

Abstract
Background
This study analyzes the predictions of a number of promoter predictors on the ENCODE regions of the human genome as part of the ENCODE Genome Annotation Assessment Project (EGASP). The systems analyzed operate on various principles and we assessed the effectiveness of different conceptual strategies used to correlate produced promoter predictions with the manually annotated 5' gene ends.
Results
The predictions were assessed relative to the manual HAVANA annotation of the 5' gene ends. These 5' gene ends were used as the estimated reference transcription start sites. With the maximum allowed distance for predictions of 1,000 nucleotides from the reference transcription start sites, the sensitivity of predictors was in the range 32% to 56%, while the positive predictive value was in the range 79% to 93%. The average distance mismatch of predictions from the reference transcription start sites was in the range 259 to 305 nucleotides. At the same time, using transcription start site estimates from DBTSS and H-Invitational databases as promoter predictions, we obtained a sensitivity of 58%, a positive predictive value of 92%, and an average distance from the annotated transcription start sites of 117 nucleotides. In this experiment, the best performing promoter predictors were those that combined promoter prediction with gene prediction. The main reason for this is the reduced promoter search space that resulted in smaller numbers of false positive predictions.
Conclusion
The main finding, now supported by comprehensive data, is that the accuracy of human promoter predictors for high-throughput annotation purposes can be significantly improved if promoter prediction is combined with gene prediction. Based on the lessons learned in this experiment, we propose a framework for the preparation of the next similar promoter prediction assessment.
Background
Complexity of the target
Accurate determination of transcription start sites (TSSs) is one of the most difficult problems in genomics. The reference genomic location from which a transcript will be generated has remained elusive for many years, mainly due to our insufficient understanding of the transcription initiation process. The transcript promoter region surrounds the TSS and serves as the docking DNA segment that binds the preinitiation complex and various transcription factors that jointly create the biochemical conditions to initiate transcription [1,2]. Consequently, the analysis of promoter regions for binding sites of transcription factors can reveal many crucial aspects of how, where and when the transcript will be generated.
The naive concept of a gene having one TSS was abandoned long ago. Current data suggest that TSSs can be found scattered across the gene loci, generally more concentrated at the 5' end, but also more downstream, sometimes in exons, introns, and interestingly in the 3' untranslated regon (UTR) [3]. Moreover, one gene region may frequently have several promoters, and within one promoter several alternative TSS locations close to each other could be found. To make this complex picture even more complicated, promoter regions are frequently shared or overlap each other, such as in sense/antisense genes and in bidirectionally promoted genes [4]. All these considerably complicate the development of strategies for attacking the problem of promoter prediction. To avoid confusion, in this report, by 'promoter prediction' we mean the prediction of the TSS locations and not the prediction of a region surrounding a TSS.
Potential use of accurate TSS locations
Promoters are among the key genomic control regions for transcriptional regulation of every gene [1,2,5]. Thus, accurate TSS location makes determination of promoters more accurate, which allows for more accurate analysis of transcriptional regulatory elements necessary for any subsequent transcriptional regulatory network analyses. Furthermore, even when there are no expressed sequence data (expressed sequence tag (EST), cDNA, mRNA or different tags such as CAGE (cap-analysis of gene expression), SAGE (serial analysis of gene expression) and so on), the computational prediction of promoters and TSSs can allow for gene discovery.
Historical perspective
Realizing the importance of predicting promoters accurately, different experimental and computational methods have been developed. The large number of gene loci in eukaryotic genes inevitably calls for high-throughput large-scale technologies for determining TSS locations. Among the most efficient ones are those based on oligo-capping [6] and CAP-trapping [7]. Another group of methods is based on the use of multiple aligned ESTs and cDNA/mRNA fragments, and an assessment of TSS location as groups of identical 5' ends or the most 5' located end within the same locus. The third group of methods is based on the assessment of the binding location of DNA-associated RNA polymerase from ChIP-chip experiments [8]. However, the TSS location cannot be determined precisely from these experiments. In summary, none of the mentioned methods is sufficiently accurate or complete; this makes it difficult to obtain a proper reference dataset - one with high coverage and accuracy - to use for evaluation of promoter predictions.
An alternative to experimental methods are computational ones, but they generally are imperfect due to our insufficient understanding of the transcription initiation process. Several reviews have been published aiming at presenting the most crucial aspects and principles used in the construction of promoter prediction systems, as well as in the assessment of performance of promoter predictors [9-14]. Solutions proposed [15-31] were based on different concepts and exhibited various degrees of performance. PromoterInspector [28] was the first study to present computational predictions with an acceptable level of false positives (FPs) with human data, after the first genome scale evaluation as part of GASP had earlier shown promising results for Drosophila [32]. Encouraged by this, several efficient methods were later proposed [15-20,23-27,29-31]. The performance of many of these solutions have been extensively evaluated in [12].
Two strategies for designing promoter predictors
There is a lot of evidence that in mammalian genomes transcription initiates at various and unusual positions, such as intergenic regions far from currently known genes, 3' UTRs of known protein-coding genes, coding exons, and introns [3,4,33]. One gene may overlap another and promoters of such genes could fall anywhere on the body of the other gene [4]. The destiny of transcripts that are initiated is decided at various levels in the post-transcriptional processing, and many such transcripts are later degraded. However, it is difficult to estimate what proportion of all transcripts that the cell generates is functional. It is also difficult to determine which TSSs generate non-functional transcripts and whether they always generate such transcripts. For a long time biologists focused on protein-coding genes and this is one of the reasons that today most of the data we have relate to that transcript group. However, non-coding transcripts have recently been recognized as important for regulation of gene expression. A significant proportion of transcripts also cannot be accurately classified as being in either the coding or the non-coding group. For all these reasons it would be valuable to make the inventory of all TSSs in one genome and to investigate their functional properties.
For some purposes, a comprehensive list of potential TSSs may be most useful, even if the list contains FPs and TSSs of non-functional transcripts; for other applications, a list containing fewer FPs and non-functional TSSs may be better, even if it systematically omits interesting TSSs whose functions are less common or less well understood. Given our current state of knowledge, we must choose; predicting all and only functional TSSs is not currently feasible. Thus, TSS prediction programs have been designed around two strategies: use only the local genomic context (that is, model some aspects of the biological transcription initiation process or look at distinguishing characteristics of the region that immediately surrounds the TSS); or also take into account possible gene presence to restrict the search to regions that are most likely to contain promoters. The latter approach may use any of the available methods of gene prediction, including de novo prediction and prediction based on aligning ESTs, cDNA sequences, and/or proteins. It is also possible to utilize the annotation of genes if it is available. Using evidence about the presence of nearby genes may considerably enhance the performance of systems that work by analyzing the local promoter context. In general, on the genome scale, such a combination will reduce sensitivity to some extent, but it will significantly reduce the total number of predictions and will increase specificity.
On the other hand, to understand biological mechanisms of regulatory regions and to cover broad spectra of such regions, we probably should not use necessarily gene identification as a part of a strategy for pinpointing TSSs. The gene finding models introduce many implicit assumptions that reduce coverage of various types of TSSs that could be of interest. Also, linking promoter predictors to gene finders does not directly model the way in which transcription is initiated in the cell. A comprehensive solution is most likely to come from modeling the information cells use to determine where to initiate transcription, including the local promoter sequence and its epigenetic state [34].
Goals of this assessment
The ENCODE Genome Annotation Assessment Project (EGASP) is explained in detail in the main EGASP report [35]. The main goal of the project has been to assess the accuracy of prediction of protein coding genes, as well as the completeness of current human genome annotations of the ENCODE regions [36] covering approximately 1% of the human genome sequence. The reference gene set against which all predictions were assessed was created by manual annotation of the ENCODE regions by the HAVANA group [37] at the Sanger Institute, within the GENCODE project [38].
In our study, we attempt to make a critical assessment of the promoter prediction field in its current state relative to the HAVANA gene annotation [39] of the ENCODE regions. Thus, we assessed the extent of correlation of promoter predictions with the 5' gene ends of the HAVANA annotation. We argue that using promoter predictors together with gene predictors or as a complement to the manual annotation of genes is a good intermediate step to improve promoter prediction performance because this constrains the search space based on information about the gene. We propose promising strategies for future development of promoter prediction systems on the basis of the current performance assessment.
Results
The method for counting correct and wrong predictions is explained in Materials and methods. We have analyzed predictions on all 44 ENCODE regions (total length 29,998,060 base-pairs (bp)), with the training set consisting of 13 regions of total length 8,538,447 bp and the remaining part as the test set with a length of 21,459,613 bp. The genomic sequences were from the human genome Build hg17. The performance results are summarized in Figures 1 and 2. Figure 1 contains results where true positive (TP) predictions were allowed to be within a maximum distance of 1,000 nucleotides from the reference TSS; Figure 2 contains results where the maximum distance allowed was 250 nucleotides. We present results within three categories: for the test ENCODE regions, for the training ENCODE regions, and for all ENCODE regions. We considered only predictions of promoters for known genes that contained coding sequence (CDS) based on the HAVANA annotation that was submitted for the EGASP workshop. In total, there were 994 unique TSSs, of which 319 were in the ENCODE training set and 675 were within the ENCODE test set. In our analysis, the reference data against which the performance of promoter predictors was evaluated were the estimated TSS locations based on the 5' ends of genes in the HAVANA annotation. It is important to note, however, that HAVANA annotation does not attempt to specifically predict TSSs but rather to best represent the exon structure, CDS and UTRs of a gene and its splice variants.
Figure 1.
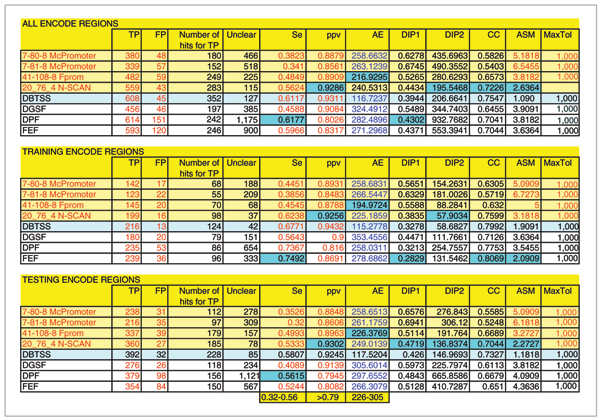
Prediction results for the distance criterion of 1,000 nucleotides. The light blue row shows the results of comparison of DBTSS+H-Invitational data to the manual HAVANA annotation. We used this as a reference to enable assessment of promoter predictor performance. The highlighted blue fields denote the score for the best performing promoter predictor. MaxTol is the maximum allowed mismatch between the predictions and the reference TSS locations. The programs with names in red officially participated in the EGASP data submission. The results shown are for the MaxTol = 1,000 nucleotides. AE is the average mismatch of predictions relative to the most close TSS location from the HAVANA annotation. It is divided by 1,000 to scale for the graph presentation. DIP1 and DIP2 are two measures representing distance from the ideal predictor as defined in [10]. ASM is the average score measure as defined in [10].
Figure 2.
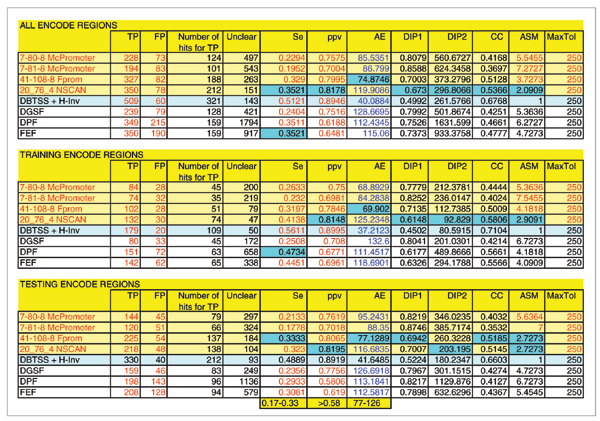
Prediction results for the distance criterion of 250 nucleotides. The light blue row shows the results of comparison of DBTSS+H-Invitational data to the manual HAVANA annotation. We used this as a reference to enable assessment of promoter predictor performance. The highlighted blue fields denote the score for the best performing promoter predictor(s). MaxTol is the maximum allowed mismatch between the predictions and the reference TSS locations. AE is the average mismatch of predictions relative to the closest TSS location from the HAVANA annotation. It is divided by 1,000 to scale for the graph presentation. DIP1 and DIP2 are two measures representing distance from the ideal predictor as defined in [10]. ASM is the average score measure as defined in [10]. The programs with names in red officially participated in the EGASP data submission. The results shown are for the MaxTol = 250 nucleotides.
In arriving at our conclusions, we used various measures of performance, as presented in [11]. The use of these different performance measures ensure that the final conclusions are less influenced by the choice of performance measures. The main reference for discussion is the current performance achieved on the ENCODE test regions. Since the ENCODE training regions have higher GC content (44.69%) than the average of the human genome, the results on the ENCODE training set and comprehensive ENCODE set are less representative.
Figure 1 shows that TSS locations compiled from DBTSS [40] and H-Invitational [41] databases, when used as predicted TSS locations and compared to the reference manual HAVANA annotation, show only 58% sensitivity (Se) and 92% positive predictive value (ppv). N-SCAN [30] has achieved a greater ppv of 93%. However, all promoter predictors had a ppv >79%, which is a considerable improvement over the last assessment [12]. The sensitivity, however, ranged from 32% to 56%. Positional mismatch of the predicted TSS locations relative to the reference ones was, on average, in the range 226 to 305 nucleotides for promoter predictors, while it was 117 nucleotides for DBTSS and H-Invitational TSS predictions. The correlation coefficient (CC; see Materials and methods) ranged from 0.52 to 0.70 for promoter predictors, and was 0.73 for DBTSS and H-Invitational TSS estimates. Figures 3 and 4 are bar graphs of different performance indicators. When the maximum allowed mismatch of the prediction from the reference TSS for counting TP predictions was 1,000 nucleotides, the best predictor, based on 11 measures of prediction success, was N-SCAN, followed by Fprom, Dragon Gene Start Finder (DGSF) [17,18], Dragon Promoter Finder (DPF) [15,16], First Exon Finder (FEF) [19], and McPromoter [23,24].
Figure 3.
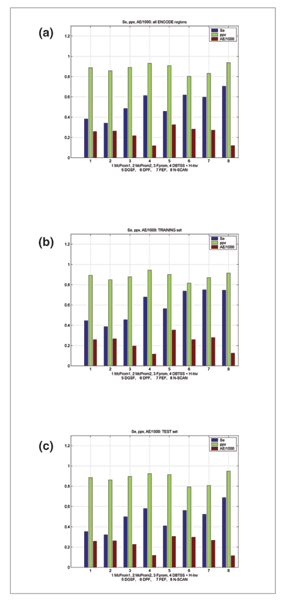
The results for different ENCODE regions. The results presented are for the maximum allowed distance of 1,000 nucleotides between the predicted TSS and the reference one. AE is the average mismatch of predictions relative to the most close TSS location from the HAVANA annotation. It is divided by 1,000 to scale for the graph presentation. Results are presented for: all ENCODE regions; the training set; and the test set. Relation of scores to the predictor performance is as follows: for Se and ppv, the higher the score, the better the performance. The scores for these two measures range from 0 to 1. For AE, the lower the score, the better.
Figure 4.
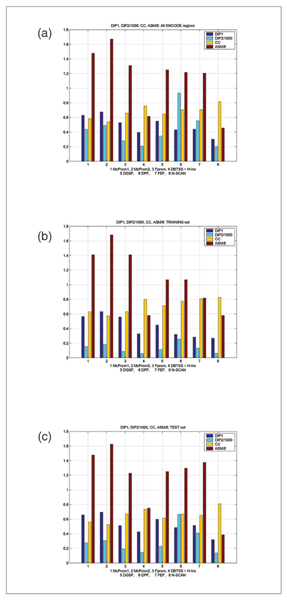
Another set of results for ENCODE regions. The results presented are for the maximum allowed distance of 1,000 nucleotides between the predicted TSS and the reference one. DIP1 and DIP2 are two measures of prediction qualities expressed as distances from the ideal predictor [10]. CC is the Pearson correlation coefficient. ASM is the average score measure as defined in [10]. DIP2 and ASM are scaled down to fit into the graph. Results are presented for all ENCODE regions, for the training set and for the test set. Relation of scores to the predictor performance is as follows: for distances from the ideal predictor (DIP1 and DIP2), as well as for ASM, the lower the score, the better. ASM represents the averaged rank position of the predictor calculated based on the individual measures of success. For CC, the greater the score, the better. CC ranges from -1 to +1.
When this maximum allowed distance was reduced to 250 nucleotides (Figure 2), the obtained sensitivity and positive predictive value were, as expected, lower. With this distance constraint, the DBTSS and H-Invitational prediction set produced a sensitivity of 49%, ppv of 89%, and an average mismatch of predictions to the reference TSS of 41 nucleotides. Promoter predictors achieved a sensitivity in the range 17% to 33%, a ppv in the range 58% to 81%, and an average positional error in the range 77 to 126 nucleotides. Correlation coefficients ranged from 0.35 to 0.51, while for the DBTSS and H-Invitational set it was 0.66. In this case, the best ranked predictors based on a cocktail of 11 measures were Fprom and N-SCAN, followed by DGSF, FEF, McPromoter (the standard system), DPF, and McPromoter (with the post-processing of shadowed predictions).
Discussion
We have analyzed four sets of promoter predictions that were submitted as a response to the EGASP call. These include McPromoter (the standard system), McPromoter (with post-processing of shadowed predictions), Fprom and N-SCAN. These submissions received internal EGASP coding 7-80-8, 7-81-8, 41-108-8, 20-76-4, respectively. The internal coding of submissions by the three numbers is explained in [42]. For the control set we used the estimated TSS locations inferred from the DBTSS and H-Invitational databases. These TSS estimates are based on flcDNAs, with those from DBTSS being derived from the oligo-capped full-length cDNAs (flcDNAs), and thus such a control set is expected to largely reflect the real TSS locations. Additionally, we also considered the predictions of three other programs, FEF, DPF and DGSF, as these were found in a recent comparative study [12] on the whole human genome to have reasonably good performance. The best performing programs in study [12] were DGSF and FEF. Thus, it was of interest to see how they would perform in EGASP. For these additional three programs, the predictions were run under the same conditions as in [12]. These collections of predictions formed the basis for the assessment of performance and promoter prediction strategies. It should be noted that all programs included in this study make assessments of the TSS locations.
Based on the results shown in Figures 1 and 2, we conclude that the best performance achieved with the ENCODE data is by programs that combine promoter prediction with gene prediction. This directly reduces the search space for promoters and minimizes the number of FP predictions since promoter searches are localized to the regions close to the estimated 5' end of genes. This also reduces the overall number of predictions. As a consequence, the accuracy of such programs (N-SCAN and Fprom) is somewhat increased compared to other programs. It is obvious that one could use the existing gene annotation to restrict the promoter search space. However, all programs evaluated in this study use ab initio predictions and do not rely on gene annotation. Moreover, programs that may rely on gene annotation to enhance promoter prediction would not work efficiently in a situation where such annotation does not exist.
The other three programs (McPromoter, DPF, DGSF) did not utilize gene structure prediction, while FEF used only a partial prediction of gene structure. In particular, McPromoter is a representative example of the aforementioned first group of successful ab initio genome-wide predictors, given that its version tuned for human data is essentially unchanged since its publication [24]. FEF uses an internal recognition of the first exon that is part of the overall gene structure, although it does not attempt to predict other parts of the gene structure. Also, DPF and DGSF use rough, simplified models of intron and exon domains in the promoter recognition process. These four programs (FEF, McPromoter, DPF, DGSF) have been tuned to search for promoters when no information except a single DNA sequence is available. This requires much tighter tuning in order to reduce FPs and maximize TPs. Still, their performance is considerable, keeping in mind that many of the FP predictions of these programs could be eliminated if some form of the complete gene structure prediction is used. One should note that the idea that promoter predictions can benefit from gene prediction is not new. One of the early suggestions in this direction was given in [9]. Although a similar idea has been contemplated by others, such as in [19], it has never been supported by comprehensive data. Our report seems to be the first one to provide such evidence on a larger scale.
The comparison analysis [12] focused on programs that do not use additional gene prediction. That study has demonstrated that a strong beneficial effect in accuracy can be achieved for many promoter predictors if masking repeats is used and the promoter search is restricted to nonmasked regions. In the current study we reach a similar general conclusion on improved accuracy when restricted search space is used, in the context of combining promoter prediction with gene prediction. Note that N-SCAN has also used masking repeats in the context of their gene prediction.
Finally, we comment on the generally better performance of promoter predictors on the training ENCODE set as opposed to the test ENCODE set. The simple explanation could be that it is a consequence of the increased GC content of the training ENCODE regions (44.69%) compared to the test ENCODE regions (42.33%). Usually, GC rich isochores represent more dense gene regions than the GC depleted isochors (at least based on current data). We also know [12] that many promoter predictors more efficiently predict GC rich promoters, which complies with the results on the ENCODE regions. However, since the DBTSS and H-Invitational TSS set shows better concordance with the HAVANA annotation data, it is also possible that part of the answer is in a more detailed and accurate annotation of the training set.
The reference TSS locations and TSS estimates from DBTSS and H-Invitational databases
We have used the HAVANA group's manual annotation of the ENCODE regions and considered the annotated 5' ends of transcripts as the reference TSS locations. As an alternative, we also used DBTSS and H-Invitational databases as a source of another collection of estimated TSS locations. Since this second collection is based on flcDNAs, of which many were oligo-capped, the TSS estimates based on this dataset should largely correspond or be close to genuine TSS locations. Actually, a recent report [43] indicates that 7% of the TSSs estimated from the oligo-capped flcDNAs of DBTSS mismatch by more than 100 bp those from the Eukaryotic Promoter Database (EPD) [44], while no precise estimates of distance mismatch are given for the remaining 93% of the DBTSS TSSs that fall within 100 bp of the corresponding EPD TSSs.
We then compared the HAVANA annotation and the TSS predictions based on the DBTSS and H-Invitational databases. It was somewhat disconcerting to find that sensitivity was only 58% with the DBTSS and H-Invitational data relative to the HAVANA reference set. Moreover, the ppv was only 92%. This estimation was done using the maximum allowed distance mismatch of 1,000 nucleotides between the estimated TSS and HAVANA annotated 5' gene ends. For those DBTSS and H-Invitational TSSs that did satisfy the distance criterion, the average positional error relative to HAVANA based estimates was 117 nucleotides, again a significant difference. Of the DBTSS and H-Invitational TSSs, 42% were more than 1,000 nucleotides apart from the closest HAVANA annotated 5' gene end. Although HAVANA gene structures may be based on the same mRNA evidence as DBTSS and H-invitational TSS predictions, HAVANA annotation may introduce a bias towards the most 5' TSS for some genes as gene structures are extended as far as other mRNAs and ESTs with identical exon structures support them (see Materials and methods). However, HAVANA annotation only uses spliced mRNA and ESTs as evidence to extend gene structures and, as such, would fail to extend the 5' end of a gene upstream where only single exon evidence supported it. Furthermore, mRNAs used by DBTSS and H-invitational to predict TSSs may not be used in HAVANA annotation to support coding genes, or possibly any gene structure, if their predicted CDSs appear questionable in its genomic context. The annotation of coding genes and splice variants supported by human ESTs and non-human mRNAs and ESTs by HAVANA may also result in 5' ends of genes being identified that are not represented in the current DBTSS and H-invitational databases. However, being aware that the experimental support for accurate TSS location is not easy to provide, we believe that this issue requires a separate and in-depth study, particularly when the CAGE data [3] have become available.
Although we used the HAVANA annotation as a reference dataset, we do not treat it as the 'gold standard' for promoter prediction. We are fully aware of the fact that there is no universally accepted genomic scale 'gold standard' for the accurate TSS locations that we could use. Different sets of experimental data bear the bias of the shortcomings of the experimental procedures used in experiments or of the postprocessing of these data. One may argue that the TSS estimates based on the DBTSS and H-Invitational database could be more reliable. However, one should not forget that TSS estimates from DBTSS and H-Invitational databases are also not guaranteed to be correct. Thus, blindly assuming that one set is good while the other is not without an in-depth evaluation of the experimental data is not justified. For this reason, we emphasize that the conclusions of our study are based on the constraints and framework defined in EGASP and those of our analysis, and they are valid to that extent.
The differences between the reference set and TSS estimates from the DBTSS and H-Invitational databases may explain the sensitivity results achieved by programs used in this study - for example, the decline in sensitivity for programs such as FEF, DPF, DGSF and McPromoter that were evaluated in [12] where DBTSS data was used as a reference. In any case, the HAVANA annotation currently represents the best gene annotation for the ENCODE regions. We believe that this has resulted in an increased ppv for promoter predictors in this study. Specifically, when we compare the ppv results from [12], we find that FEF, DPF, DGSF and McPromoter all have a much higher ppv on the ENCODE data and the associated HAVANA annotation, likely because of the more accurate annotation of gene loci regions.
Comparison with a previous study on the whole human genome
The direct comparison of the results of this study and the one performed recently on the whole human genome [12] is not possible simply for the reason that the reference data against which assessments are made are different. In [12] we used the whole human genome and the data from DBTSS; in the current study we used HAVANA annotation as the reference and focus only on ENCODE regions that make up about 1% of the whole human genome. In addition, the two datasets are not very similar, as we have already shown.
However, in spite of these differences in the reference dataset, we are still in a position to make some global observations. Compared to the previous whole human genome analysis [12], in this study we used a more stringent distance constraint: the maximum allowed mismatch of the predicted TSS from the reference TSS was 1,000 nucleotides. In [12] as the maximum allowed was 2,000 nucleotides. Because of this, one would expect the decrease in ppv, but we observe the opposite trend for all programs that were evaluated in [12] (FEF, DPF, DGSF and McPromoter). In [12] the reported ppv was in the range 25% to 67%. In the current study, with the stringent distance criterion, the ppv for these programs is in the range 79% to 91%, which is a positive surprise. For N-SCAN and Fprom, which were not included in [12], the ppv is also very high at 94% and 89%, respectively. Sensitivities for FEF, DPF, DGSF and McPromoter were, in [12], in the range 54% to 80% and in this study, as expected, they have been reduced, falling to the range 32% to 56%. However, one should be cautious in drawing conclusions as the DBTSS and H-Invitational TSS set shows only 58% sensitivity and a 92% ppv relative to the HAVANA annotation.
Another positive surprise is the positional accuracy of promoter predictors. Note that for experimental DBTSS and H-Invitational TSSs the positional error is 117 nucleotides. All promoter predictors in the current study achieved an average positional error in the range 226 to 305 nucleotides relative to the HAVANA annotation. This is only two- to three-fold larger than the average positional error of the DBTSS and H-Invitational experimental data.
Future developments
The lessons from EGASP relative to promoter predictions is that it is beneficial to combine the TSS/promoter predictors with gene finding programs irrespective how gene prediction is done. Using such an approach it will be possible to retune promoter predictors and also to partly change their design philosophy since more relaxed conditions will be required due to the restricted search space.
However, this cannot be a final solution as it will inevitably bias the predictions to only those towards the 5' gene end, or, at best, extend predictions to cover the whole body of the gene. The intergenic space will be covered only to the extent provided by the abilities of gene finding programs to detect new genes by ab initio methods.
Although most of the promoter predictors today can detect TSSs on the basis of an ab initio approach, we need to enhance their predictive ability. The ultimate solution will be to mimic the cellular transcription initiation process through technical implementation in promoter predictors. That is likely to allow efficient detection of a broad range of genuine TSSs in arbitrary genomic sequence irrespective of the support from experimental data or gene predictions. This is a challenging task and requires more sophisticated technical solutions that take advantage of the molecular biology of promoter regulation.
We also observe that the positional accuracy of promoter predictors requires further improvement. A recent review [14] proposed that the next goal in positional accuracy of promoter predictors is a 20 nucleotides mismatch relative to the experimental TSS locations, that is, on the same scale as naturally observed variation in the initiation process. However, this leaves the open issue of a good reference dataset. But, if we intend to achieve this goal, we have to incorporate more of the relevant biological information in the recognition algorithms. Related to this is also the following problem. Due to the massive expressed data (EST/cDNA/mRNA) available, annotation naturally uses such sources of information. Promoter prediction programs that utilize expressed sequences should generate predictions most close to the annotation based reference dataset, as this is more or less how the reference annotation is derived as well. This brings into focus an issue of circularity that will just confirm that promoter predictors that use such strategies comply well with the annotated data.
Scenario for promoter prediction for future experiments
Lessons from the current experiment motivate us to propose a framework for future promoter prediction assessment. It is absolutely necessary to conduct promoter prediction experiments within different categories of conditions that programs utilize, so as to be in a position to compare individual contributions of different types of information used. Two broad scenarios are of interest: one that assesses the genomic context within which the predictions are made, and another that assesses types of data/information used in deriving predictions.
In the first group, it will be helpful to consider separately methods that utilize only the immediate region surrounding a TSS (say [-200, +200]), as opposed to those that use a much broader genomic context. The reason for this is to evaluate the contribution of global and local signals in promoter predictions. The latter methods can include those that make use of gene structure prediction.
The second group could include: ab initio predictions based exclusively on the use of genomic sequence from one genome; ab initio predictions that use only genomic sequences from multiple genomes; predictions that utilize different support information (that is, known protein mapping, and so on), but not transcript data (that is, mRNA/EST mapping); and predictions that use information from mapping transcript data, as well as any other information. The comparison of programs would make sense only within categories, but not across various categories.
Conclusion
The current study argues in favor of combining promoter predictions with gene structure predictions as an intermediate improvement for promoter prediction accuracy. The long term goal has to be the development of a positionally accurate ab initio promoter prediction solution. For the next EGASP or similar project, different categories of promoter predictions should be provided, to enable the comparison of approaches differing on a large scale and the assessment of contributions of different types of information used in solutions. These in return would allow for more efficient promoter prediction programs.
Materials and methods
EGASP participants
We analyzed the following prediction sets provided in response to the EGASP call for submissions: 7-80-8 (McPromoter, the standard system), 7-81-8 (McPromoter with post-processing of shadowed predictions), 41-108-8 (Fprom), 20_76_4 (N-SCAN).
Additional prediction sets
To make the assessment of promoter predictions more complete, we also added four additional set of predictions, the TSSs estimated based on the DBTSS and H-Invitationsl data, which represent a large-scale experimental TSS dataset based on capped flcDNA, and those from FEF, DGSF, and DPF.
McPromoter
McPromoter is an ab initio system for predicting transcription start sites and was among the first fully probabilistic approaches to this problem. It uses a sequence of six Markov chain models for different subregions and elements within a core promoter spanning position -250 to +50, such as TATA-box, spacer, and initiator regions. As the core promoter is considerably different for distantly related eukaryotes, we have trained two separate models on vertebrate (mammalian) and invertebrate (fly) sequences. The Drosophila system has been under constant development [23], motivated by the identification of additional core promoter elements such as DPE (reviewed in [2]). The mammalian system has essentially remained constant throughout several years, including the data set it is trained on (a set of 565 sequences taken from the EPD) [24]. Small differences result from different strategies for the post-processing of the initial posterior probabilities of the predictor: For instance, submission 7-81-8 addressed the issue of shadow predictions, that is, simultaneous predictions on both strands of a core promoter caused by a strong signal in base composition. Here, we removed a lower scoring prediction if it fell within 1 kb of a higher scoring prediction of the standard system (7-80-8) on the opposite strand. However, as the results clearly show, this simple strategy actually decreased the performance slightly, indicating that a fraction of TPs is accompanied by stronger scoring predictions on the opposite strand in close proximity. The version of the McPromoter program used is MM:II, with a threshold of +0.005. The program can be found at [45].
Fprom: Softberry Pol-II promoter recognition approach
The task of finding eukaryotic polymerase II promoter involves two internal issues: finding the exact position of TSSs within long upstream regions of eukaryotic genes; and avoiding FP predictions within exon and intron sequences. To resolve the second part of this task some authors of promoter finding software include some recognition procedures of gene coding parts inside promoter prediction programs [15,28]. However, gene finding software such as Genscan [46] or Fgenesh [47] provides a much better accuracy in coding exon-intron identification than any such empirical procedures. We think that the best promoter identification strategy is to predict all gene components in one program. In creating such a program, it has currently been decided to use some intermediate variant, which includes the following steps: compute the gene annotation using a gene prediction pipeline and run promoter prediction on 5'-regions upstream of the annotated coding regions of predicted genes.
For promoter location within the selected regions, we used the Fprom (find promoter) program, which is the development of an algorithm realized earlier in the TSSW/TSSG programs [48]. For each potential TSS position of a given sequence, the Fprom program evaluates its possibility to be a TSS using two linear discriminant functions (for TATA+ and TATA- promoters) with characteristics computed in the [-200, +50] region around the given position. For TATA promoter recognition we consider the following features selected by discriminant analysis on the learning set of known promoters: hexamers in region [-200, -45]; hexamers in region [0:+40]; triplets in region [-200, -45]; triplets in region [0, +40]; TATA box maximal weight in interval [-45, -25]; TATA box average score on interval [-45, -25]; CpG-content; position triplet matrix in the [TSS-50, TSS+30] region; similarities between [-200, -100] and [-100, -1] regions; protein-DNA twist; protein-induced deformability; regulatory motif density in region [-200, -101] in the direct chain; and regulatory motif density in region [-100, -1] in the reverse chain.
If we find a TATA-box (using TATA-box weight matrix) in the positions [-45, -25] of the analyzed region, then we compute the value of LDF for TATA+ promoters, otherwise the value of the linear discriminant function (LDF) for TATA-less promoters. Only one prediction, with the highest LDF score and greater than some threshold, is selected within any 300 bp region. We run Fprom on 5' regions extracted from the predicted genes. For each such region, we selected the closest to the CDS predicted promoter and presented it in our results. The Fprom program can be found at the Softberry's web site at [49] and contains no user adjustable parameters.
N-SCAN
N-SCAN [50,51] is an extension of TWINSCAN [52]. N-SCAN's DNA sequence modeling is identical to TWINSCAN with the addition of states modeling 5' UTR exons and introns [30] and the capability to include conserved non-coding states in intergenic regions. N-SCAN's method of incorporating alignment information is quite different from TWINSCAN's method. TWINSCAN utilizes alignment information from one informant genome through a conservation sequence. A conservation sequence is generated by assigning each target sequence base a match, mismatch/gap or unaligned symbol based on a BLASTN alignment of the two genomes. N-SCAN replaces TWINSCAN's conservation sequence with a multiple genome alignment that represents the evolutionary relationships among the target and multiple informants with a Bayesian network rooted at the target genome along with a richer alphabet representing a more detailed modeling of substitution rates, insertions, and deletions across all informants. N-SCAN does not predict TSSs as isolated features, but rather as the 5' boundary of the first exon in a gene structure.
N-SCAN's human gene predictions employed human genome Build hg17 (May 2004), the corresponding RefSeq mappings, and a whole-genome, 8-way, MULTIZ alignment, which were all downloaded from UCSC [53]. The particular alignment subset chose human (hg17) as the target genome and mouse (mm5), rat (rn3), and chicken (galGal2) as informants, with all gaps in the target removed. Build hg17 was masked for interspersed repeats, but not low-complexity or simple repeats as identified by UCSC. The human sequence was further pseudogene masked (MJ van Baren and MR Brent 2005, submitted). The RefSeq mappings were filtered to remove probable errors; parameters were trained on three-quarters of the filtered RefSeq mappings. The program design and setting is explained in the companion article [54].
First Exon Finder
The main idea implemented in FEF [19] is that promoter prediction should be derived from prediction of the first exon. This is implemented by splitting the first exons into two groups, one that is GC rich and another that is GC poor. Several types of compositional features are used in the recognition process that is implemented as a rule-based solution with several quadratic discriminant functions. In [12], FEF was found to be among the best ab initio promoter predictors. It was also found that its performance benefits if combined with masking repeats by RepeatMasker. The recommendations from [12] were implemented with the default FEF parameter setting: a cutoff value for the first-exon a posteriori probability of 0.5, a cutoff value for the promoter a posteriori probability of 0.4, and a cutoff value of the splice-donor a posteriori probability of 0.4. We used the download version of the program. The web-server implementation can be found at [55].
Dragon Promoter Finder
DPF [15,16] uses three types of models for promoter regions, exonic regions and intronic regions. It utilizes position weight matrices of overlapping pentamers in these three regions to derive its predictions. The program uses separation of promoters to GC rich and GC poor groups and uses five different prediction models for different levels of sensitivity. It uses only 200 nucleotides DNA segments to make predictions. In this study, it was used with the default parameters and according to recommendations from [12], which combine predictions with masking repeats by RepeatMasker and uses clustering of its predictions. This means that predictions are clustered if the distance between the neighboring predictions is 1,000 nucleotides or less. Such clusters are represented by the average position of predictions in the cluster. The program version 1.5 was run with the expected sensitivity of 0.65 and according to recommendations from [12]. The program can be found at [56].
Dragon Gene Start Finder
DGSF [17,18] uses predictions of DPF in the region it assesses to be a CpG island. The program is aimed at finding the approximate start of gene loci. It first localizes the CpG island and then identifies the most likely DPF prediction within that region. Version 1.0 of the program was run with its default threshold parameter of 0.994 and according to recommendations from [12]. The program can be found at [57].
Counting predictions and other performance measures
The counting of TP and FP predictions is illustrated in Figure 3. If the maximum allowed distance of the prediction form the closest reference TSS on the same strand is D nt, then, if one or more predictions fall on the region [-D, +D] relative to the reference TSS location and on the same strand where the TSS resides, the TSS is counted as TP. If the reference TSS is missed based on this type of counting, then such a TSS is a false negative (FN). All reference TSS locations that were missed by this counting of TP predictions represent true negatives (TN). Every other prediction that falls on the annotated part of the gene loci in the segment [+D+1, EndOfTheGene] at the same strand where TSS resides counts as a FP. One has to be aware that some real TSSs/promoters could be in the regions [+1001, EndOfTheGene]. The other predictions were not taken for the determination of TPs and FPs. Figure 5 illustrates the counting method.
Figure 5.
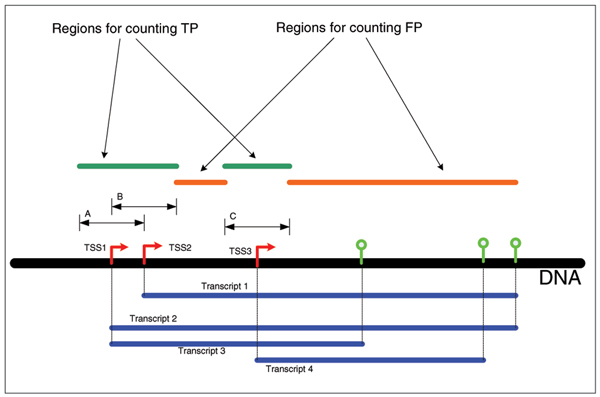
The counting method for TPs and FPs. All hits to the 'orange' segments count as FPs. Only one hit within A, B, or C counts as a TP for a unique position of TSS (for example, three hits within C will count only as one TP). Note that all TSS locations that were mutually different were considered as valid reference TSSs. So, alternative TSSs were considered different TSSs. Each of these had to be predicted. If one prediction falls on the intersection of A and B, then that prediction identifies two TSS locations (one that correspond to TSS related to A, and the other corresponding to TSS related to B). In other words, one prediction correctly identifies all reference TSS locations within the distance criterion.
The measures of performance were those used in [12]. In determining the average distance of predictions, only the minimum distance of one prediction from all reference TSSs was considered. Sensitivity is the proportion of correct predictions of TSSs relative to all experimental TSSs, defined as:
Se = TP/(TP + FN)
A ppv is the proportion of correct predictions of TSSs out of all counted positive predictions, defined as:
ppv = TP/(TP + FP)
The CC is the Pearson correlation coefficient, defined as:
CC = (TP × TN - FP × FN)/((TP + FP)(TP + FN)(TN + FP)(TN + FN))1/2
Data
The ENCODE regions mapped at the human genome Build hg17 (May 2005) were used. Out of the HAVANA annotation for ENCODE regions we analyzed only the category of known genes with CDS (category 2). After eliminating the redundant TSS locations, we obtained 994 unique TSSs for all ENCODE regions, 319 unique TSSs in the ENCODE 'training' set (13 regions), and 675 unique TSSs in the ENCODE test set. Note that the region ENr313 does not have any annotation. The length of DNA sequences in these regions is: all regions 29,998,060 bp; 'training' regions 8,538,447 bp; and testing regions 21,459,613 bp.
Reference TSS locations: HAVANA annotated 5' end of gene objects
All HAVANA/GENCODE annotation is based on primary EST, mRNA and protein evidence and structures are only extended as far as the supporting evidence allows. No automated predictions are used to support gene objects. Main gene structures are based on human (and where a novel structure with canonical splicing is supported) non-human mRNA and EST evidence identified in the nucleotide sequence databases and aligned by wuBLASTN [58]. Significant hits are re-aligned to the unmasked genomic sequence using est2genome [59] and proteins aligned by wuBLASTX. All evidence is navigated using the Blixem alignment viewer [60]. The 5' ends of gene structures are extended using only splicing human mRNA and EST evidence that agrees completely with the structure of the gene object that it is used to extend. As such, where mRNAs and ESTs support an identical gene structure but have different length 5' UTRs, they are merged into a single gene structure that is extended as far as the longest supported 5' UTR, that is to the most 5' aligned base of the most 5' EST or mRNA. Where sequence from the 5' end of mRNA and EST evidence is missing from the Est2genome alignment, visual inspection of the dot-plot output from the Dotter tool [60] is used in an attempt to identify any alignment with the genomic sequence upstream of the identified end of homology. Where a very short length of sequence (<15 bases) is missing from the 5' end of the alignment, a dot-plot is unsuitable due to the difficulty in seeing very short alignments at the edge of the display and the AcedB Restriction Analysis tool (essentially a pattern matching tool) [61] is used to try and identify any alignment with the genome. As such, the annotated 5' ends of gene objects are specified according to the best possible alignment of transcriptional evidence to the genome rather than specifically identifying TSSs in the genomic sequence. As new transcript evidence is added to the databases, so novel 5' exons and 5' extensions of existing exons continue to be identified.
TSS estimates from DBTSS and H-Invitational databases
Using DBTSS data (version 4.2, 11 Jan 2005), we obtained 12,763 TSS estimates for hg17, and of these, 286 were mapped to ENCODE regions. These were complemented by H-Invitational TSS data. We used 95% identity and 90% homology in BLAST mapping of H-Invitational data to hg17. This provided us with 20,116 TSS estimates. Within the ENCODE regions we found 325 TSS estimates not overlapping with DBTSS data. In total, the DBTSS and H-Invitational datasets provided 611 experimental TSS locations. These are provided as Additional data files 1 and 2.
Supplementary Material
Acknowledgments
Acknowledgements
This article has been published as part of Genome Biology Volume 7, Supplement 1, 2006: EGASP '05. The full contents of the supplement are available online at http://genomebiology.com/supplements/7/S1.
References
- Weinzierl ROJ. Mechanisms of Gene Expression: Structure, Function, and Evolution of the Basal Transcriptional Machinery. London: Imperial College Press; 1999. [Google Scholar]
- Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- FANTOM Consortium; RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- RIKEN Genome Exploration Research Group, Genome Science Group (Genome Network Project Core Group) and FANTOM Consortium Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Pedersen AG, Baldi P, Chauvin Y, Brunak S. The biology of eukaryotic promoter prediction - a review. Computers Chem. 1999;23:191–207. doi: 10.1016/S0097-8485(99)00015-7. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Sugano S. Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene. 1994;138:171–174. doi: 10.1016/0378-1119(94)90802-8. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics. 1996;37:327–336. doi: 10.1006/geno.1996.0567. [DOI] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Qu C, Van Calcar S, Trinklein ND, Cooper SJ, Luna RM, Glass CK, Rosenfeld MG, Myers RM, Ren B. Direct isolation and identification of promoters in the human genome. Genome Res. 2005;15:830–839. doi: 10.1101/gr.3430605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickett JW, Hatzigeorgiou AG. Eukaryotic promoter recognition. Genome Res. 1997;7:861–878. doi: 10.1101/gr.7.9.861. [DOI] [PubMed] [Google Scholar]
- Prestridge DS. Computer software for eukaryotic promoter analysis. Methods Mol Biol. 2000;130:265–295. doi: 10.1385/1-59259-686-x:265. [DOI] [PubMed] [Google Scholar]
- Bajic VB. Comparing the success of different prediction software in sequence analysis: A review. Brief Bioinform. 2000;1:214–228. doi: 10.1093/bib/1.3.214. [DOI] [PubMed] [Google Scholar]
- Bajic VB, Tan SL, Suzuki Y, Sugano S. Promoter prediction analysis on the whole human genome. Nat Biotechnol. 2004;22:1467–1473. doi: 10.1038/nbt1032. [DOI] [PubMed] [Google Scholar]
- Ohler U, Frith M. Models for complex eukaryotic regulatory DNA sequences. In: Bajic VB, Tan TW, editor. Information Processing and Living Systems. London, UK: Imperial College Press; 2005. pp. 575–610. [Google Scholar]
- Bajic VB, Werner T. Promoter prediction. In: Dunn MJ, Jorde LB, Little PF, Subramaniam S, editor. Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics Part 4 Bioinformatics, 42 Gene Finding and Gene Structure. John Wiley and Sons, Ltd; Hoboken, New Jersey; 2005. DOI: 10.1002/047001153X.g402301. [Google Scholar]
- Bajic VB, Seah SH, Chong A, Zhang G, Koh JLY, Brusic V. Dragon Promoter Finder: recognition of vertebrate RNA polymerase II promoters. Bioinformatics. 2002;18:198–199. doi: 10.1093/bioinformatics/18.1.198. [DOI] [PubMed] [Google Scholar]
- Bajic VB, Seah SH, Chong A, Krishnan SP, Koh JL, Brusic V. Computer model for recognition of functional transcription start sites in RNA polymerase II promoters of vertebrates. J Mol Graphics Model. 2003;21:323–332. doi: 10.1016/S1093-3263(02)00179-1. [DOI] [PubMed] [Google Scholar]
- Bajic VB, Seah SH. Dragon Gene Start Finder identifies approximate locations of the 5' ends of genes. Nucleic Acids Res. 2003;31:3560–3563. doi: 10.1093/nar/gkg570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic VB, Seah SH. Dragon Gene Start Finder: an advanced system for finding approximate locations of the start of gene transcriptional units. Genome Res. 2003;13:1923–1929. doi: 10.1101/gr.869803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri RV, Grosse I, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nature Genetics. 2001;29:412–417. doi: 10.1038/ng780. [DOI] [PubMed] [Google Scholar]
- Down TA, Hubbard TJ. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002;12:458–461. doi: 10.1101/gr.216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Computers Chem. 2001;26:51–56. doi: 10.1016/S0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- Knudsen S. Promoter2.0: for the recognition of PolII promoter sequences. Bioinformatics. 1999;15:356–361. doi: 10.1093/bioinformatics/15.5.356. [DOI] [PubMed] [Google Scholar]
- Ohler U, Liao GC, Niemann H, Rubin GM. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 2002;3:RESEARCH0087. doi: 10.1186/gb-2002-3-12-research0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U, Stemmer G, Harbeck S, Niemann H. Stochastic segment models of eukaryotic promoter regions. Proc Pacific Sym Biocomputing. 2000;5:380–391. doi: 10.1142/9789814447331_0036. [DOI] [PubMed] [Google Scholar]
- Ponger L, Mouchiroud D. CpGProD: identifying CpG islands associated with transcription start sites in large genomic mammalian sequences. Bioinformatics. 2002;18:631–633. doi: 10.1093/bioinformatics/18.4.631. [DOI] [PubMed] [Google Scholar]
- Hannenhalli S, Levy S. Promoter prediction in the human genome. Bioinformatics. 2001;17(Suppl):S90–S96. doi: 10.1093/bioinformatics/17.suppl_1.s90. [DOI] [PubMed] [Google Scholar]
- Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands. Nat Genet. 2000;26:61–63. doi: 10.1038/79189. [DOI] [PubMed] [Google Scholar]
- Scherf M, Klingenhoff A, Werner T. Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: a novel context analysis approach. J Mol Biol. 2000;297:599–606. doi: 10.1006/jmbi.2000.3589. [DOI] [PubMed] [Google Scholar]
- Solovyev VV, Shahmuradov IA, Prom H. Promoters identification using orthologous genomic sequences. Nucleic Acids Res. 2003;31:3540–3545. doi: 10.1093/nar/gkg525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RH, Gross SS, Brent MR. Begin at the beginning: predicting genes with 5' UTRs. Genome Res. 2005;15:742–747. doi: 10.1101/gr.3696205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, States DJ. Consensus promoter identification in the human genome utilizing expressed gene markers and gene modeling. Genome Res. 2002;12:462–469. doi: 10.1101/gr.198002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SEL. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- Guigó R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E, et al. EGASP: The human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006;7(Suppl 1):S2. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- The HAVANA Team http://www.sanger.ac.uk/HGP/havana/
- The GENCODE Project http://genome.imim.es/gencode/
- Harrow J, Denoeud F, Frankish A, Reymond A, Chen C-K, Chrast J, Lagarde J, Gilbert JGR, Storey R, Swarbreck D, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl 1):S4. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Yamashita R, Sugano S, Nakai K. DBTSS, DataBase of Transcriptional Start Sites: progress report 2004. Nucleic Acids Res. 2004;32:D78–D81. doi: 10.1093/nar/gkh076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Itoh T, Suzuki Y, O'Donovan C, Fukuchi S, Koyanagi KO, Barrero RA, Tamura T, Yamaguchi-Kabata Y, Tanino M, et al. Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biol. 2004;2:e162. doi: 10.1371/journal.pbio.0020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EGASP Submissions ftp://genome.imim.es/pub/projects/gencode/data/egasp05/egasp_submissions_20050503/submissions_bysubmitter.pdf
- Yamashita R, Suzuki Y, Wakaguri H, Tsuritani K, Nakai K, Sugano S. DBTSS: DataBase of Human Transcription Start Sites, progress report 2006. Nucleic Acids Res. 2006;(34 Database):D86–89. doi: 10.1093/nar/gkj129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praz V, Perier R, Bonnard C, Bucher P. The Eukaryotic Promoter Database, EPD: new entry types and links to gene expression data. Nucleic Acids Res. 2002;30:322–324. doi: 10.1093/nar/30.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPromoter MM:II http://genes.mit.edu/McPromoter.html
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev VV, Salamov AA. The Gene-Finder computer tools for analysis of human and model organisms genome sequences. In: Rawling C, Clark D, Altman R, Hunter L, Lengauer T, Wodak S, editor. Proceedings of the Fifth International Conference on Intelligent Systems for Molecular Biology: 21-25 June; Halkidiki, Greece. AAAI Press; Menlo Park, CA, USA; 1997. pp. 294–302. [PubMed] [Google Scholar]
- Fprom http://www.softberry.com/berry.phtml?topic=fprom&group=programs&subgroup=promoter
- Gross SS, Brent MR. Using multiple alignments to improve gene prediction. In: Miyano S, Mesirov JP, Kasif S, Istrail S, Pevzner PA, Waterman MS, editor. Research in Computational Molecular Biology: Proceedings of the 9th Annual International Conference, RECOMB 2005; Boston. Cambridge, MA, Springer; 2005. pp. 374–388. [Google Scholar]
- Gross SS, Brent MR. Using multiple alignments to improve gene prediction. J Comput Biol. 2006;13:379–393. doi: 10.1089/cmb.2006.13.379. [DOI] [PubMed] [Google Scholar]
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;17(Suppl 1):S140–S148. doi: 10.1093/bioinformatics/17.suppl_1.s140. [DOI] [PubMed] [Google Scholar]
- UCSC Browser http://genome.ucsc.edu/
- Arumugam M, Wei C, Brown RH, Brent MR. Pairagon+N-SCAN_EST: a model-based gene annotation pipeline. Genome Biol. 2006;7(Suppl 1):S5. doi: 10.1186/gb-2006-7-s1-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First Exon Finder http://rulai.cshl.edu/tools/FirstEF/
- Dragon Promoter Finder http://research.i2r.a-star.edu.sg/promoter/promoter1_5/DPF.htm
- Dragon Gene Start Finder http://research.i2r.a-star.edu.sg/promoter/dragonGSF1_0/genestart.htm
- wuBLASTN http://blast.wustl.edu
- Mott R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput Appl Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Wootton JC. Integrated graphical analysis of protein sequence features predicted from sequence composition. Proteins. 2001;45:262–273. doi: 10.1002/prot.1146. [DOI] [PubMed] [Google Scholar]
- Durbin R, Griffiths E. In: Acedb genome database Genetics, Genomics, Pro-teomics and Bioinformatics Online Bioinformatics Modern Programming Paradigms in Biology. Peter Clote, editor. Vol. 4. Boston College, Massachusetts, USA: Wiley Interscience; 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.