. [DOI] [PubMed] [Google Scholar]
- 28.Bonneterre J, Thurlimann B, Robertson JFR, Krzakowski M, Mauriac L, Koralewski P, Vergote I, Webster A, Steinberg M, von Euler M. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: Results of the tamoxifen or arimidex randomized group efficacy and tolerability study. J Clin Oncol. 2000;18:3748–3757. doi: 10.1200/JCO.2000.18.22.3748. [DOI] [PubMed] [Google Scholar]
- 29.Boccardo F, Rubagotti A, Puntoni M, Guglielmini P, Amoroso D, Fini A, Paladini G, Mesiti M, Romeo D, Rinaldini M, Scali S, Porpiglia M, Benedetto C, Restuccia N, Buzzi F, Franchi R, Massidda B, Distante V, Amadori D, Sismondi P. Switching to anastrozole versus continued tamoxifen treatment of early breast cancer: Preliminary results of the Italian tamoxifen anastrozole trial. J Clin Oncol. 2005;23:5138–5147. doi: 10.1200/JCO.2005.04.120. [DOI] [PubMed] [Google Scholar]
- 30.Mouridsen H, Gershanovich M, Sun Y, Perez-Carrion R, Boni C, Monnier A, Apffelstaedt J, Smith R, Sleeboom HP, Jaenicke F, Pluzanska A, Dank M, Becquart D, Bapsy PP, Salminen E, Snyder R, Chaudri-Ross H, Lang R, Wyld P, Bhatnagar A. Phase iii study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: Analysis of survival and update of efficacy from the international letrozole breast cancer group. J Clin Oncol. 2003;21:2101–2109. doi: 10.1200/JCO.2003.04.194. [DOI] [PubMed] [Google Scholar]
- 31.Mouridsen H, Gershanovich M, Sun Y, Perez-Carrion R, Boni C, Monnier A, Apffelstaedt J, Smith R, Sleeboom HP, Janicke F, Pluzanska A, Dank M, Becquart D, Bapsy PP, Salminen E, Snyder R, Lassus M, Verbeek JA, Staffler B, Chaudri-Ross HA, Dugan M. Superior efficacy of letrozole versus tamoxifen as first-line therapy for postmenopausal women with advanced breast cancer: Results of a phase III study of the international letrozole breast cancer group. J Clin Oncol. 2001;19:2596–2606. doi: 10.1200/JCO.2001.19.10.2596. [DOI] [PubMed] [Google Scholar]
- 32.Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, Sahmoud T. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: First results of the ATAC randomised trial. Lancet. 2002;359:2131–2139. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 33.Howell A, Cuzick J, Baum M, Buzdar A, Dowsett M, Forbes JF, Hoctin-Boes G, Houghton J, Locker GY, Tobias JS. Results of the ATAC (arimidex, tamoxifen, alone or in combination) trial after completion of 5 years' adjuvant treatment for breast cancer. Lancet. 2005;365:60–62. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- 34.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Cameron DA, Palmer MJ, Pater JL. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: Updated findings from NCIC CTG MA.17. J Natl Cancer Inst. 2005;97:1262–1271. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- 35.Carlson RW. The history and mechanism of action of fulvestrant. Clin Breast Cancer 6 Suppl. 2005;1:S5–8. doi: 10.3816/cbc.2005.s.008. [DOI] [PubMed] [Google Scholar]
- 36.Gradishar WJ, Sahmoud T. Current and future perspectives on fulvestrant. Clin Breast Cancer. 2005;6(Suppl 1):S23–29. doi: 10.3816/cbc.2005.s.011. [DOI] [PubMed] [Google Scholar]
- 37.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure (Camb) 2001;9:145–153. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 38.Howell A, Robertson JFR, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, Vergote I, Erikstein B, Webster A, Morris C. Fulvestrant, formerly ici 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. 2002;20:3396–3403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- 39.Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, Gertler SZ, May JT, Burton G, Dimery I, Webster A, Morris C, Elledge R, Buzdar A. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J Clin Oncol. 2002;20:3386–3395. doi: 10.1200/JCO.2002.10.058. [DOI] [PubMed] [Google Scholar]
- 40.Gottardis MM, Robinson SP, Jordan VC. Estradiol-stimulated growth of MCF-7 tumors implanted in athymic mice: A model to study the tumoristatic action of tamoxifen. J Steroid Biochem. 1988;30:311–314. doi: 10.1016/0022-4731(88)90113-6. [DOI] [PubMed] [Google Scholar]
- 41.Gottardis MM, Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res. 1988;48:5183–5187. [PubMed] [Google Scholar]
- 42.Gottardis MM, Jiang SY, Jeng MH, Jordan VC. Inhibition of tamoxifen-stimulated growth of an mcf-7 tumor variant in athymic mice by novel steroidal antiestrogens. Cancer Res. 1989;49:4090–4093. [PubMed] [Google Scholar]
- 43.Wolf DM, Jordan VC. A laboratory model to explain the survival advantage observed in patients taking adjuvant tamoxifen therapy. Recent Results Cancer Res. 1993;127:23–33. doi: 10.1007/978-3-642-84745-5_4. [DOI] [PubMed] [Google Scholar]
- 44.Wolf DM, Jordan VC, William L. Mcguire Memorial Symposium. Drug resistance to tamoxifen during breast cancer therapy. Breast Cancer Res Treat. 1993;27:27–40. doi: 10.1007/BF00683191. [DOI] [PubMed] [Google Scholar]
- 45.Wolf DM, Jordan VC. Characterization of tamoxifen stimulated MCF-7 tumor variants grown in athymic mice. Breast Cancer Res Treat. 1994;31:117–127. doi: 10.1007/BF00689682. [DOI] [PubMed] [Google Scholar]
- 46.Wolf DM, Langan-Fahey SM, Parker CJ, McCague R, Jordan VC. Investigation of the mechanism of tamoxifen-stimulated breast tumor growth with nonisomerizable analogues of tamoxifen and metabolites. J Natl Cancer Inst. 1993;85:806–812. doi: 10.1093/jnci/85.10.806. [DOI] [PubMed] [Google Scholar]
- 47.Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, O'Regan RM, Jordan VC. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res. 2000;6:2028–2036. [PubMed] [Google Scholar]
- 48.Osipo C, Gajdos C, Liu H, Chen B, Jordan VC. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J Natl Cancer Inst. 2003;95:1597–1608. doi: 10.1093/jnci/djg079. [DOI] [PubMed] [Google Scholar]
- 49.Osipo C, Gajdos C, Cheng D, Jordan VC. Reversal of tamoxifen resistant breast cancer by low dose estrogen therapy. J Steroid Biochem Mol Biol. 2005;93:249–256. doi: 10.1016/j.jsbmb.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 50.O'Regan RM, Osipo C, Ariazi E, Lee ES, Meeke K, Morris C, Bertucci A, Sarker MA, Grigg R, Jordan VC. Development and therapeutic options for the treatment of raloxifene-stimulated breast cancer in athymic mice. Clin Cancer Res. 2006;12:2255–2263. doi: 10.1158/1078-0432.CCR-05-2584. [DOI] [PubMed] [Google Scholar]
- 51.Liu H, Lee E-S, Gajdos C, Pearce ST, Chen B, Osipo C, Loweth J, McKian K, De Los Reyes A, Wing L, Jordan VC. Apoptotic action of 17{beta}-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95:1586–1597. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 52.Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94:131–141. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 53.Lewis JS, Meeke K, Osipo C, Ross EA, Kidawi N, Li T, Bell E, Chandel NS, Jordan VC. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97:1746–1759. doi: 10.1093/jnci/dji400. [DOI] [PubMed] [Google Scholar]
- 54.O'Regan RM, Cisneros A, England GM, MacGregor JI, Muenzner HD, Assikis VJ, Bilimoria MM, Piette M, Dragan YP, Pitot HC, Chatterton R, Jordan VC. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. J Natl Cancer Inst. 1998;90:1552–1558. doi: 10.1093/jnci/90.20.1552. [DOI] [PubMed] [Google Scholar]
- 55.Gottardis MM, Wagner RJ, Borden EC, Jordan VC. Differential ability of antiestrogens to stimulate breast cancer cell (MCF-7) growth in vivo and in vitro. Cancer Res. 1989;49:4765–4769. [PubMed] [Google Scholar]
- 56.Jiang SY, Wolf DM, Yingling JM, Chang C, Jordan VC. An estrogen receptor positive MCF-7 clone that is resistant to antiestrogens and estradiol. Mol Cell Endocrinol. 1992;90:77–86. doi: 10.1016/0303-7207(92)90104-e. [DOI] [PubMed] [Google Scholar]
- 57.Robertson JF, Erikstein B, Osborne KC, Pippen J, Come SE, Parker LM, Gertler S, Harrison MP, Clarke DA. Pharmacokinetic profile of intramuscular fulvestrant in advanced breast cancer. Clin Pharmacokinet. 2004;43:529–538. doi: 10.2165/00003088-200443080-00003. [DOI] [PubMed] [Google Scholar]