

Abstract
A MUC1-based vaccine was used in a preclinical model of colon cancer. The trial was conducted in a MUC1-tolerant immune competent host injected with MC38 colon cancer cells expressing MUC1. The vaccine included: MHC class I-restricted MUC1 peptides, MHC class II-restricted pan helper peptide, unmethylated CpG oligodeoxynucleotide, and granulocyte macrophage-colony stimulating factor. Immunization was successful in breaking MUC1 self-tolerance, and in eliciting a robust anti-tumor response. The vaccine stimulated IFN-γ-producing CD4+ helper and CD8+ cytotoxic T cells against MUC1 and other undefined MC38 tumor antigens. In the prophylactic setting, immunization caused complete rejection of tumor cells, while in the therapeutic regimen, tumor burden was significantly reduced.
Keywords: MUC1, CpG ODN, GM-CSF, and colon cancer
Introduction
Colorectal cancer is the third most common malignancy in the US with 75% of cases diagnosed early and treated effectively with surgery, chemotherapy, radiation therapy, or a combination of these modalities. However, 25% of patients that present with metastatic disease have a five-year survival of only 10%. Complications from metastatic disease are the leading causes of cancer-related deaths. In recent years there has been great interest in cancer vaccines, which have the potential of controlling metastatic disease, prolonging time to recurrence, and ultimately serving as a preventive measure. MUC1 (CD227) is a membrane-tethered mucin glycoprotein expressed on the apical surfaces of normal glandular epithelia and it is over expressed and aberrantly glycosylated in >70% of human colon cancer [1–3]. Recent description of MUC1 as a target for cytotoxic T lymphocytes (CTLs) has raised interest in using this protein as a target for immunotherapy [4–14]. Several preclinical and clinical trials targeting the tumor-associated MUC1 antigen have elicited strong anti-tumor CTLs [1,2,12,14–24]. Importantly, there have been several reports of two HLA-A2 binding peptides derived from the MUC1 protein [25–27]. MUC1-specific CTLs have also been induced in vivo after vaccination of breast, ovarian, and metastatic renal cancer patients with peptide-pulsed dendritic cells (DCs) [16,28,29]. However, these approaches have generated only modest and infrequent clinical responses. One reason may be that traditionally, cancer-specific vaccines have focused on the activation of the CD8+ CTLs, often involving direct stimulation of immunity using HLA-class I binding peptide epitopes. Recently it has become clear that activation of the CTL immune effector arm alone is insufficient to mediate an effective anticancer response. A major problem is that CD8+ T cells alone cannot be sustained without the concomitant activation of CD4+ T helper (Th) cells. In fact, it is now widely recognized that the Th cell regulates nearly all aspects of the adaptive immune response [30]. In addition, Th cells can recruit the innate immune system during immune augmentation. Therefore, the focus of the immune response in cancer has shifted away from activating CTL immunity alone to activating Th cell immunity concurrently with CTLs. In this study, in addition to the MHC class I MUC1 epitopes, we have utilized a MHC class II pan T helper peptide, TPPAYRPPNAPIL, derived from the hepatitis B virus core antigen sequence that is capable of activation of Th cells [30]. This vaccine formulation achieves an effective antitumor immunity.
In addition to the peptide vaccine, we have used adjuvants that include synthetic oligonucleotides containing CpG motifs and GM-CSF. CpG ODN are recognized by cells of the innate immune system through the Toll-like receptor 9 (TLR9), leading to potent stimulation of both the innate and the adaptive immune responses [31–33]. The net effect of TLR9 activation is to induce Th1 biased cellular and humoral effector functions with strong CTL generation [33]. In murine models of cancer and in phase I/II clinical trials in patients with metastatic melanoma and non-small cell lung cancer, CpG ODN has improved tumor responses to conventional therapies such as chemotherapy, radiotherapy, and immunotherapy [34–41]. GM-CSF is a commercially available cytokine currently used in patients undergoing chemotherapy to shorten the duration of post-chemotherapy neutropenia. Recently published evidence also suggests that GM-CSF plays an important role as an immune adjuvant [42,43]. The following observations illustrate the mechanisms by which GM-CSF can potentiate the immunogenicity of an antigen: 1) GM-CSF is a key mediator of DC maturation and function [44]; 2) GM-CSF increases surface expression of class I and II MHC molecules as well as co-stimulatory molecules of DCs in vitro [44]; 3) GM-CSF enhances antibody responses to known immunogens in vivo [45]; 4) tumor cells transfected with genes encoding/expressing GM-CSF are able to induce long lasting, specific anti-tumor immune responses in vivo [46]; and 5) GM-CSF encapsulated in biodegradable microspheres mixed with whole tumor cells resulted in systemic anti-tumor immune responses comparable to those of GM-CSF transfected tumor cells [47]. Addition of GM-CSF to a peptide vaccine has been successfully utilized in patients with metastatic melanoma [48]. Thus, GM-CSF was included in our MUC1-peptide vaccine formulation with the expectation that it would enhance immunogenicity of the peptides. Based on existing data suggesting the potent immune adjuvant properties of CpG ODN and GM-CSF, we elected to test the efficacy of both these agents in the setting of a MUC1/pan T helper peptide vaccine immunization in a MUC1-tolerant colon cancer mouse model. To optimally enhance the antigenicity, the peptides and immune adjuvants were delivered emulsified in incomplete Freund’s adjuvant (IFA). In addition, the local inflammatory properties of IFA may play an important role in attracting antigen presenting cells to the site of injection [49,50].
Herein we present results of the vaccination strategy containing MHC class I-binding MUC1 peptides along with a MHC class II-binding pan helper peptide and adjuvants in a colon cancer mouse model. Subcutaneous tumors were elicited by injecting the syngeneic MC38 colon cancer cell line expressing full-length human MUC1 into human MUC1.Tg mice. These mice exhibit T and B cell tolerance and are immune competent [51]. Both clinical and immunological responses were achieved in the preclinical study with induction of Th cells and CTLs against MUC1 and other undefined tumor antigens presented by the MC38 colon cancer cells.
Materials and Methods
Tumor Generation
MC38 colon cancer cells transfected with full length human MUC1 (MC38.MUC1) or empty vector containing the neomycin resistance gene (MC38.neo) were maintained in DMEM medium supplemented with 5% fetal calf serum (FCS), 100U of penicillin, 0.1μg of streptomycin, 2mM L-glutamine, and 150μg/ml G418. Cells were maintained in log phase at 37°C with 5% CO2. Eight to ten-week-old MUC1.Tg mice were injected subcutaneously in the left flank with 1 x 106 MC38 colon cancer cells in 100μl of PBS. For prophylactic experiments, MC38 cells were injected 7 days after the first immunization (Figure 1A). For therapeutic experiments, tumors were allowed to grow for 7 days prior to the first immunization (Figure 1B).
Figure 1.
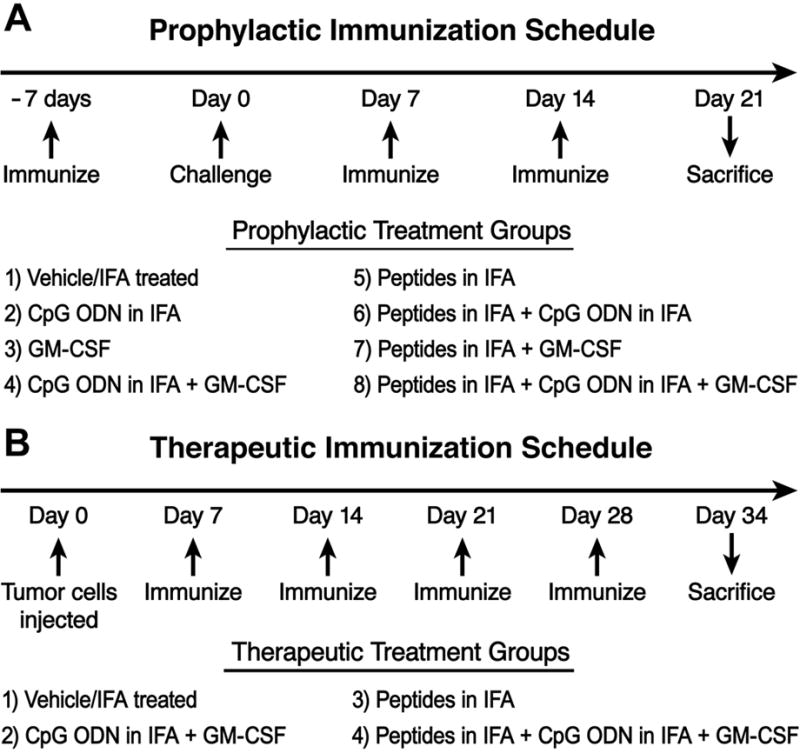
A) Schematic of the experimental design for prophylactic study, B) Schematic of experimental design for therapeutic study
Vaccination Strategy
The vaccine included: a) two MHC class I-restricted MUC1 peptides, APGSTAPPA and SAPDTRPAP (100μg); b) one MHC class II helper peptide TPPAYRPPNAPIL (140μg) (hepatitis B virus core antigen sequence 128–140 [52]); c) mouse unmethylated CpG oligodeoxynucleotide constructs (CpG ODN, 100μg) (Coley Pharmaceuticals, Canada); and d) granulocyte macrophage-colony stimulating factor (GM-CSF, 10,000 Units or 2μg, Cell Sciences, Canton, MA). All peptides and adjuvants were emulsified in IFA and administered subcutaneously at a site away from the tumor injection. Schematic representation of the treatment schedules for the prophylactic and therapeutic studies are shown in Figure 1.
Tumor Palpation
Mice were palpated every day starting at day 7 post-tumor cell injection until sacrifice. At time of sacrifice, tumors in the vehicle treated mice were approximately one gram in weight and were well vascularized. MC38.MUC1 tumors stained positive for human MUC1 and MC38.neo tumors were negative by immunohistochemistry (data not shown). Palpable tumors were measured by calipers and tumor weight was calculated according to the formula: grams = (length in cm) x (width) 2/2 [53]. In accordance with IACUC regulations, all surviving mice were sacrificed when tumors reached 10% of body weight. Mice were carefully observed for signs of ill health, including lethargy, abdominal distention, failure to eat or drink, marked weight loss and hunched posture.
IFNγ Elispot Assay
From the tumor draining lymph nodes (TDLNs), CD4+ and CD8+ T cells were isolated using the magnetic activated cell sorter (MACS, Miltenyi Biotech, Auburn, CA). IFN-γ ELISPOT assay was performed using capture IFN-γ antibody as recommended by the manufacturer (Mabtech, Stockholm, Sweden). Autologous irradiated DCs pulsed with the immunizing peptides (class I-restricted H2-Db binding APGSTAPPA + H2-Kb binding SAPDTRPAP + class II-restricted pan-helper-peptide TPPAYRPPNAPIL) were used as stimulator cells at a responder to stimulator ratio of 10:1. DCs were prepared according to the protocol published previously [10,54]. In some experiments, DCs were pulsed with either the MUC1 peptides or the helper peptide separately. Control wells contained T cells stimulated with DCs pulsed with irrelevant peptide (vesicular stomatitis virus peptide, RGYKYQGL). Spot numbers were determined using computer assisted video image analysis by Zellnet Consulting, Inc. (FortLee, NJ). DCs pulsed with irrelevant peptides were used as control. Splenocytes from a C57BL/6 mice stimulated with concavalin A (Con A) was used as positive control (data not shown).
51Chromium (Cr)-release assay
Determination of CTL activity was performed using a standard 51Cr-release method. Splenocytes, CD4+ and CD8+ T cells from TDLN served as effector cells. Autologous irradiated DCs pulsed with immunizing peptide were used as stimulator cells and co-incubated with the effector cells at an effector : stimulator ratio of 10:1 for 48h. Effector cells were then recovered and incubated with 51Cr-labeled tumor target cells at effector : target ratios of 100:1, 50:1 and 25:1. Target cells included MC38.MUC1, MC38.neo, B16.MUC1, and B16.neo cell lines. Epithelial tumor targets express low levels of MHC class I on the cell surface. Thus, target cells were treated with 5ng/ml IFN-γ (Amersham, Piscataway, NJ) one day prior to the assay to up-regulate MHC class I surface expression and loaded with radioactivity (100μCi 51Cr (Amersham) per 106 target cells) for 3h. Effectors and targets were co-incubated for 6h. Radioactive 51Cr released at the end of 6h was determined using the Topcount Micro-scintillation Counter (Packard Biosciences, Shelton, CT). Specific lysis was calculated according to the following formula: (experimental cpms- spontaneous cpms/complete cpms - spontaneous cpms) x 100. Spontaneous 51Cr release in all experiments was 10–15% of complete 51Cr release.
Statistical Analysis
Biostatisticians at the Mayo Clinic Biostatistics Core Facility conducted statistical analysis for all data. A two-factor ANOVA was used to generate significant differences between experimental groups. For the ELISPOT analysis, data were adjusted for operator (different days at which assays were conducted).
Results
Complete prevention of MC38.MUC1 tumor growth in MUC1.Tg mice immunized with the vaccine formulation
In a prophylactic setting, MC38.MUC1 or MC38.neo tumor cells (1 X 106 cells per mouse) were injected 7 days post first immunization. Two more immunizations were administered at days 7 and 14 post-tumor challenge and mice were sacrificed on day 21 (Figure 1A). We observed complete rejection of MC38.MUC1 tumors in MUC1.Tg mice receiving MUC1-specific immunization in combination with CpG ODN and GM-CSF (p < 0.001 compared to all other groups) (Figure 2A). Tumor burden for all experimental groups is represented as percent of tumor burden in vehicle-treated mice. All 8 mice in the peptide + adjuvant group remained tumor free. Mice in all other treatment groups developed tumor but with lower incidence and lower tumor weight compared to mice treated with vehicle (Figure 2A, p < 0.001 for peptides, peptides + CpG ODN and peptides + GM-CSF groups compared to vehicle; p < 0.04 for GM-CSF and CpG + GM-CSF groups compared to vehicle). Mice treated with CpG ODN alone was the only group that did not show lower tumor burden compared to vehicle-treated mice. Four of the eight mice in the full treatment group (peptide + GM-CSF + CpG ODN) were re-challenged with MC38.MUC1 tumor cells two months post primary vaccination. All 4 mice rejected the tumor cells, suggesting a strong memory response against MC38.MUC1 cells (data not shown).
Figure 2. A) Complete inhibition of MC38.MUC1 colon cancer tumor cells in a MUC1.Tg mice post peptide immunization.
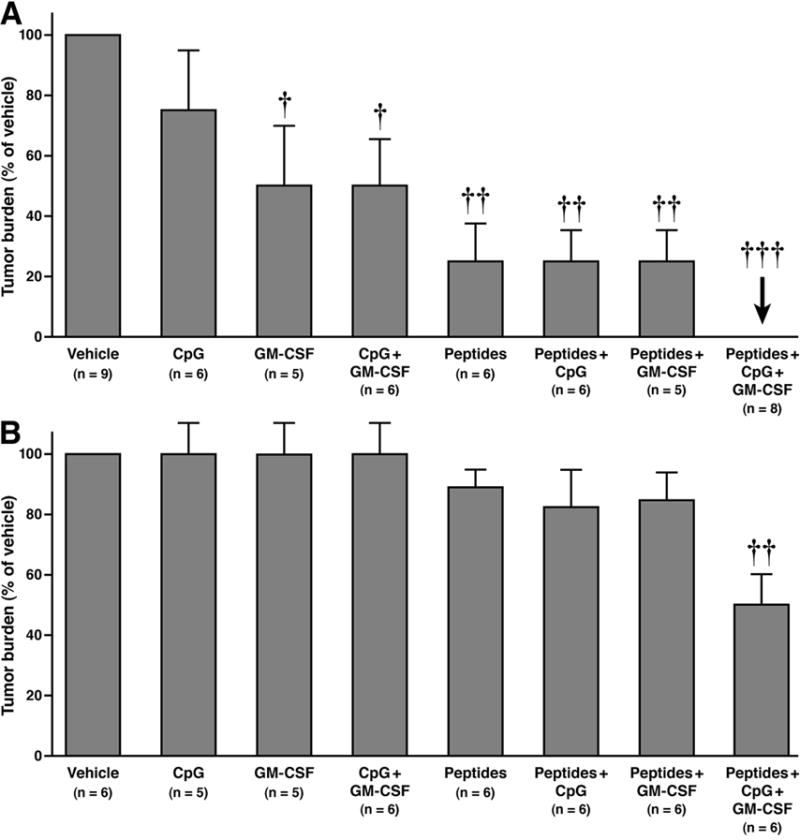
MUC1.Tg mice immunized with MUC1-based peptide vaccine in combination with pan-helper-peptide, GM-CSF and CpG ODN. Immunizations were given 1 week prior to tumor challenge with MC38.MUC1 cells followed by two boosts one week apart. Tumors were measured using digital calipers and final weight calculated as (L x W2)/2. Each point represents data from one mouse. The vehicle group was significantly different from the other groups (p < 0.001 compared to all other groups). Other significant differences were also determined (p < 0.001 for peptides, peptides + CpG ODN and peptides + GM-CSF groups compared to vehicle; p < 0.04 for GM-CSF and CpG + GM-CSF groups compared to vehicle). B) Reduced tumor burden of MC38.neo colon cancer tumor cells in a MUC1.Tg model post peptide immunization. MUC1.Tg mice immunized with MUC1-based peptide vaccine in combination with pan-helper-peptide, GM-CSF and CpG ODN. Immunizations were given 1 week prior to tumor challenge with MC38.MUC1 cells followed by two boosts one week apart. Tumors were measured using digital calipers and final weight calculated as (L x W2)/2. Each point represents data from one mouse. Immunization with peptides + CpG ODN + GM-CSF was successful in significantly but partially reducing MC38.neo tumor growth in MUC1.Tg mice (p < 0.01 compared to all other groups). †p < 0.04, ††P < 0.01, †††P < 0.001.
Partial effect of the vaccine on MC38.neo tumor growth in MUC1.Tg mice
Interestingly, the immunization strategy with peptides + CpG ODN + GM-CSF was also successful in partially reducing MC38.neo tumor growth in MUC1.Tg mice (Figure 2B, p < 0.01 compared to all other groups), presumably due to an immune response elicited by the pan-helper-peptide immunization, CpG ODN and GM-CSF. Mice in all other treatment groups had slightly lower tumor weights but were not significantly different from mice treated with vehicle (Figure 2B). Thus, the vaccine formulation not only elicited a MUC1-specific immune response but also an MC38 tumor-specific immune response.
In vivo stimulation of IFN-γ-producing CD4+ helper T cells and CD8+ cytotoxic T cells in MUC1.Tg mice immunized with the vaccine formulation
To elucidate the mechanism of anti-tumor response, CD4+ and CD8+ T cells were isolated from lymph nodes of immunized and non-immunized mice and analyzed for MUC1-specific IFN-γ spot formation (ELISPOT). CD4+ and CD8+ T cells were stimulated overnight with syngeneic DCs pulsed with the three immunizing peptides. In the MC38.MUC1 challenged mice, both IFN-γ-producing CD4+ and CD8+ T cells were observed (Figure 3A and B). An irrelevant peptide, vesicular stomatitis virus peptide (RGYKYQGL, VSV NP52–59) showed numbers of spots similar to vehicle (data not shown). Compared to CD4+ T cells, we observed lower numbers of IFN-γ spot-forming CD8+ T cells (Figure 3A and B, note the difference in scale for the y-axis). A strong CD4+ helper T cell response was elicited, possibly due to the presence of the pan-helper-peptide and CpG ODN, both known to target the CD4+ helper T cell population [33,55–57]. A significant increase in IFN-γ-producing CD4+ T cells was noted in peptide + CpG ODN and peptide + CpG ODN + GM CSF groups compared to the vehicle group (p < 0.05 for peptides + CpG ODN, and p < 0.001 for peptides + CpG ODN + GM CSF). The only group that showed significant increase in IFN-γ-producing CD8+ T cells were mice treated with peptides + both adjuvants compared to mice treated with vehicle and all other groups including the adjuvants alone group (p < 0.05). We therefore selected to confirm the ELISPOT data with an in vitro CTL assay only from the two groups (vehicle versus peptides + adjuvants). Tumor targets were MC38.MUC1 and MC38.neo tumor cells and effectors were either splenic T cells, or sorted CD4+ or CD8+ T cells from TDLNs. Compared to vehicle treated mice, splenic T cells from mice immunized with peptides + adjuvants showed the highest CTL activity against MC38.MUC1 targets (data not shown). Moderate levels of CTL activity were also observed with MC38.neo target cells, suggesting that the immunization elicited CTLs not only against MUC1 tumor antigen but also against other unknown MC38 tumor antigens (data not shown). This result substantiates the moderate decrease in tumor burden observed with the immunization in the MC38.neo challenged mice (Figure 2B).
Figure 3. Induction of IFN-γ production by CD4+ and CD8+ T cells in response to MUC1 and pan-helper-peptides, which was enhanced by GM-CSF and CpG ODN in MC38.MUC1 challenged mice.
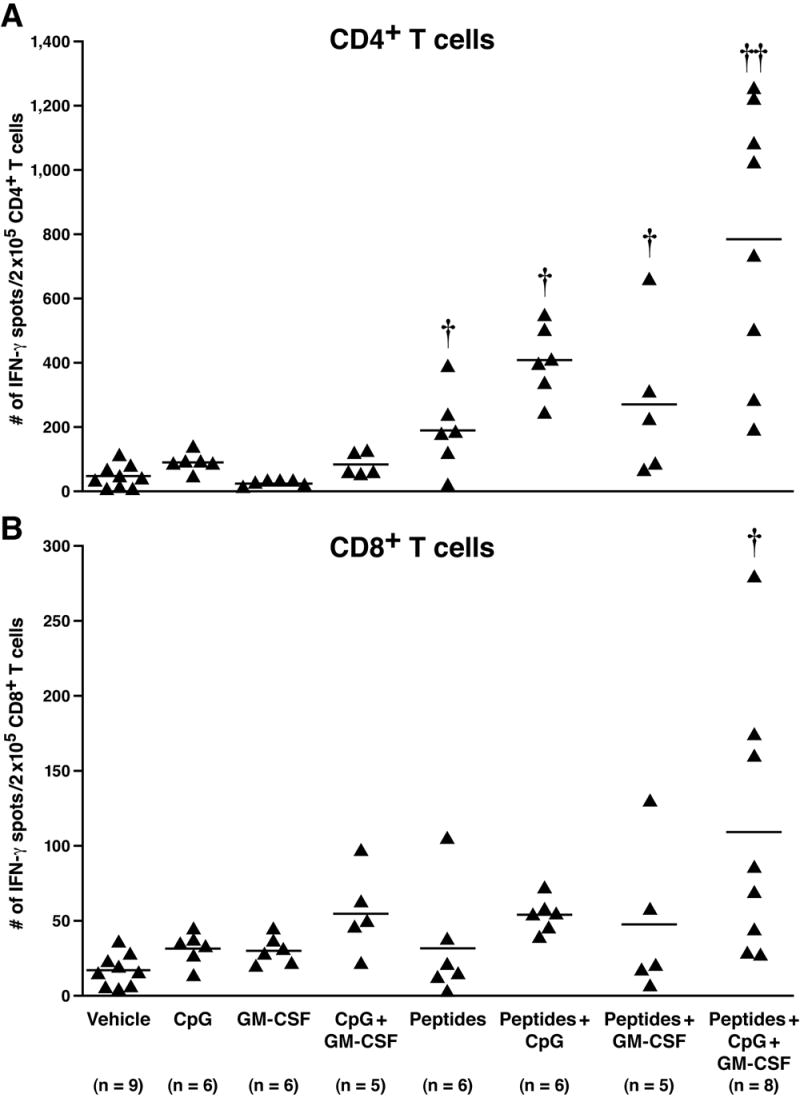
At sacrifice, CD4+ and CD8+ T cells were sorted from lymph nodes. Cells were stimulated for 24h with irradiated DCs pulsed with the immunizing peptides on an ELISPOT plate. Each point represents data from one mouse. A) Number of IFN-γ spots in CD4+ T cells: a significant increase was noted in peptides + CpG ODN and peptides + CpG ODN + GM CSF groups compared to the vehicle group (p < 0.05 for peptides + CpG ODN, and p < 0.001 for peptides + CpG ODN + GM CSF). B) Number of IFN-γ spots in CD8+ T cells: the only group that showed significant increase in IFN-γ producing CD8+ T cells were mice treated with peptides + adjuvants compared to mice treated with vehicle (P < 0.05). †† p < 0.001, † p < 0.05 compared to vehicle treatment. Note that the y-axes differ in A and B.
In the MC38.neo challenged mice, immunization stimulated the CD4+ T cells to produce IFN-γ in all groups compared to vehicle. However, significantly higher spot formation was detected in the groups treated with peptides + CpG ODN and peptides + CpG ODN + GM CSF (p < 0.05 and p < 0.01, respectively). These groups were also significantly different from adjuvants alone or peptide alone groups suggesting the important role of pan-helper-peptide and CpG ODN. GM-CSF by itself did not have much effect. The CD8+ T cells showed only modest increase in spot formation with no statistical significance between groups (Figure 4A and B). The modest increase is once again due to the adjuvants (particularly CpG ODN), since groups that received CpG ODN in the vaccine formulation showed increased spot formation. The data suggest that the pan-helper-peptide + CpG ODN can elicit a reasonable immune response against MC38 tumor cells and mount a partial anti-tumor response regardless of the presence of MUC1 antigen. Furthermore, the data imply that both adjuvants administered with the peptide vaccination may be better than one adjuvant alone in eliciting a robust anti-tumor response (Figure 2). It should be noted that in Figure 4, the mice were challenged with MC38.neo cells and not MC38.MUC1 cells; therefore there is no MUC1-specific response but the response is due to the adjuvants and helper peptide. If you compare Figure 4 with Figure 3 (in which the mice were challenged with MC38.MUC1 cells), there is a significant increase in the CD8+ T cell response with peptides + adjuvants versus all other groups, suggesting that the response is clearly MUC1 peptide-specific.
Figure 4. Induction of IFN-γ production by CD4+ but not CD8+ T cells in response to MUC1 and pan-helper-peptides, was enhanced by GM-CSF and CpG ODN in MC38.neo challenged mice.
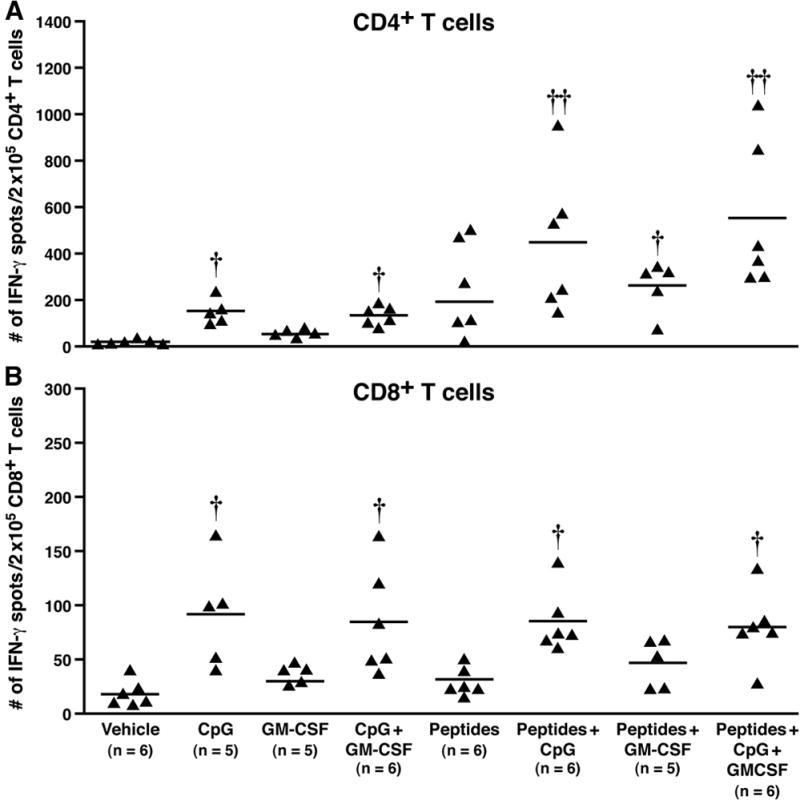
At sacrifice, CD4+ and CD8+ T cells were sorted from lymph nodes. Cells were stimulated for 24h with irradiated DCs pulsed with the immunizing peptides on an ELISPOT plate. Each point represents data from one mouse. A) Number of IFN-γ spots in CD4+ T cells: significantly higher spot formation was detected in the groups treated with peptides + CpG ODN and peptides + CpG ODN + GM CSF (p < 0.05 and p < 0.01, respectively). These groups were also significantly different from adjuvants alone or peptide alone groups (p < 0.05). B) Number of IFN-γ spots in CD8+ T cells: a modest increase in spot formation was noted with no statistical significance between groups. †† p<0.01, † p<0.05 compared to vehicle treatment. Note that the y-axis differs in A and B.
Significant reduction in established MC38.MUC1 tumors in MUC1.Tg mice immunized with the vaccine formulation
To test the efficacy of the vaccine formulation in a therapeutic setting, MC38.MUC1 tumor cells (1 x 106 cells/mouse) were established in MUC1.Tg mice for 7 days. Tumors were well-vascularized and palpable by this time. Immunization was administered on days 7, 14, 21, and 28 post-tumor injection and mice were sacrificed on day 34 (Figure 1B). We opted to conduct the therapeutic part of the study using MC38.MUC1 tumor cells with 4 groups of mice. The experimental groups included: a) peptides + CpG ODN + GM-CSF, b) peptides, c) CpG ODN + GM-CSF, and d) vehicle. A significant reduction in tumor growth was observed in the group treated with peptides + adjuvants compared to mice treated with vehicle (Figure 5, p = 0.003). Although tumor burden in the peptide alone group was not significantly different from the control (p=0.12), the adjuvants alone group (GM-CSF + CpG ODN) showed a significant reduction in tumor burden as compared to vehicle (p=0.011), suggesting that the adjuvants were strong enough to cause a reduction in tumor burden even without the tumor associated antigen (TAA)-specific peptides. The MUC1 peptide vaccine did not significantly affect the growth of the established MC38.neo tumors compared to peptides alone or adjuvants (data not shown).
Figure 5. MUC1 peptide vaccine significantly reduced burden of established MC38.MUC1 tumors in MUC1.Tg mice.
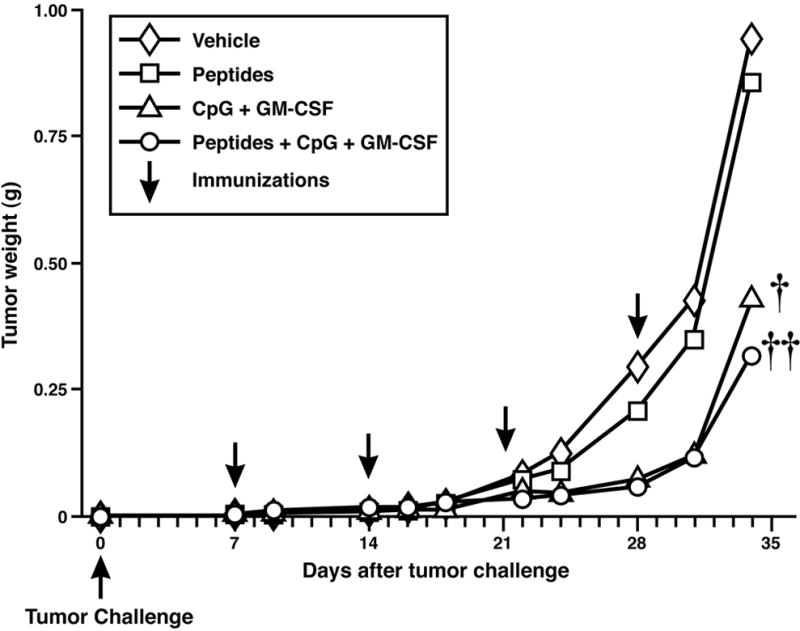
Tumors were established for 7 days by subcutaneous injection of MC38.MUC1 cells (1 x 106) into the flanks of MUC1.Tg mice. Tumor-bearing mice were immunized weekly with MUC1-based peptide vaccine in combination with pan-helper-peptide, CpG ODN and GM-CSF. Tumors were measured (cm) and final weight (gm) calculated as (L x W2)/2. A significant reduction in tumor growth was observed in the vaccine treatment compared to vehicle control or peptide alone (††p = 0.003). The adjuvants alone (CpG ODN + GM-CSF) also showed a significant reduction in tumor burden compared to vehicle or peptides only (†p = 0.01).
Immunization elicited a strong IFN-γ response in CD4+ T cells isolated from mice bearing established MC38.MUC1 tumors
At sacrifice, CD4+ and CD8+ T cells were sorted from TDLNs. Cells were stimulated for 24 hours with irradiated DCs pulsed with the immunizing peptides on an ELISPOT plate. To delineate the effect of the MUC1 peptide from the pan-helper-peptide, the DCs were pulsed with either the MUC1 peptides, or the pan helper peptide or both. Compared to the vehicle-treated group, CD4+ T cells from mice treated with adjuvants, peptides, or peptides + adjuvants showed significantly increased IFN-γ spot formation (Figure 6A, p < 0.02 for adjuvants, p < 0.001 for peptides and p < 0.001 for peptides + adjuvants). Significant differences were also achieved between adjuvants and peptides groups (p < 0.001 except for the DCs pulsed with MUC1 peptide arm) and between adjuvants and peptides + adjuvant group (p < 0.001). Similarly a significant difference was noted between peptides and peptides + adjuvants groups in all three DC arms (p < 0.001). Thus, an additive effect of adjuvants + peptides is detected in the IFN-γ-producing CD4+ T cell parameter with our immunization strategy in a therapeutic setting. As can be appreciated from Figure 6A, IFN-γ production in the CD4+ T cells was stimulated primarily by the pan-helper-peptide since there was no difference in spot formation between DCs stimulated with pan-helper-peptide alone or pan-helper-peptide + MUC1 peptide in any of the experimental groups. DCs pulsed with MUC1 peptide alone showed significantly lower spot formation than DCs pulsed with pan-helper-peptide in mice treated with peptides or peptides + adjuvants (p < 0.001), once again confirming that the pan-helper-peptide is the main stimulator of IFN-γ production by CD4+ T cells. CD8+ T cells showed modest albeit significant increase in IFN-γ production in the adjuvants group as well as in the peptides + adjuvants group compared to the vehicle regardless of the peptides used to pulse the DCs (Figure 6B, p = 0.02). The peptides group did not show significant difference in IFN-γ-spot formation compared to vehicle and like the vehicle group, showed significantly lower spot formation than the other two groups (adjuvants alone or peptides + adjuvants, p = 0.02) except for the DCs pulsed with pan-helper-peptide alone arm (p = 0.6, Figure 6B), suggesting that the CD8+ T cells were less influenced by the pan-helper-peptides compared to the CD4+ T cells. Thus, the immunization strategy stimulated a strong CD4+ IFN-γ-producing T cell response primarily to the pan-helper-peptide.
Figure 6. IFN-γ production by CD4+ T cells was induced in response to pan-helper-peptides, which was enhanced by CpG ODN and GM-CSF.
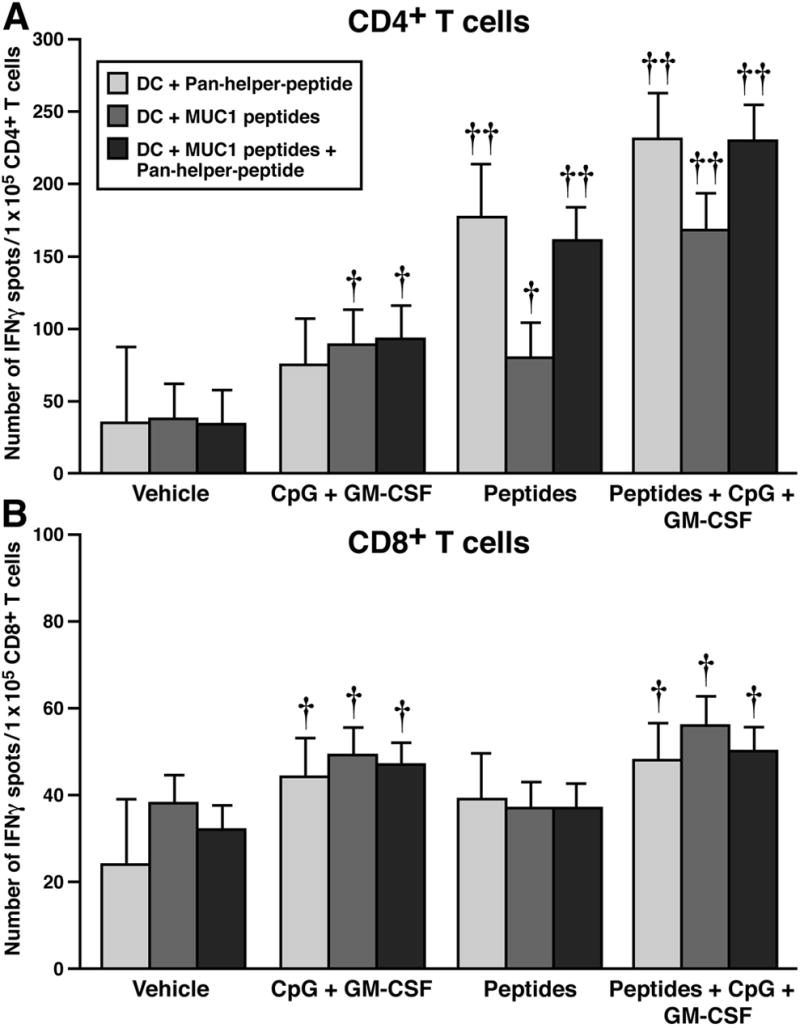
At sacrifice, CD4+ and CD8+ T cells were sorted from TDLN. Cells were stimulated for 24 hours with irradiated DCs pulsed with the immunizing peptides on an ELISPOT plate. A) Number of IFN-γ spots from CD4+ T cells: Compared to vehicle treated, mice treated with adjuvants, peptides, or peptides + adjuvants showed significantly increased IFN-γ spot formation (p < 0.02 for adjuvants, p < 0.001 for peptides and p < 0.001 for peptides + adjuvants). Significant differences were also achieved between adjuvants and peptides group (p < 0.001 except for the DCs pulsed with the MUC1 peptide arms) and between adjuvants and peptides + adjuvant groups (p < 0.001). Significant differences were noted between peptides and peptides + adjuvants group in all three DC arm (p < 0.001). B) Number of IFN-γ spots from CD8+ T cells: There was a significant increase in IFN-γ production in the adjuvants group as well as in the peptides + adjuvants group compared to the vehicle regardless of the peptides used to pulse the DCs (p =0.02). The peptides group did not show significant difference in IFN-γ-spot formation compared to vehicle but showed significantly lower spot formation than the other two groups (adjuvants alone or peptides + adjuvants, p = 0.02) except for the DCs pulsed with pan-helper-peptide alone (p = 0.6). †p ≤ 0.02, ††p < 0.001 compared to vehicle. Note that y-axis differs in A and B.
Immunization elicited CTLs against MC38 and MUC1 tumor antigens in mice bearing established MC38.MUC1 tumors
Although the CD8+ T cells did not produce much IFN-γ in the ELISPOT assay, we determined if splenic T cells from immunized mice showed lytic activity against the tumor, since we did observe a significant decrease in the tumor burden with immunization (Figure 5). To further establish if the CTLs were specific against MUC1 and/or the tumor cell itself, we utilized two different tumor cell targets, one specific for the immunized mice, MC38 colon cancer cells, and the other, an irrelevant melanoma cell line, namely, B16. Both cell lines expressed either full-length MUC1 or empty vector (neo). The results are summarized in Figure 7. When B16 cells were utilized as the target, the CTLs were lytic only against the cells that expressed MUC1 but not against B16.neo (p < 0.001). Note that there is no cytolysis from mice immunized with vehicle or adjuvants alone when B16 was used as the target. Lytic activity was only observed when mice were immunized with peptides alone (p < 0.001 compared to vehicle) or peptides + adjuvants (p < 0.001 compared to vehicle) confirming that the immunization elicited CTLs specifically against the MUC1 antigen and not against a non-specific tumor cell. In contrast, when MC38 cells were used as targets, the lytic activity was not only against the MC38.MUC1 but also against MC38.neo cells (p < 0.001 for all experimental groups compared to the vehicle group). Another important data that emerged from this experiment was that mice treated with adjuvants alone elicited a cytolytic response against the MC38 tumors regardless of whether MUC1 was expressed or not. These results explain clearly the lower tumor burden in the adjuvants group as well as begin to explain the anti-tumor effect on MC38.neo tumors (Figure 2). The peptides alone group generated CTLs only against the MC38.MUC1 but not against MC38.neo (p < 0.001 between MC38.MUC1 and MC38.neo targets). Mice treated with vehicle showed no lytic activity against any tumor target.
Figure 7. Immunization with MUC1-peptide vaccine increased MUC1-specific CTL activity in mice with established MC38.MUC1 tumors.
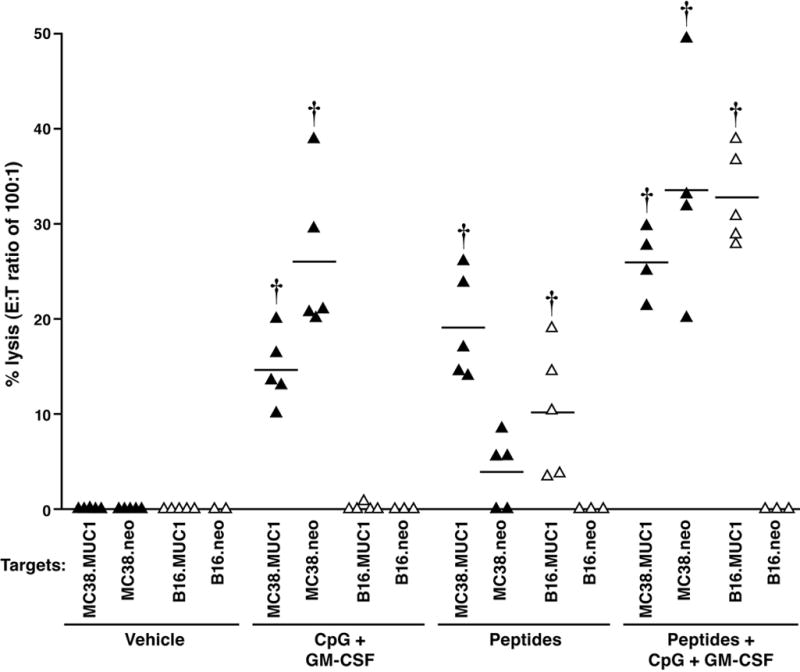
Effector cells (splenocytes) from immunized mice were subjected to a standard 51Cr-release assay. Targets were MC38.MUC1, MC38.neo, B16.MUC1, and B16.neo cells. N = 4 to 6 mice per group. MC38 targets are designated with ▴ and B16 targets with Δ. CTLs were lytic against the B16.MUC1 cells but not against B16.neo (p < 0.001). Note that there is no cytolysis from mice immunized with vehicle or adjuvants alone when B16 was used as the target. Lytic activity was only observed when mice were immunized with peptide alone (p < 0.001) or peptides + adjuvants (p < 0.001). With MC38 tumor targets, lytic activity was against MC38.MUC1 and MC38.neo cells (p < 0.001 for all experimental groups compared to the vehicle group). The peptides alone group generated CTLs only against the MC38.MUC1 but not against MC38.neo cells (p < 0.001). Mice treated with vehicle showed no lytic activity against any tumor target. ††P < 0.001 compared to vehicle. E : T ratios of 100:1, 50:1 and 25:1 were conducted and titrations were observed. Due to multiple targets and multiple animals per experimental group, we selected to show E : T ratio of 100:1.
Discussion
Although MUC1-based immunotherapy has been able to induce MUC1-specific CTL responses in a substantial number of cancer patients, the response has not been long lasting, and has mostly remained without a significant clinical response. We hypothesize that this may be because less focus has been given to the generation of Th cells that are required to sustain a long-lasting anti-tumor response. We have developed a MUC1-based vaccine strategy that generates a robust CD4+ Th cell response with sustained antigen-specific CD8+ CTL response that clearly translated to a successful anti-tumor response. The vaccine consisted of peptides derived from MHC class I-restricted MUC1 epitopes and MHC class II restricted pan-helper T cell epitope. CpG ODN and GM-CSF were used as adjuvants. The vaccine was tested both in a prophylactic and a therapeutic setting in an appropriate mouse model of MUC1 tolerance. The MUC1.Tg mice were challenged with MC38 colon cancer cell lines transfected with full-length human MUC1 (MC38.MUC1) or empty vector containing the neomycin resistance gene (MC38.neo). In the prophylactic setting, the vaccine specifically caused rejection of the MC38.MUC1 tumors but not the MC38.neo tumors, although a 50% reduction in tumor burden was observed in these tumors (Figure 2). This apparently was due to the robust CD4+ Th cell response elicited by the pan-helper-peptide and the adjuvants, CpG ODN and GM-CSF, present in the vaccine formulation (Figure 4). This Th response facilitated the generation of a moderate CD8+ CTL response accompanied by a modest anti-tumor response. In contrast, both the CD4+ Th and CD8+ CTL responses were highly significant in the immunized mice challenged with the MC38.MUC1 tumor cells compared to mice treated with vehicle (Figure 3). This immune response translated to a significant anti-tumor response (Figure 2). Even more important is that the vaccination strategy generated a strong memory response against the MC38.MUC1 tumor cells, such that mice that were tumor free after primary immunization remained tumor free for months after re-challenge with the same MC38.MUC1 tumor cells (data not shown). Since the peptide vaccine was utilized in the study, we monitored the humoral response by testing for MUC1 antibody levels in the serum of immunized mice using a specific ELISA. The levels of MUC1 antibody were found to be below the detectable levels (data not shown). Similarly, we tested for Th1 versus Th2 cytokines in the T cell supernatant collected from the ELISPOT plates at 24h post stimulation with DCs pulsed with the immunizing peptide. Cytokine bead array (CBA) analysis revealed an increase in IFN-γ and TNF-α levels, although IL-4, IL-5 and IL-10 were undetectable (data not shown). Together, these data suggest a predominantly Th1 type cellular immune response with minimal Th2 response. This is not surprising since both CpG ODN and GM-CSF are known to promote a Th1-type response [48,49,55].
In the therapeutic setting, the vaccination was effective in significantly reducing the tumor burden compared to the vehicle or peptides alone treated groups (Figure 5). To our surprise there was no significant difference between the adjuvant and the adjuvant + peptides group (Figure 5), suggesting that the adjuvants alone were potent enough to cause a clinically significant anti-tumor response. This was most prominent in the therapeutic setting and was much less obvious in the prophylactic setting (compare Figure 2A with Figure 5). In fact, in the prophylactic setting the peptides alone group had a much more significant anti-tumor response than the adjuvants alone group. These data raise several questions regarding the differences in immune status of tumor-bearing mice versus non-tumor bearing mice and how the two respond to the same vaccination. Tumor-bearing mice (in this case the mice in the therapeutic arm with 7-day established tumor) may have existing low-level anti-MUC1 or anti-MC38 responses, which were accentuated with the addition of the adjuvants. In the prophylactic setting the immunization was given prior to tumor challenge and therefore the response to the peptides was stronger than to the adjuvants alone. Thus, the global immune status of the host receiving the vaccination impacts greatly on the host’s ability to generate CTL immune responses to the peptide vaccines. It is clear from the therapeutic study that CTLs were generated not only against MUC1 but also against other tumor antigens presented by the MC38 colon cancer cells (Figure 7). This represents an important mechanism by which the immune system can efficiently eliminate malignant cells and suggests the induction of epitope spreading.
Although patients with advanced colorectal cancer have thus far derived no substantial clinical benefit from peptide vaccination, we do not know enough regarding the global immune status of the patients to abandon our efforts in this direction. We agree with those who find it premature to give up on active cancer vaccines, because much work remains [58–61]. For example, we must gain a better understanding of T cell functions, such as T cell avidity for tumor cells, T cell homing to the tumor site, durability of the T cell response, and activation of more than one effector mechanism.
To address the limitation of previous peptide-based therapeutic antitumor vaccine strategies and low clinical efficacy in the clinic, scientists have used several approaches to improve vaccine potency. These include selection of more immunogenic peptides, modifying naturally occurring peptides by increasing affinity to MHC class I molecules, combining peptides derived from multiple tumor associated antigens, combining MHC class I and class II binding peptides from the same antigen, and by using novel immune adjuvants. We have successfully combined the last two strategies and as a result increased the Th cell response and therefore the CTL response in our model system.
MUC1 occurs naturally as a heavily glycosylated protein which contains two well-known tumor-associated carbohydrate antigens (TACA): the disaccharide Thomsen-Friedenreich (TF) antigen (β-Gal-(1->3)-α-GalNAc-O-serine/threonine and its precursor, the monosaccharide Tn (GalNAc-O-serine/threonine) [62,63]. TACA-containing glycopeptides are appealing CTL-based vaccines as they are widely expressed in a variety of tumors including colon cancer, their expression is largely tumor specific, they are induced early in neoplastic transformation, and they elicit a high affinity, degenerate carbohydrate-specific response [64]. Future studies will focus on generating glycosylated and/or anchor-improved MUC1 peptides that will have higher MHC binding and thus greater ability to activate T cells [65]. The idea that the glycopeptides from highly glycosylated proteins can serve as CTL-based vaccines has been pioneered by Olivera Finn [66], Vasso Apostolopoulos [67] and Alexandra Franco [52,64] and colleagues. Eliciting immunity to the novel anchor-modified glycosylated MUC1 peptides may result in robust anti-tumor immunity and long-term immune memory.
Acknowledgments
This work was supported by the SPORE in Gastrointestinal Cancers NIH P50 CA95060 and by the Mayo Foundation. We would like to thank Drs. D. Kufe and J. Schlom for providing the MC38.MUC1 and MC38.neo cell lines. We also thank Marvin Ruona in the Visual Communications core for preparing the figures in the correct format, Irene Beauvais for her assistance in submitting the manuscript, Dr. Joe Hentz and Noble Brie in the Biostatistics core for performing the statistical analysis and all technicians in the animal core facility for help in maintaining our mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zotter S, Hageman PC, Lossnitzer A, Mooi WJ, Hilgers J. Tissue and tumor distribution of human polymorphic epithelial mucin. Cancer Reviews. 1988;11–12:55–101. [Google Scholar]
- 2.Croce MV, Isla-Larrain MT, Rua CE, Rabassa ME, Gendler SJ, Segal-Eiras A. Patterns of MUC1 tissue expression defined by an anti-MUC1 cytoplasmic tail monoclonal antibody in breast cancer. J Histochem Cytochem. 2003;51(6):781–788. doi: 10.1177/002215540305100609. [DOI] [PubMed] [Google Scholar]
- 3.Girling A, Bartkova J, Burchell J, Gendler S, Gillet C, Taylor-Papadimitriou J. A core protein epitope of the polymorphic epithelial mucin detected by the monoclonal antibody SM-3 is selectively exposed in a range of primary carcinomas. Int J Cancer. 1989;43:1072–1076. doi: 10.1002/ijc.2910430620. [DOI] [PubMed] [Google Scholar]
- 4.Domenech N, Henderson RA, Finn OJ. Identification of an HLA-A11-restricted epitope from the tandem repeat domain of the epithelial tumor antigen mucin. J Immunol. 1995;155(10):4766–4774. [PubMed] [Google Scholar]
- 5.Agrawal B, Reddish MA, Longenecker BM. In vitro induction of MUC-1 peptide-specific type 1 T lymphocyte and cytotoxic T lymphocyte responses from healthy multiparous donors. J Immunol. 1996;157(5):2089–2095. [PubMed] [Google Scholar]
- 6.Apostolopoulos V, Haurum JS, McKenzie IFC. Muc1 Peptide Epitopes Associated With Five Different H-2 Class I Molecules. Eur J Immunol. 1997;27(10):2579–2587. doi: 10.1002/eji.1830271017. [DOI] [PubMed] [Google Scholar]
- 7.Apostolopoulos V, Karanikas V, Haurum JS, McKenzie IF. Induction of HLA-A2-restricted CTLs to the mucin 1 human breast cancer antigen. J Immunol. 1997;159(11):5211–5218. [PubMed] [Google Scholar]
- 8.Reddish M, MacLean GD, Koganty RR, et al. Anti-MUC1 class I restricted CTLs in metastatic breast cancer patients immunized with a synthetic MUC1 peptide. Int J Cancer. 1998;76(6):817–823. doi: 10.1002/(sici)1097-0215(19980610)76:6<817::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Mukherjee P, Ginardi AR, Madsen CS, et al. Mice with spontaneous pancreatic cancer naturally develop MUC1-specific CTLs that eradicate tumors when adoptively transferred. J Immunol. 2000;165(6):3451–3460. doi: 10.4049/jimmunol.165.6.3451. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee P, Madsen CS, Ginardi AR, et al. Mucin 1-specific immunotherapy in a mouse model of spontaneous breast cancer. J Immunother. 2003;26(1):47–62. doi: 10.1097/00002371-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee P, Ginardi AR, Tinder TL, Sterner CJ, Gendler SJ. MUC1-specific CTLs eradicate tumors when adoptively transferred in vivo. Clin Can Res. 2001;7:848s–855s. [PubMed] [Google Scholar]
- 12.North SA, Graham K, Bodnar D, Venner P. A pilot study of the liposomal MUC1 vaccine BLP25 in prostate specific antigen failures after radical prostatectomy. J Urol. 2006;176(1):91–95. doi: 10.1016/S0022-5347(06)00494-0. [DOI] [PubMed] [Google Scholar]
- 13.Kohlgraf KG, Gawron AJ, Higashi M, et al. Contribution of the MUC1 tandem repeat and cytoplasmic tail to invasive and metastatic properties of a pancreatic cancer cell line. Cancer Res. 2003;63(16):5011–5020. [PubMed] [Google Scholar]
- 14.Ramanathan RK, Lee KM, McKolanis J, et al. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immunother. 2005;54(3):254–264. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treon SP, Mollick JA, Urashima M, et al. MUC1 core protein is expressed on multiple myeloma cells and is induced by dexamethasone. Blood. 1999;93(4):1287–1298. [PubMed] [Google Scholar]
- 16.Brossart P, Schneider A, Dill P, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001;61(18):6846–6850. [PubMed] [Google Scholar]
- 17.Loveland BE, Zhao A, White S, et al. Mannan-MUC1-pulsed dendritic cell immunotherapy: a phase I trial in patients with adenocarcinoma. Clin Cancer Res. 2006;12(3):869–877. doi: 10.1158/1078-0432.CCR-05-1574. Pt 1. [DOI] [PubMed] [Google Scholar]
- 18.Apostolopoulos V, Pietersz GA, Tsibanis A, et al. Pilot phase III immunotherapy study in early-stage breast cancer patients using oxidized mannan-MUC1 [ISRCTN71711835] Breast Cancer Res. 2006;8(3):R27. doi: 10.1186/bcr1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pantuck AJ, van Ophoven A, Gitlitz BJ, et al. Phase I trial of antigen-specific gene therapy using a recombinant vaccinia virus encoding MUC-1 and IL-2 in MUC-1-positive patients with advanced prostate cancer. J Immunother. 2004;27(3):240–253. doi: 10.1097/00002371-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 20.North S, Butts C. Vaccination with BLP25 liposome vaccine to treat non-small cell lung and prostate cancers. Expert Rev Vaccines. 2005;4(3):249–257. doi: 10.1586/14760584.4.3.249. [DOI] [PubMed] [Google Scholar]
- 21.Pecher G, Haring A, Kaiser L, Thiel E. Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother. 2002;51(11–12):669–673. doi: 10.1007/s00262-002-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kontani K, Taguchi O, Ozaki Y, et al. Dendritic cell vaccine immunotherapy of cancer targeting MUC1 mucin. Int J Mol Med. 2003;12(4):493–502. [PubMed] [Google Scholar]
- 23.Scholl S, Squiban P, Bizouarne N, et al. Metastatic Breast Tumour Regression Following Treatment by a Gene-Modified Vaccinia Virus Expressing MUC1 and IL-2. J Biomed Biotechnol. 2003;2003(3):194–201. doi: 10.1155/S111072430320704X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder LA, Goletz TJ, Gunn GR, et al. A MUC1/IL-18 DNA vaccine induces anti-tumor immunity and increased survival in MUC1 transgenic mice. Vaccine. 2006;24(16):3340–3352. doi: 10.1016/j.vaccine.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 25.Brossart P, Heinrich KS, Stuhler G, et al. Identification of HLA-A2-restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood. 1999;93(12):4309–4317. [PubMed] [Google Scholar]
- 26.Heukamp LC, van der Burg SH, Drijfhout JW, Melief CJ, Taylor-Papadimitriou J, Offringa R. Identification of three non-VNTR MUC1-derived HLA-A*0201-restricted T-cell epitopes that induce protective anti-tumor immunity in HLA-A2/K(b)-transgenic mice. Int J Cancer. 2001;91(3):385–392. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1051>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 27.Tsang KY, Palena C, Gulley J, Arlen P, Schlom J. A human cytotoxic T-lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC-1. Clin Cancer Res. 2004;10(6):2139–2149. doi: 10.1158/1078-0432.ccr-1011-03. [DOI] [PubMed] [Google Scholar]
- 28.Wierecky J, Mueller M, Brossart P. Dendritic cell-based cancer immunotherapy targeting MUC-1. Cancer Immunol Immunother. 2006;55(1):63–67. doi: 10.1007/s00262-005-0673-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wierecky J, Muller MR, Wirths S, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66(11):5910–5918. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 30.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374(6522):546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 32.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 33.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5(6):471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 34.Ballas ZK, Krieg AM, Warren T, et al. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J Immunol. 2001;167(9):4878–4886. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]
- 35.Heckelsmiller K, Rall K, Beck S, et al. Peritumoral CpG DNA elicits a coordinated response of CD8 T cells and innate effectors to cure established tumors in a murine colon carcinoma model. J Immunol. 2002;169(7):3892–3899. doi: 10.4049/jimmunol.169.7.3892. [DOI] [PubMed] [Google Scholar]
- 36.Milas L, Mason KA, Ariga H, et al. CpG oligodeoxynucleotide enhances tumor response to radiation. Cancer Res. 2004;64(15):5074–5077. doi: 10.1158/0008-5472.CAN-04-0926. [DOI] [PubMed] [Google Scholar]
- 37.Brunner C, Seiderer J, Schlamp A, et al. Enhanced dendritic cell maturation by TNF-alpha or cytidine-phosphate-guanosine DNA drives T cell activation in vitro and therapeutic anti-tumor immune responses in vivo. J Immunol. 2000;165(11):6278–6286. doi: 10.4049/jimmunol.165.11.6278. [DOI] [PubMed] [Google Scholar]
- 38.Schneeberger A, Wagner C, Zemann A, et al. CpG motifs are efficient adjuvants for DNA cancer vaccines. J Invest Dermatol. 2004;123(2):371–379. doi: 10.1111/j.0022-202X.2004.23208.x. [DOI] [PubMed] [Google Scholar]
- 39.Bourquin C, Schreiber S, Beck S, Hartmann G, Endres S. Immunotherapy with dendritic cells and CpG oligonucleotides can be combined with chemotherapy without loss of efficacy in a mouse model of colon cancer. Int J Cancer. 2006;118(11):2790–2795. doi: 10.1002/ijc.21681. [DOI] [PubMed] [Google Scholar]
- 40.Krieg AM. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr Oncol Rep. 2004;6(2):88–95. doi: 10.1007/s11912-004-0019-0. [DOI] [PubMed] [Google Scholar]
- 41.Speiser DE, Lienard D, Rufer N, et al. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005;115(3):739–746. doi: 10.1172/JCI23373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones T, Stern A, Lin R. Potential role of granulocyte-macrophage colony-stimulating factor as vaccine adjuvant. Eur J Clin Microbiol Infect Dis. 1994;13:S47–53. doi: 10.1007/BF01973602. Suppl 2. [DOI] [PubMed] [Google Scholar]
- 43.Jager E, Ringhoffer M, Dienes HP, et al. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int J Cancer. 1996;67(1):54–62. doi: 10.1002/(SICI)1097-0215(19960703)67:1<54::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 44.Fagerberg J. Granulocyte-macrophage colony-stimulating factor as an adjuvant in tumor immunotherapy. Med Oncol. 1996;13(3):155–160. [PubMed] [Google Scholar]
- 45.Carlsson T, Struve J. Granulocyte-macrophage colony-stimulating factor given as an adjuvant to persons not responding to hepatitis B vaccine [letter] Infection. 1997;25(2):129. doi: 10.1007/BF02113594. [DOI] [PubMed] [Google Scholar]
- 46.Pardoll DM. Paracrine cytokine adjuvants in cancer immunotherapy. Annu Rev Immunol. 1995;13:399–415. doi: 10.1146/annurev.iy.13.040195.002151. [DOI] [PubMed] [Google Scholar]
- 47.Golumbek PT, Azhari R, Jaffee EM, et al. Controlled release, biodegradable cytokine depots: a new approach in cancer vaccine design. Cancer Res. 1993;53(24):5841–5844. [PubMed] [Google Scholar]
- 48.Markovic SN, Suman VJ, Ingle JN, et al. Peptide vaccination of patients with metastatic melanoma: improved clinical outcome in patients demonstrating effective immunization. Am J Clin Oncol. 2006;29(4):352–360. doi: 10.1097/01.coc.0000217877.78473.a4. [DOI] [PubMed] [Google Scholar]
- 49.Disis ML, Bernhard H, Shiota FM, et al. Granulocyte-macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood. 1996;88(1):202–210. [PubMed] [Google Scholar]
- 50.Kaplan C, Morel-Kopp MC, Verdy E, Pron B, Tchernia G. Fetal and neonatal immune thrombocytopenias. Study group "Mother-child immune thrombopenias". Presse Med. 1992;21(36):1717–1724. [PubMed] [Google Scholar]
- 51.Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998;58:315–321. [PubMed] [Google Scholar]
- 52.Franco A, Yokoyama T, Huynh D, Thomson C, Nathenson SG, Grey HM. Fine specificity and MHC restriction of trinitrophenyl-specific CTL. J Immunol. 1999;162(6):3388–3394. [PubMed] [Google Scholar]
- 53.Simpson-Herrerns L, Lloyd HH. Kinetic parameters and growth curves for experimental tumor systems. Cancer Chemother Rep. 1970;54:143–174. [PubMed] [Google Scholar]
- 54.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miconnet I, Koenig S, Speiser D, et al. CpG are efficient adjuvants for specific CTL induction against tumor antigen-derived peptide. J Immunol. 2002;168(3):1212–1218. doi: 10.4049/jimmunol.168.3.1212. [DOI] [PubMed] [Google Scholar]
- 56.Davila E, Celis E. Repeated administration of cytosine-phosphorothiolated guanine-containing oligonucleotides together with peptide/protein immunization results in enhanced CTL responses with anti-tumor activity. J Immunol. 2000;165(1):539–547. doi: 10.4049/jimmunol.165.1.539. [DOI] [PubMed] [Google Scholar]
- 57.Davila E, Velez MG, Heppelmann CJ, Celis E. Creating space: an antigen-independent, CpG-induced peripheral expansion of naive and memory T lymphocytes in a full T-cell compartment. Blood. 2002;100(7):2537–2545. doi: 10.1182/blood-2002-02-0401. [DOI] [PubMed] [Google Scholar]
- 58.Finn O. History of tumour vaccines and novel approaches for preventive cancer vaccines. Dev Biol (Basel) 2004;116:3–12. [PubMed] [Google Scholar]
- 59.Finn OJ. Tumor immunology at the service of cancer immunotherapy. Curr Opin Immunol. 2004;16(2):127–129. doi: 10.1016/j.coi.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 60.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3(8):630–641. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 61.Pure E, Allison JP, Schreiber RD. Breaking down the barriers to cancer immunotherapy. Nat Immunol. 2005;6(12):1207–1210. doi: 10.1038/ni1205-1207. [DOI] [PubMed] [Google Scholar]
- 62.Springer GF. T and Tn, general carcinoma autoantigens. Science. 1984;224(4654):1198–1206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]
- 63.Muller S, Hanisch FG. Recombinant MUC1 probe authentically reflects cell-specific O-glycosylation profiles of endogenous breast cancer mucin. High density and prevalent core 2-based glycosylation. J Biol Chem. 2002;277(29):26103–26112. doi: 10.1074/jbc.M202921200. [DOI] [PubMed] [Google Scholar]
- 64.Xu Y, Gendler SJ, Franco A. Designer glycopeptides for cytotoxic T cell-based elimination of carcinomas. J Exp Med. 2004;199(5):707–716. doi: 10.1084/jem.20031865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sidney J, Grey HM, Kubo RT, Sette A. Practical, biochemical and evolutionary implications of the discovery of HLA class I supermotifs. Immunol Today. 1996;17(6):261–266. doi: 10.1016/0167-5699(96)80542-1. [DOI] [PubMed] [Google Scholar]
- 66.Vlad AM, Muller S, Cudic M, et al. Complex carbohydrates are not removed during processing of glycoproteins by dendritic cells: processing of tumor antigen MUC1 glycopeptides for presentation to major histocompatibility complex class II-restricted T cells. J Exp Med. 2002;196(11):1435–1446. doi: 10.1084/jem.20020493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Apostolopoulos V, Yuriev E, Ramsland PA, et al. A glycopeptide in complex with MHC class I uses the GalNAc residue as an anchor. Proc Natl Acad Sci U S A. 2003;100(25):15029–15034. doi: 10.1073/pnas.2432220100. [DOI] [PMC free article] [PubMed] [Google Scholar]