

Abstract
Studies of the neurobehavioural components of borderline personality disorder (BPD) have shown that symptoms and behaviours of BPD are partly associated with disruptions in basic neurocognitive processes, in particular, in the executive neurocognition and memory systems. A growing body of data indicates that the glutamatergic system, in particular, the N-methyl-D-aspartate (NMDA) subtype receptor, plays a major role in neuronal plasticity, cognition and memory and may underlie the pathophysiology of multiple psychiatric disorders. In this paper, we review the literature regarding BPD and its cognitive deficits and the current data on glutamatergic and NMDA neurotransmission. We propose that multiple cognitive dysfunctions and symptoms presented by BPD patients, like dissociation, psychosis and impaired nociception, may result from the dysregulation of the NMDA neurotransmission. This impairment may be the result of a combination of biological vulnerability and environmental influences mediated by the NMDA neurotransmission.
Medical subject headings: borderline personality disorder, cognitive dysfunction, NMDA neurotransmission.
Abstract
Des études portant sur les éléments neurocomportementaux du trouble de la personnalité limite (TPL) ont montré qu'il y a un lien partiel entre les symptômes et les comportements du TPL et des perturbations des processus neurocognitifs fondamentaux, et en particulier des systèmes de la neurocognition exécutive et de la mémoire. De plus en plus de données indiquent que le système glutamatergique, et en particulier le récepteur du sous-type N-méthyl-D-aspartate (NMDA), joue un rôle majeur dans la plasticité des neurones, la cognition et la mémoire et peut sous-tendre la pathophysiologie de multiples troubles psychiatriques. Dans cette communication, nous passons en revue les publications sur le TPL et ses déficits cognitifs, ainsi que les données de l'heure sur la neurotransmission glutamatergique et par NMDA. Nous posons en hypothèse que de multiples symptômes et dysfonctionnements de la cognition que présentent des patients atteints de TPL, comme la dissociation, la psychose et le déficit de la nociception, peuvent découler de la dysrégulation de la neurotransmission par NMDA. Ce déficit peut résulter d'une vulnérabilité biologique conjuguée à des influences environnementales médiées par la neurotransmission par NMDA.
Introduction
In the DSM-IV,1 personality disorders are differentiated from other major mental disorders in the axis II category. This categorization implies more biological vulnerability in axis I disorders and more psychosocial and developmental sequelae in the axis II disorders. However, the demarcation of axis I versus axis II disorders is owing mostly to our limited knowledge of the neurobiology of personality disorders. In fact, neurobiological vulnerability and developmental insults are critical for both axis I and II disorders. Borderline personality disorder (BPD) is a good example of the connection between the biological and psychosocial arenas. Biological vulnerability and developmental insults combined determine the presentation of BPD. BPD is a common, disabling condition characterized by a pervasive pattern of disability in affect regulation, impulse control, interpersonal relationships and self-image.2,3 Approximately 1%–2% percent of the US general population has BPD.4 BPD is associated with high comorbidity with other psychiatric disorders, including anxiety, mood, and posttraumatic stress disorders (PTSDs); substance abuse and dependence; other personality disorders; and psychosis.5 BPD diagnostic criteria have been organized into 4 sectors of psychopathology: affective disturbance, disturbed cognition, impulsivity and intense, unstable relationships.6 Patients with BPD may also present with dissociative episodes and temporary failure of reality testing.7,8 Hence, BPD may be understood as a syndrome, with patients presenting with variable constellations of symptoms (Table 1).
Table 1
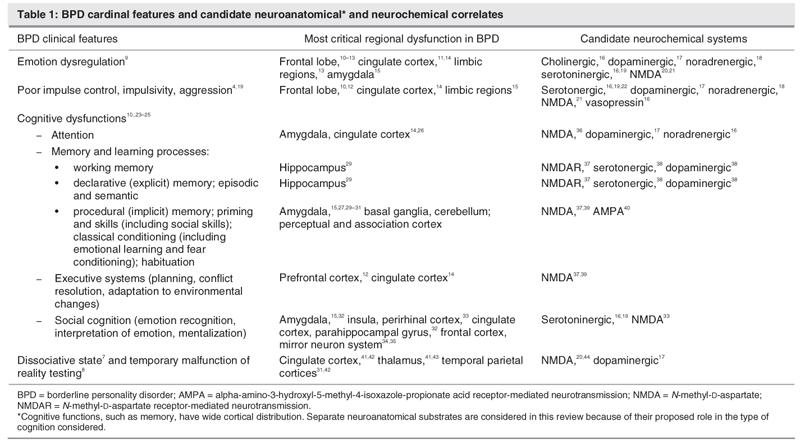
Study of the neurobehavioural components of BPD is still in its infancy. However, clinical theoreticians and researchers have proposed that the symptoms and behaviours of BPD are partly associated with disruptions in basic neurocognitive processes, in particular, in the executive neurocognition and memory systems.10 Mentalization (or theory of mind [TOM])45 deficits have also recently been proposed as a major component that is impaired in BPD psychopathology.23 Manualized psychotherapeutic treatments9,23,46 have demonstrated their efficacy compared with placebo, yet we still lack precise guidelines to direct patients to a specific modality of psychotherapy or pharmacological treatment. Above all, a fundamental neurobiological understanding of BPD is missing.
This paper reviews recent neurobiological data related to BPD, the concepts of neuroplasticity and mentalization or TOM, glutamatergic and N-methyl-D-aspartate (NMDA) neurotransmission and the interplay between the environment and the developing central nervous system (CNS). We propose relations between these mechanisms and BPD at etiological and symptomatic levels. Multiple neurotransmitters, including dopamine and serotonin, are involved in many of the functions considered in this paper. Further, interactions between the glutamatergic system and the different aminergic systems, although well-known at the cellular level, are not clearly elucidated at the functional level; therefore, this matter is outside the scope of this review. Finally, we explore new therapeutic modalities to modify the maladaptation of NMDA neurotransmission. Recent research has investigated the potential therapeutic effect of new pharmacological agents hypothesized to improve (or suppress) learning through their enhancement of NMDA neurotransmission.47–49 We propose that similar pharmacological agents could be used in addition to the conventional BPD treatments.
Risk and vulnerability factors of BPD
BPD likely arises from complex interactions between genetic, neurobiological and environmental factors.50 Multiple studies support the notion that sexual abuse and, to a lesser degree, physical abuse are major contributing factors to the etiology of BPD.51 Neglect, as well as chaotic and inconsistent home environments during childhood, also appear to be significant risk factors.52 Other vulnerability factors for BPD include premature birth, higher rate of psychiatric disorder in the family, separation from parents and unfavourable parental rearing styles.53
These risk factors for BPD are in line with findings of attachment research. Contemporary attachment researchers have proposed that the quality of early attachment organization can impact on the development of cognitive capacities.23 Chronic stress and failure of the caregiver to perceive and respond adequately to the child's distress would trigger regressive, nonmentalizing functioning. Hence, internal experience remains unlabelled and chaotic, and the uncontained affect generates further dysregulation and symbolization deficits.54 This combination of trauma and disrupted attachment security may instigate a developmental pathway leading to adult BPD. Other findings point to potential gene–environment interactions that impact both neurobiological vulnerability and the cognitive and attachment organization relevant to BPD. For instance, Fan and colleagues26 showed how genetic polymorphism may be related to the performance of attentional networks that are recognized as impaired in BPD patients.24,55
Cognitive deficits in BPD
Maladaptive cognition has recently been recognized as one of the cardinal features of BPD (Table 1). Current theories of BPD emphasize emotional dysregulation9 but are not as explicit about the potential for mutually interacting impairment in executive neurocognition and memory. BPD patients display deficits on tasks that require controlled information processing, such as sustained attention, spatial working memory and executive functioning.55 In an extensive review, Fertuck and colleagues23 concluded that people with BPD are more compromised than are control subjects in all measured executive neurocognition and memory domains. Ruocco,10 reporting a meta-analysis of the available cognitive literature in BPD, posits a widespread neuropsychological deficit linked largely to functioning of the frontal lobes. Further, Posner and colleagues11 reported that BPD patients are specifically deficient in attentional networks involved in conflict resolution and in the voluntary inhibition of thought and behaviour.
Mentalization processes, or TOM, are an advanced component of emotional cognition, and it has been proposed that they are impaired in BPD psychopathology.23 TOM refers to the ability to internally represent the mental states of others, for example, their thoughts, desires, beliefs, intentions and knowledge.45 In cognitive terms, mentalization is a feature of procedural memory that is created in the context of secure attachment. Mentalization is the ability to interpret the expression on another person's face and to have a sense of what that person is feeling without extensive conscious effort to figure out the meaning of the facial expression; it is a critical component of empathy. The development of mentalization occurs mostly between the ages of 4 and 6 years, and it is only from 6 years of age onward that a normally developing child gains full and explicit awareness of mental states and their role in the explanation and prediction of other people's behaviour.45
Recent studies show that the emotional cognition as revealed by the ability to recognize and interpret facial expressions of emotion is impaired in patients with BPD.56 Studies demonstrate that physically abused children display a response bias for angry facial expressions, whereas neglected children have more difficulty discriminating expression than do control subjects or physically abused children.57 Using functional magnetic resonance imaging, Donegan and others15 noted increased left amygdala activation in BPD patients when presented with a neutral face; this may be interpreted as amygdala hypereactivity. Overall, subjects with BPD have a biased perception or interpretation of emotional signals.
Another important part of emotional cognition is reversal learning, that is, the ability to learn a new and different behaviour in a similar setting, after learning a first behaviour (“reversing” the prior learning). There is no direct study of reversal learning in patients with BPD, but reversal-learning deficits have been proposed in sociopathy, a diagnosis with similar phenomenology and developmental and psychosocial vulnerability to BPD.58
During the last decade, Fonagy59 and Bateman and Fonagy23 established that patients with BPD suffer from poor mentalization processes. In psychodynamic terms, they suggest that inhibition of mentalization may be a prototypic response to trauma. Clinical and experimental evidence supports the view that disorganizing effects of trauma on attention and stress regulation may bring a partial and temporary collapse of mentalization.60
Finally, in light of the growing data on the molecular features of neuroplasticity, learning and memory and the important role of NMDA neurotransmission (see below), we hypothesize that mentalization processes may rely partly on neuronal plasticity and memory processes that are mediated, among others, by the NMDA subtype receptor (NMDAR) of glutamatergic neurotransmission. If this proposition is correct, it might lead to innovative research paths in the etiology and treatment of BPD.
Neurobiological basis of BPD
Neurotransmitter systems
The biological underpinning of BPD is complex and poorly understood. Previous studies have emphasized the aminergic neurotransmission and hypothalamic-pituitary-adrenal (HPA) axis hyperactivity in relation to the pathophysiology of BPD in terms of vulnerability, temperament and stress imprinting.16 BPD patients, as with other individuals with a long history of excessive social stress, show HPA axis hyperactivity.18 Figueroa and Silk22 proposed a model in which the effect of trauma interacts with an underlying predisposition to serotonergic dysfunction. Biological, neuroendocrine and imaging studies also provide evidence for the involvement of serotonergic activity in impulsive aggression19 and in working memory.38 Friedel17 proposed that dopamine dysfunction may be associated with 3 dimensions of BPD: emotional dysregulation, impulsivity and cognitive perceptual impairment. His hypothesis is limited by the current absence of studies that directly demonstrate dopamine dysfunction in BPD.
In summary, BPD does not consist of impairment in a single neurotransmitter system. The glucocorticoid and aminergic neurotransmission are part of the pathophysiology of BPD. In addition to glucocorticoid and aminergic neurotransmission, NMDA neurotransmission might also play a critical role in neurodevelopment, synaptic plasticity, learning and memory, all phenomena potentially relevant to the vulnerability and pathophysiology of BPD. To date, no clear articulation has been sought between BPD and NMDA neurotransmission.
Functional anatomy
Neuroimaging studies have consistently demonstrated reduced hippocampal and amygdalar volumes in patients with BPD versus control subjects.27 There are at least 2 known psychological mechanisms that can induce the structural diminution found in BPD: stress and neglect. Animals and human studies show that a high level of stress is associated with HPA axis activation, and elevated glucocorticoid levels can be toxic to the neuronal population in the hippocampus.61 Elevated glucocorticoids are also accompanied by glutamatergic activation which, itself, is neurotoxic (i.e., excitotoxicity).62 In addition, neglect and poor environmental stimulation result in poor synaptic density and decreased volumes of brain regions enriched in NMDARs.63 These 2 mechanisms together may directly affect the brain development of patients with BPD who are known to experience deprivation, abuse and chronic stress during critical periods of development.
The human hippocampus is important for spatial and episodic or autobiographical memory.64 Consistent with the hippocampal atrophy found in neuroimaging studies of BPD patients,12,27 BPD subjects present impairment in their explicit and autobiographical memory.55 The amygdala plays an important role in modulating vigilance and generating negative emotional states.30 Amygdala dysfunction has been proposed to be involved in emotional dysregulation15 and conditioned fear65 and in implicit memory processes (e.g., consolidation of directly activated memories).66 Recent research has shown that the lateral nucleus of the amygdala is a region specifically implicated in the formation of memories for stressful experiences. Johnson and others67 proposed that, in the lateral amygdala, newly discovered glucocorticoid receptors may have a specialized role in modulating synaptic transmission plasticity related to fear and emotional memory.
In rodents, chronic stress induced by immobilization produces an enhanced dendritic arborization in the amygdala. This is in striking contrast to the degenerative effects demonstrated in the hippocampus as a result of the same stressor.28 These paradoxical stress-induced changes are consistent with the different roles of the hippocampus and amygdala in the neural circuitry of stress. It may demonstrate that chronic stress can cause different types of dendritic remodelling, depending on the neural structure. These anatomic findings are consistent with the functional studies showing that, compared with healthy subjects, BPD patients present a greater activation of their amygdala in response to aversive stimuli68 or facial expression.15 One of the characteristics of patients developing BPD is a repeated history of mistreatments; it is possible that these types of early chronic stress impact dendritic arborization differentially in the hippocampus and amygdala. This could be one neurobiological underpinning of BPD symptoms (i.e., a vulnerable, hyperreactive amygdala and an underdeveloped, dysfunctional hippocampus).
Neuroimaging studies demonstrate that TOM is a special domain of cognition that involves widespread corticolimbic regions, including bilateral regions of the temporo-parietal junction; the posterior and superior cortex; the temporal pole, cingulate cortex, dorsolateral prefrontal cortex; and the temporal lobe.69 Supporting their role in TOM, these brain regions are involved in several disorders for which TOM deficits have been documented, including schizophrenia,70 bipolar disorder,71 autistic spectrum disorders,72 adult patients with frontal lobe damage73 and antisocial personality disorder.
In addition to these findings, various functional imaging techniques have documented aberrant functioning in the cingulate cortex in BPD.13 Impulsivity14 and deficits in attentional control, response facilitation/inhibition and conflict monitoring25 are mediated by anterior cingulate and are often observed in BPD subjects. Evidence also suggests that the corpus callosum is reduced in subjects experiencing early trauma or neglect or both. Teicher and others74 proposed that these patients present a reduced integration between the right and left hemispheres. He suggests that the dysfunctional integration between hemispheres may predispose patients to shift abruptly from left-to right-dominated states with very different emotional perception and memories. This finding could be the result of impaired NMDA-mediated neurodevelopment.75
At a cellular level, the recent discovery of mirror neurons34 and their likely role in the ability to perceive, understand and feel emotional states observed in others,35 (Table 1) make them a prospective key actor in mentalization processes76 and in neural mechanisms possibly involved in the development of BPD. Mirror neurons fire when a primate performs an action and when it observes the same action performed by another (especially conspecific) primate. For this reason, mirror neurons might help to understand several human features, from imitation to empathy, mindreading (i.e., TOM) and language learning.
Fundamentals of NMDA neurotransmission
Synaptic plasticity refers to the variability of the strength of a signal transmitted via a synapse. It is part of the Hebbian theory about the neurochemical foundation of memory and learning. Introduced by Donald Hebb77 in 1949, the idea is that “cells that fire together wire together.” Although oversimplified, his proposal was further established when, in 1970, Bliss and Lomo78 identified long-term potentiation (LTP) as the long-lasting strengthening of the synaptic connection between 2 neurons after a series of conditioning trains of impulses. In 1983, Collingridge and colleagues79 showed that the induction of associative LTP was dependent on NMDA neurotransmission.
Neuroplasticity subsumes diverse processes of vital importance by which the brain perceives, adapts and responds to various internal and external stimuli. Since the seminal work of Wiesel and Hubel,80 multiple studies have confirmed that brain development is an experience-dependent process. The same researchers proposed the notion of critical periods in neurodevelopment, for example, heightened epochs of brain plasticity, during which sensory experiences produced long-lasting and large-scale change in neuronal circuits. During that critical time, appropriate stimulations are required for normal development. A classic example is the need for visual stimulation in developing ocular dominance columns.81 This activity-dependent neuroplasticity is modulated by multiple systems, including the NMDA neurotransmission.82 Disruption of any of these components during key periods will alter normal neurodevelopment.
Glutamate was recognized as a neurotransmitter in the 1970s, and the subtypes of glutamate receptors were differentiated in the early 1980s. Today we know that glutamate is the primary excitatory neurotransmitter in the mammalian brain: 60% of brain neurons use glutamate as their primary neurotransmitter.83 Ionotropic receptors for glutamate are divided into NMDA and non-NMDA receptors, including AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate) and kainate subtypes. The involvement of NMDAR in working memory has been shown in primate studies where NMDA antagonists impair their working memory,36 and potentiation of NMDA neurotransmission can correct the memory deficits. Similar results have been established in humans.84
The distribution of NMDAR within different brain regions varies developmentally.20 In humans, the NMDA neurotransmission functions as networks that sustain the associative function of the cortex and hippocampus, the sensory relay operation of the hypothalamus, the danger alarming processing function of the amygdala and basal forebrain and motivation response system.44 The NMDAR channel has several properties that are different from all other receptors. For example, NMDAR can “detect” the coincidence of 2 events (associativity). Hence, it can encompass different forms of plasticity, such as LTP and long-term depression (LTD).85
Regulation of NMDAR is complex; it depends on developmental stages and brain regions.21 NMDAR also plays an important role in another crucial step in the organization of our brain circuitry, named synaptic scaling.86 Synaptic scaling, or pruning, is a critical step of brain maturation, involving the restriction of axonal fields and a rearrangement and refinement of the synapse. NMDA neurotransmission has also been proposed to be involved in metaplasticity.87 Metaplasticity refers to a higher form of synaptic plasticity where prior synaptic activity leads to a persistent change in the direction or magnitude of subsequent activity-dependent plasticity without affecting actual synaptic efficacy. As a result, prior activity may shift the threshold for LTP and LTD induction. It has been demonstrated that the environment factors influence a shift in plasticity threshold in the somatosensory, piriform and motor cortices during development and learning.21 The NMDA systems could be one platform for transmitting external signals into molecular events.88,89 Recent studies have implicated NMDA neurotransmission in stress-induced hippocampal atrophy and cell death in the hippocampus, which has a very high concentration of glutamate.62
The function of the NMDA synapse can be modulated in different ways. Depending on the quality and magnitude of the stimuli, neurobiological and clinical consequences may vary. For instance, overactivation of NMDAR via the glutamate binding site results in neurotoxicity and cell death. Conversley, studies indicate that D-serine, a full agonist of the glycine site, or sarcosine, acting as an antagonist on the glycine transporter-1 (GlyT-1) and enhancing NMDA function, can improve positive, negative and cognitive symptoms of schizophrenia.90
Finally, the overlap and convergence of both dopaminergic and glutamatergic projections in the mammalian brain91 are well studied, but the study of their potential functional interactions are not examined in this review.
Interplay between environmental influences and CNS adaptive mechanisms of NMDA neurotransmission
Multiple animal and human studies have investigated the neurobiological consequences of environmental influence of stress, most critically for BPD at the anatomic, molecular and functional levels.92,93 A widely held view is that the combination of genetic vulnerability, early life stress, and ongoing stress may ultimately determine individual responsiveness to life events and vulnerability to psychiatric disorders, including BPD. At a molecular level, stress hormones have potent growth-inhibiting effects on the CNS via the interactions between the environment and NMDA neurotransmission. Their putative consequences at a neurobiological and clinical level are of prime importance.
Stress is often used as a broad term applied to external and internal stimuli, or lack of stimulus, that may alter the physical and mental homeostasis of a person.94 Contrary to the initial emphasis on physical threat, psychosocial or interpersonal stress, novelty, isolation, reward, withholding of reward and anticipation of punishment are among the most potent activators of the physiologic stress system.95 Research on the relation between specific stressors and specific psychological outcomes found little evidence for the notion that particular risk factors are uniquely related to particular outcomes.96 This heterogeneity in stressors and variability in response to trauma indicates a complex interaction between biological vulnerability and environmental impact. An early stressful environment can detrimentally and, at times, irreversibly impact multiple neuronal systems, including aminergic neurotransmission. A key intermediary for these mechanisms is the NMDA neurotransmission system that trophically regulates the critical period of growth and metabolism of widespread corticolimbic areas. The cascade of stress-induced neuronal death involves multiple systems, such as the catecholamine, glucocorticoid, free radicals and glutamate mechanisms. Stress activates catecholamine responses and NMDA neurotransmission. Excessive NMDAR stimulation is a common pathway, leading to free radicals, which are themselves associated with oxidative stress and may ultimately destroy the cells.
In their rodent study of the consequences of chronic stress on the dendritic arborization in the amygdala and hippocampus, Vyas and others28 suggest further research to examine the involvement of NMDA neurotransmission-dependent mechanisms. Animal studies show that important forms of learning in both the conditioning and extinction of fear are dependent on the proper function of NMDA neurotransmission in the amygdala.97 Other studies provided morphological evidence that glutamate plays a role in excitatory neurotransmission at synapses in the lateral nucleus of the amygdala.98 These findings support other data implicating excitatory amino acid-mediated synaptic plasticity in emotional learning and memory processes at the level of the amygdala. Continuation of or reexposure to stress results in unregulated excitation of glutamate neurons,99 and a growing body of data have implicated glutamatergic neurotransmission in stress-induced hippocampal atrophy and death.62
Multiple studies have demonstrated that maternal care influences the neurobiology of brain development.100 Early maternal separation appeared to exert a dramatic suppressive effect on synaptic overproduction in the early development of the hippocampus but did not appear to affect this process in other brain regions. These observations might explain why childhood abuse appears to be associated with reduced hippocampal volume.101 Isolation and stress are also known to decrease the survival rate of dentate gyrus cells of the hippocampus, whereas cognitive training, antidepressant drugs, mastication and mossy fibre stimulation increase it.102 Therefore, the environmental signals can be translated into diverse anatomic and functional outcomes in the hippocampus through the glutamatergic neurotransmission; both NMDA and non-NMDA receptors activation.
Deprivation of empathic care, through chronic excessive arousal intensification or reduction, creates a growth-inhibiting environment that produces an immature, physiologically undifferentiated orbitofrontal cortex (OFC) affect-regulatory system.103 The OFC receives input from all sensory areas of the posterior cortex, including projections from the face and head regions and from temporal regions related to vision, as well as outputs to motor areas. OFC also uniquely projects extensive pathways to limbic areas in the temporal pole and central nucleus of the amygdala and to glutamate receptors of the mesocorticolimbic dopamine neurons.103 Hence, the OFC articulates between motor, sensory and limbic interactions. It matures in the last half of the second year and is known to be critically and directly involved in attachment functions,104 such as mentalization105 and cognitive emotional interaction.106 Research also provides evidence for the potential involvement of NMDA receptor dependent plasticity in the orbital prefrontal cortex (OPFC). Bohn and colleagues107 demonstrated that intra-OPFC blockade of NMDA receptors impaired reversal learning. These anatomic underpinnings of OPFC can underscore the potential significance of NMDA neurotransmission in the neurodevelopment of attachment, emotion and cognition.
Other than hazardous signals, environmental factors may also have positive or healing effects on neurodevelopment. Bredy and others63 showed that maternal care in the rodent influences the development of cognitive function in the offspring through neural systems known to mediate activity-dependent synaptic plasticity. The offspring of mothers that exhibited increased levels of pup licking and grooming showed increased hippocampal NMDA subunit mRNA expression, enhanced synaptogenesis and improved hippocampal-dependent spatial learning, compared with animals reared by mothers who exhibited low levels of licking or grooming. They also showed that the effects of reduced maternal care on cognitive function can be reversed with peripubertal environmental enrichment.63,100 These findings imply that, to a certain extent, prevention and therapeutic interventions can rely on the plasticity of substrate and may involve NMDA neurotransmission.
In conclusion, NMDA-glutamatergic neurotransmission might represent a major interface between the environment and neurobiological plasticity and could be a critical mediator at the level of the hippocampus, amygdala and frontal cortex. Neuronal damage mediated by NMDA neurotransmission may vary depending on the intensity, duration and type of stress. The extent and long-term consequences of the stress-induced neuronal changes also depend on the time of occurrence within the lifespan. The age between 18 months and 4 years is a critical period, during which the human brain may be more vulnerable to increased risk of developing later BPD pathology. It is a time when parts of the CNS, such as the hippocampus and frontal cortex, are still going through important structural changes (pruning and myelination); it is also a time when explicit memory appears and cognitive processes involving mentalization capacities develop. During this period of neurodevelopment, vulnerability, as mediated by NMDA neurotransmission on hazardous environmental stress, may play a role in the development of BPD. Conversley, it may also be a time when therapeutic intervention could be especially effective because of the plasticity of the developing brain.
BPD symptoms and NMDA neurotransmission
The distinctive symptoms and cognitive dysfunction presented by BPD are likely to be related to some level of dysfunction with NMDA neurotransmission. At an etiological level, we know that trauma, abuse, neglect, chaos and abandonment are key factors in the later emergence of BPD. Through metaplasticity, NMDA activity is believed to set early in life, for different brain structures, the plasticity thresholds in the CNS during development and learning.21 By potentially modulating the neurotransmission threshold level regarding, for instance, the type of neurobiological response to stress and its physiological consequences, NMDA-mediated neuroplasticity might be a key factor for later vulnerability and reaction to a traumatic situation (Table 2).
Table 2
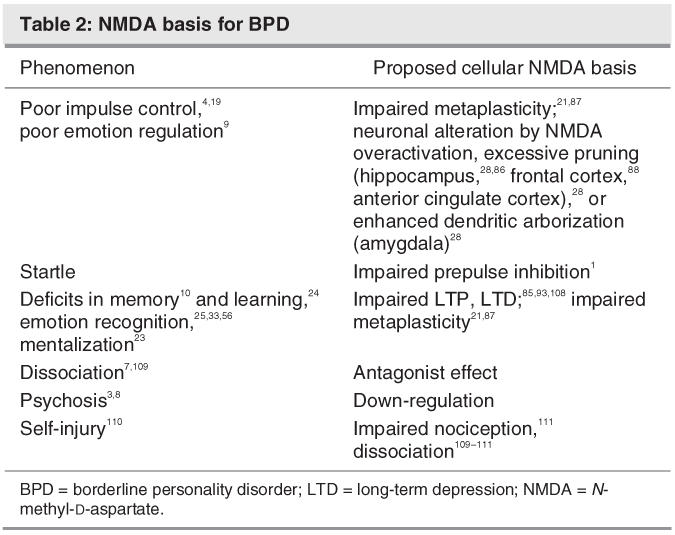
During the first years of life, the level of stimulation or lack of stimulation of individuals, through the NMDA neurotransmission, may influence the way they apprehend their environment, from pain, fear, response to a threat, or interpretation of facial emotions.
BPD arises out of an interaction between multiple neurobehavioural systems, in particular, NMDA neurotransmission, which plays a critical role in experience-dependent neuroplasticity. Due to its specific developmental insults in the critical period of neurodevelopment, BPD emerges as a phenotype reflective of a highly disorganized emotional cognition, which presents with reactive negative affectivity system, high levels of social fear, low levels of positive affect and low levels of nonaffective constraint. To the extreme, the vulnerability may result in psychosis and dissociation.
Memory, learning and cognition
Multiple animal studies have demonstrated that exposure to stress may influence neurogenesis in the dentate gyrus of hippocampus via regulation of the NMDAR activation.93 Fear conditioning and extinction, both considered forms of new learning, are dependent on proper function of NMDA neurotransmission in the amygdala. Consolidation of the memories for these associative learning processes may also involve interplay of the NMDA-mediated plasticity in prefrontal-amygdala circuits. Supporting the role NMDA neurotransmission plays in learning and cognition, all antagonists, acting at the level of the NMDA receptors, block learning processes.85 Infusion of NMDA antagonists have shown to impair response reversal learning in rodents.108 In primates, NMDAR antagonists produce working memory deficits.
From a clinical perspective, recent reviews propose that patients with BPD present specific cognitive deficits, including poor spatial and executive memory, poor autobiographical memory and poor mentalization processes. Animal studies have demonstrated that dysfunction of NMDA neurotransmission plays a significant role in the first 2 conditions. Similarly, learning and reversal learning deficits and poor attention are common in BPD, and these deficits are at least partly mediated by NMDA neurotransmission.108 Recently, the involvement of NMDA neurotransmission has also been demonstrated in the ability to recognize objects33 face recognition is another aspect of possibly dysfunctional cognition in patients with BPD.
Dissociation
Pathological dissociation is conceptualized as a disturbance in the integrative functions of identity, memory and consciousness. Animal43 and human41,42 research has suggested that certain dissociative states may be an expression of dysfunctional cortico-thalamic connectivity, which is abundant in NMDA neurotransmission. Brunner and colleagues109 reported that dissociation scores are significantly higher for BPD patients, compared with healthy subjects and patients with schizophrenic disorders. Consequences of this disturbance are psychopathological symptoms, such as identity diffusion7 or confusion, amnesic episodes, feelings of estrangement, and impairment or loss of sensory and motor function. That NMDA antagonists (such as the dissociative anesthetic, ketamine and phencyclidine) can produce dissociative symptoms with compelling similarities to those that occur frequently in BPD7 (such as dissociation and derealized or depersonalized states) suggests that dysfunction or downregulation of NMDA receptor-mediated neurotransmission may play a role in the core psychopathology of the dissociation of BPD. It is also known that high doses of NMDAR antagonists lead to toxic effect, particularly in frontal and cingulate brain regions during a specific developmental age.112 Attenuated NMDA function owing to neglect or poor environmental stimulation could thus lead to structural changes and downregulation of neurotransmisison critical for cognition in BPD. This underdevelopment of NMDA neurotransmission likely lays the ground for dissociation later in life.
Psychosis
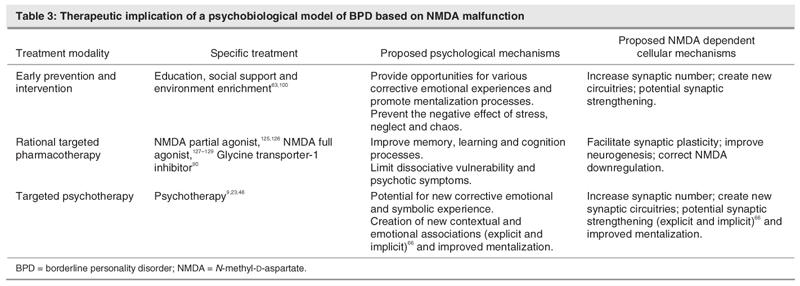
In addition to dissociation phenomena, other psychotic symptoms or lapses in reality testing (paranoid experiences, hallucinations, magical thinking or ideas of reference have been frequently documented in patients with BPD.3 The psychosis is often refractory to the antipsychotic treatments, based on D2 or D2/5-HT2 blockade. This is not surprising if the hypofunction of NMDA neurotransmission is the fundamental basis of the psychosis. Consistent with this, NMDA antagonists generate psychotic symptoms in healthy volunteers and exacerbate symptoms of patients with psychosis,42,113 while NMDA-enhancing agents can improve the symptoms of psychosis.114 NMDA antagonist treatment also changes neuronal activity in anterior cingulated, prefrontal cortex and other limbic regions115 implicated in the cognitive deficits of BPD (Table 1). Therefore, the NMDA-enhancing agents could address the fundamental NMDA deficits in patients with BPD and psychotic symptoms (Table 3).
Table 3
Mentalization
To discuss the TOM of BPD, we can learn from research on autism, which focused on various symptoms and cognitive dysfunction that are shared with BPD, such as decreased pain sensitivity, depersonalization, and a well-documented impairment in TOM.72 As result of his research with patients with autism, Carlsson116 proposed that a contributing factor in the context of TOM is a deficient transmission in glutamatergic intracortical “association pathways.” He suggests that autism may be a handicap that is formed because of a lack of social training, during a critical “time window.”117 In parallel with Carlsson's hypothesis, we propose that the BPD etiopathology might involve defective glutamatergic NMDA neurotransmission owing to a lack of appropriate social nurturing or, paradoxically, an excess of hazardous stimulation during NMDA-mediated plasticity, which results in failed mentalization during the developmental age. Carlsson notes that individuals with autism receiving NMDA antagonist treatment express a feeling of “emptiness inside,” which is also recognized by clinicians as a typical subjective feature of BPD patients.
In mentalization deficit research, a major issue is the lack of well-established, comprehensive, validated tools to assess the quality of mentalization. Mentalization abilities are complex and involve multiple perceptive and cognitive mechanisms. Researchers need a validated test or tool to appropriately compare different types of mentalization deficits within various pathologies. For now, we propose that the growth of mentalization ability is on a continuum, and its development likley depends on multiple genetic systems and their interactions with the environmental signals. We propose that NMDA neurotransmission may play a crucial role in the development of mentalization capacities because of its role in metaplasticity, memory and learning processes, and early associative learning.
A difference in the mentalization impairments presented by patients with BPD or patients with psychotic or autistic disorders may be in the degree and quality of impairment and in the potential for improvement. It is estimated that genetics account for approximately 80% of the etiological factors determinant of schizophrenia118 and autism, whereas minimal genetic determinants have been elucidated in BPD. Thus, if NMDA neurotransmission is involved in both schizophrenia and BPD, even if they share some phenomenology, distinct pathophysiologies are involved in these 2 conditions. Also, BPD patients appear to be more amenable to significant improvements with or without treatment, possibly because of less impairment to begin with or because of different metaplasticity potential.
Sensory perception
Self-injurious behaviours are frequent in patients with BPD.110 Schmahl and colleagues111 proposed that the nociceptive deficits in BPD may be caused by altered intracortical processes similar to certain meditative/dissociative states. They suggested a disturbance of the affective motivational or cognitive-evaluative pain component in self-injurious behaviours. Anatomically, the anterior cingulate cortex (ACC) has been shown to encode an affective component of painful stimulus119; in animal studies, NMDAR antagonists have been shown to attenuate the activity of ACC neurons involved in pain perception.120 The same part of the ACC (rostral cingular zone) involved in pain perception overlaps with one brain region involved in mentalization.45 NMDA antagonists have been extensively studied for their anesthetic properties, and animal studies show the importance of the NMDAR in modulating sensitivity to pain and morphine tolerance.121 Hence, the lack or dysfunctional sensitivity to pain in BPD may be partly mediated through NMDA processes. Consistent with this, NMDA antagonists like ketamine can prevent the experience of physical pain.
BPD specificity
Since NMDA neurotransmission is widely distributed in CNS, it is not surprising that it is involved in many neuropsychiatric disorders. Similarly, serotonergic and dopaminergic neurotransmission are involved in multiple CNS disorders. The specificity of the disorders, however, comes from a complex combination of genetic, neurobiological vulnerability and the timing and characteristics of the environmental insults. Although schizophrenia, autism and BPD are different disorders, NMDA neurotransmission is involved in all 3. They have some shared facets, including psychotic symptoms and cognitive dysfunctions, which may be due to shared biological substrate/components. Corresponding to the shared NMDA substrate, in a psychoanalytic theorization of BPD, major defence mechanisms used by patients with BPD (such as paranoia, splitting, projective identification) are considered “primitive defense mechanisms” and shared by both borderline and psychotic organizations.8 A low dose of antipsychotic medications is a common treatment for autism and patients with BPD with transitory psychotic symptoms. However, in BPD pathology, these symptoms are generally temporary and often subside with time and treatment. Thus, the differences in their intensity, occurrence and temporality may be due to variability in vulnerability, level of dysfunction of the cognitive systems and ability to benefit from environmental influence.
Currently, there is a debate regarding the DSM classification of BPD as an axis I or axis II pathology. It has been shown that, with time and appropriate environmental input, BPD patients are able to acquire and develop new cognitive and emotional abilities and, with time (10–15 years), most of them no longer meet BPD diagnostic criteria.2,5 Although BPD patients likely present a lifetime vulnerability and typical response to certain type of stress (which can be associated with the idea of an axis II), the occurrence of borderline pathology may be considered a prolonged “state of decompensation” amenable to treatment; most often, total recovery is achieved. For this reason, we propose that BPD pathology should be considered a developmental disorder.
The NMDA component in BPD etiopathology and treatment could give insight into the complexity of classifying this disorder in the DSM categorization. NMDA plays an early role in metaplasticity and could influence the “axis II/vulnerability” aspect. Its particular properties as a mediator between environmental influences and neurodevelopment may also influence life-time vulnerability and capability for learning and recovery. Whereas the psychosis and cognitive impairments in schizophrenia and autism could be caused by impairments in the NMDA system in a more fundamental level, for example, prenatal insults and macroscopically maldevelopment of CNS and is much less amendable, as in BPD.
NMDA neurotransmission is implicated at several complex levels of neurodevelopment and cognition. We have postulated that various symptoms and cognitive deficits found in BPD can be related to NMDA dysfunctions. This impairment is the result of the conjunction of biological predisposition and environmental factors, both of which express their impacts and effects through NMDA neurotransmission. However, little is known about the role that NMDA-relevant genetic predisposition plays in individual response to stress and psychological trauma. Overall, the developing brains of children are more vulnerable to BPD precipitants. Excessive and long-term stress caused by trauma, abuse, chaos or neglect will impact metaplasticity and neuroplasticity, especially during early childhood.
Implications for a new preventive and therapeutic approach
Prevention and treatment of BPD can certainly benefit from the neurobiological understanding of the relevant dynamics of NMDA neurotransmission. Successful treatment for BPD typically involves long-term psychotherapy. Three modes of psychotherapy have demonstrated their efficacy for BPD: transference-focused psychotherapy,46 dialectical–behavioural psychotherapy9 and mentalization-based therapy.23 In general, psychotherapy is a controlled form of learning that occurs in the context of a therapeutic relationship, and from this perspective, the neurobiology of psychotherapy can be understood as a special process of learning.122,123 We hypothesize that the comparable efficacy of these 3 forms of psychotherapy in BPD may reside in learning processes occurring within a structured and reliable environment. Therapists help patients to identify emotions and social cues, enhance interpersonal and emotion regulation skills, and increase the ability to mentalize through clarification, interpretation, education, validation, teaching and exercise according to the theoretical models. Once the patients can identify and appropriately interpret their own and others' state of mind, they learn how to prevent or modulate previously pathological or detrimental, often impulsive behaviours. If the learning hypothesis is correct, particularly for psychiatric disorders, such as BPD, which respond to psychotherapeutic treatment, a way to improve the therapeutic outcome, in addition to psychotherapy, is to enhance the learning that occurs during the psychotherapeutic process.48
Pharmacological treatments are generally recommended in BPD as an adjunct to psychotherapy to address specific target symptoms of BPD or to treat comorbid axis I conditions.4 There is some evidence that combined use of psychotherapy and pharmacotherapy can lead to a better outcome for patients with BPD.4 However, to date, pharmacotherapy lacks specific medication that could enhance the acquisition and retention of new learning, such as the development of psychosocial skills and improved mentalization or symbolization abilities.
NMDA neurotransmission influences and is influenced by experience and has a clear impact on cognition in general, specifically on emotion, affect, motivation, appraisal and evaluation of environmental stimuli. Neuronal plasticity is affected by the environment not only at the initial establishment of neuronal circuitries of a developing brain, but also during our entire life through memory retrieval, consolidation and rewiring of the synaptic connections.124 This potential of neuroplasticity has considerable implications for such rehabilitation processes as psychotherapy for BPD. Many neurobiology studies demonstrate that activation of the NMDA receptor is involved in the processes of learning and memory. NMDA (partial) agonists have been shown to improve learning and memory in animals.125,126 Full agonists of NMDA-glycine site, including D-serine, glycine and D-alanine, have also been shown to improve the cognitive and negative symptoms of schizophrenia.127–129 Other studies indicate that sarcosine, which acts as an antagonist on the GlyT-1 and enhances the NMDA function by making more glycine available for the glycine co-agonist site of the NMDA receptor, can improve positive, negative and cognitive symptoms of schizophrenia.130 Supporting the hypothesis that NMDA enhancers can improve cognition, D-cycloserine given to patients with Alzheimer's disease is associated with modest cognitive improvement.131 Recently, Ressler and colleagues48 showed that pharmacological agent D-cycloserine, acting as a partial coagonist at the NMDA receptor, improves the outcome of psychotherapy of diverse anxiety disorders, by enhancing cognitive processes or fear extinction. Similarly, Hofmann and colleagues49 proposed to enhance the treatment effects of exposure therapy for social anxiety disorder with D-cycloserine.
It is unclear whether psychotherapy can modulate the NMDA neurotransmission directly. If so, there can be an additive or synergistic effect when agent-enhancing NMDA neurotransmission is applied with psychotherapy. Despite, or because of, the potential enhancement of NMDA neurotransmission and facilitation of the learning processes of both the acquisition and extinction of the fear response, we suggest that the use of the NMDA-modulating agents needs to be carefully evaluated. As we have seen, the function of the NMDA synapses can be modulated in different ways. The timing and administration of any treatment may be crucial in determining the outcome of its cognitive modulations. As we know in the treatment of PTSD and acute stress disorder, recollection and verbalization can either relieve or reinforce the effect of a traumatic experience.95 That is, we must be cautious about the time of intervention. The goal of therapy is not to reinforce the traumatic experiences/memories and “pathological reflexes” (e.g., fear, anxiety) engraved at the amygdalar level but to enhance new, nonpathological learning or reflex through new neuronal circuitry between the frontal cortex and the limbic system, including the hippocampus. Consistent with this, D-cycloserine treatment resulted in significant improvements in numbing, avoidance, and anxiety symptoms and reduction in the perseverative error scores as measured by the Wisconsin Card Sorting Test in patients with PTSD.132 Additionally, as noted, seemingly paradoxical findings93 in the role of glucocorticoids modulating neurogenesis, appropriate timing and the intensity of therapeutic intervention needs to be titrated and targeted at the amygdala versus the hippocampus. It raises the possibility that the partial agonist could be indicated for fear extinction, and full agonists or Gly-T1 inhibitors for enhancement of learning and memory. A partial agonist, like D-cycloserine, could be indicated for patients early in the disease course, when they present predominantly with fear and anxiety; the agonist would be for the recovery and skill-building phase or for patients with significant cognition deficit, dissociation or psychosis.
Increasing evidence suggests that dysfunction of NMDA neurotransmission may play an important role in the pathophysiology of dissociative states.31,44 NMDA agonists could be used to limit the level of dissociation, in particular, in potentially stressful circumstances, such as a psychotherapy setting when transference and attachment mechanisms reactivate intense fear and cognitive bias in patients with BPD. NMDA activation could also improve the psychotic symptoms presented by patients with BPD, which are similar to the symptomatology of schizophrenia and other psychotic disorders but which are often refractory to antipsychotic drugs that are predominantly D2/5-HT2 antagonists. Hence, by limiting dissociation or psychotic symptoms, we may improve patients' ability to mentalize and to learn to integrate new, less threatening, socialization patterns. This hypothesis could be tested with a double blind study comparing 2 groups of patients treated by the same modality of psychotherapy and being given either placebo or NMDA agonists. If the hypothesis is correct, patients receiving the latter should show a quicker or qualitatively different improvement, for instance, in some of their cognitive and mentalization capacities.
The newly identified NMDA-therapeutic target can be integrated into the 3 psychotherapeutic models currently recognized as efficacious in treating BPD pathology. Of course, further research is required to identify more rigorously defined dimensions of BPD (e.g., impulsivity, dissociation, mood regulation, specific cognitive deficit, mentalization problems, psychosis). By identifying specific types of cognitive and mentalization deficits, we may be able to recognize more specifically the treatment from which individuals may benefit the most. Consequently, we could provide these patients with attunement and a nontraumatic environment in which to reorganize and find an improved, “neuromental” state.
A better understanding of the neurobiological effects of neglect, stress and trauma and their long-term effect on young brains should help to promote, determine and implement biological and psychosocial preventions. Neurosciences' new insights into the complex and subtle interactions between biology and environment should not be seen as a simplistic and reductivist view of the wholesome biopsychosocial human. On the contrary, when translating clinical observations into biological terms, neuroscience can only facilitate the integration of studies from multiple disciplines. An integrative view and the use of diverse theoretical and clinical data should help to more accurately identify multifactorial vulnerabilities and to determine the most suitable ways to prevent and treat pathology.
Acknowledgments
The authors thank Drs. Anthony Bateman, Kyle Boone, Ira Lesser and Michael Makhinson for their critical review of the manuscript. Dr. Tsai is supported in part by Los Angeles Biomedical Institute, Torrance, California and an Independent Investigator Award from NARSAD. In memory of Jambur Ananth M.D. for his unconditional support.
Footnotes
Contributors: Drs. Grosjean and Tsai designed the study. Dr. Grosjean acquired and analyzed the data. Drs. Grosjean and Tsai wrote the article and critically reviewed it. Both authors gave approval for the final version of the article to be published.
Competing interests: None declared.
Correspondence to: Dr. Bernadette Grosjean, Department of Psychiatry, Harbor-UCLA Medical Center, 1000 West Carson Street, Box 497, Torrance 90509, CA, USA; bernaharbor@yahoo.com
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington: The Association; 1994.
- 2.Zanarini MC. Borderline personality disorders. Boca Raton: Taylor & Francis; 2005:19-40.
- 3.Gunderson JG. Borderline personality disorder: a clinical guide. 1st ed. Washington, DC: American Psychiatric Press; 2001:1-33.
- 4.Torgersen S. Epidemiology. In Oldham JM, Skodol AE, Bender DS eds. The American Psychiatric Publishing textbook of personality disorders. 1st ed. Washington, DC: American Psychiatric Pub; 2005:129-141.
- 5.Zanarini MC, Frankenburg FR, Vujanovic AA, et al. Axis II comorbidity of borderline personality disorder: description of 6-year course and prediction to time-to-remission. Acta Psychiatr Scand 2004;110:416-20. [DOI] [PubMed]
- 6.Lieb K, Zanarini MC, Schmahl C, et al. Borderline personality disorder. Lancet 2004;364:453-61. [DOI] [PubMed]
- 7.Zanarini MC, Ruser T, Frankenburg FR, et al. The dissociative experiences of borderline patients. Compr Psychiatry 2000;41:223-7. [DOI] [PubMed]
- 8.Kernberg O. Borderline personality organization. J Am Psychoanal Assoc 1967;15:641-85. [DOI] [PubMed]
- 9.Linehan M. Cognitive-behavioral treatment of borderline personality disorder. New York: Guilford Press; 1993:558 p.
- 10.Ruocco AC. The neuropsychology of borderline personality disorder: a meta-analysis and review. Psychiatry Res 2005;137:191-202. [DOI] [PubMed]
- 11.Posner MI, Rothbart MK, Vizueta N, et al. An approach to the psychobiology of personality disorders. Dev Psychopathol 2003;15:1093-106. [DOI] [PubMed]
- 12.Tebartz van Elst L, Hesslinger B, Thiel T, et al. Frontolimbic brain abnormalities in patients with borderline personality disorder: a volumetric magnetic resonance imaging study. Biol Psychiatry 2003;54:163-71. [DOI] [PubMed]
- 13.Juengling FD, Schmahl C, Hesslinger B, et al. Positron emission tomography in female patients with borderline personality disorder. J Psychiatr Res 2003;37:109-15. [DOI] [PubMed]
- 14.Hazlett EA, New AS, Newmark R, et al. Reduced Anterior and Posterior Cingulate Gray Matter in Borderline Personality Disorder. Biol Psychiatry. 2005. [DOI] [PubMed]
- 15.Donegan NH, Sanislow CA, Blumberg HP, et al. Amygdala hyperreactivity in borderline personality disorder: implications for emotional dysregulation. Biol Psychiatry 2003;54:1284-93. [DOI] [PubMed]
- 16.Gurvits IG, Koenigsberg HW, Siever LJ. Neurotransmitter dysfunction in patients with borderline personality disorder. Psychiatr Clin North Am 2000;23:27-40. [DOI] [PubMed]
- 17.Friedel RO. Dopamine dysfunction in borderline personality disorder: a hypothesis. Neuropsychopharmacology 2004;29:1029-39. [DOI] [PubMed]
- 18.Rinne T, de Kloet ER, Wouters L, et al. Hyperresponsiveness of hypothalamic-pituitary-adrenal axis to combined dexamethasone/corticotropin-releasing hormone challenge in female borderline personality disorder subjects with a history of sustained childhood abuse. Biol Psychiatry 2002;52:1102-12. [DOI] [PubMed]
- 19.Coccaro EF, Siever LJ, Klar HM, et al. Serotonergic studies in patients with affective and personality disorders. Correlates with suicidal and impulsive aggressive behavior. Arch Gen Psychiatry 1989;46:587-99. [DOI] [PubMed]
- 20.Watanabe M, Inoue Y, Sakimura K, et al. Distinct spatio-temporal distributions of the NMDA receptor channel subunit mRNAs in the brain. Ann N Y Acad Sci 1993;707:463-6. [DOI] [PubMed]
- 21.Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci 2005;28:229-38. [DOI] [PubMed]
- 22.Figueroa E, Silk KR. Biological implications of childhood sexual abuse in borderline personality disorder. J Personal Disord 1997;11:71-92. [DOI] [PubMed]
- 23.Bateman A, Fonagy P. Psychotherapy for borderline personality disorder: mentalization-based treatment. Oxford; New York: Oxford University Press; 2004: 381 p.
- 24.Posner MI, Rothbart MK, Vizueta N, et al. Attentional mechanisms of borderline personality disorder. Proc Natl Acad Sci U S A 2002;99:16366-70. [DOI] [PMC free article] [PubMed]
- 25.Milham MP, Banich MT. Anterior cingulate cortex: an fMRI analysis of conflict specificity and functional differentiation. Hum Brain Mapp 2005;25:328-35. [DOI] [PMC free article] [PubMed]
- 26.Fan J, Fossella J, Sommer T, et al. Mapping the genetic variation of executive attention onto brain activity. Proc Natl Acad Sci U S A 2003;100:7406-11. [DOI] [PMC free article] [PubMed]
- 27.Driessen M, Herrmann J, Stahl K, et al. Magnetic resonance imaging volumes of the hippocampus and the amygdala in women with borderline personality disorder and early traumatization. Arch Gen Psychiatry 2000;57:1115-22. [DOI] [PubMed]
- 28.Vyas A, Mitra R, Shankaranarayana Rao BS, et al. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 2002;22:6810-8. [DOI] [PMC free article] [PubMed]
- 29.Rusch N, van Elst LT, Ludaescher P, et al. A voxel-based morphometric MRI study in female patients with borderline personality disorder. Neuroimage 2003;20:385-92. [DOI] [PubMed]
- 30.Birbaumer N, Veit R, Lotze M, et al. Deficient fear conditioning in psychopathy: a functional magnetic resonance imaging study. Arch Gen Psychiatry 2005;62:799-805. [DOI] [PubMed]
- 31.Lange C, Kracht L, Herholz K, et al. Reduced glucose metabolism in temporo-parietal cortices of women with borderline personality disorder. Psychiatry Res 2005;139:115-26. [DOI] [PubMed]
- 32.Lobaugh NJ, Gibson E, Taylor MJ. Children recruit distinct neural systems for implicit emotional face processing. Neuroreport 2006;17:215-9. [DOI] [PubMed]
- 33.Winters BD, Bussey TJ. Glutamate receptors in perirhinal cortex mediate encoding, retrieval, and consolidation of object recognition memory. J Neurosci 2005;25:4243-51. [DOI] [PMC free article] [PubMed]
- 34.Rizzolatti G, Fadiga L. Grasping objects and grasping action meanings: the dual role of monkey rostroventral premotor cortex (area F5). [discussion 95-103]. Novartis Found Symp 1998;218:81-95. [DOI] [PubMed]
- 35.Iacoboni M, Molnar-Szakacs I, Gallese V, et al. G. Grasping the intentions of others with one's own mirror neuron system. PLoS Biol 2005;3:e79. [DOI] [PMC free article] [PubMed]
- 36.Tsukada H, Nishiyama S, Fukumoto D, et al. Chronic NMDA antagonism impairs working memory, decreases extracellular dopamine, and increases D1 receptor binding in prefrontal cortex of conscious monkeys. Neuropsychopharmacology 2005;30:1861-9. [DOI] [PubMed]
- 37.Homayoun H, Stefani MR, Adams BW, et al. Functional Interaction Between NMDA and mGlu5 Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharmacology 2004;29:1259-69. [DOI] [PubMed]
- 38.Ellis KA, Nathan PJ. The pharmacology of human working memory. Int J Neuropsychopharmacol 2001;4:299-313. [DOI] [PubMed]
- 39.Stefani MR, Moghaddam B. Transient N-methyl-D-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry 2005;57:433-6. [DOI] [PubMed]
- 40.Rumpel S, LeDoux J, Zador A, et al. Postsynaptic receptor trafficking underlying a form of associative learning. Science 2005;308:83-8. [DOI] [PubMed]
- 41.Lanius RA, Williamson PC, Bluhm RL, et al. Functional connectivity of dissociative responses in posttraumatic stress disorder: a functional magnetic resonance imaging investigation. Biol Psychiatry 2005;57:873-84. [DOI] [PubMed]
- 42.Lahti AC, Holcomb HH, Medoff DR, et al. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 1995;6:869-72. [DOI] [PubMed]
- 43.Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience 2000;101:243-76. [DOI] [PubMed]
- 44.Chambers RA, Bremner JD, Moghaddam B, et al. Glutamate and post-traumatic stress disorder: toward a psychobiology of dissociation. Semin Clin Neuropsychiatry 1999;4:274-81. [DOI] [PubMed]
- 45.Frith U, Frith CD. Development and neurophysiology of mentalizing. Philos Trans R Soc Lond B Biol Sci 2003;358:459-73. [DOI] [PMC free article] [PubMed]
- 46.Kernberg OF. Psychodynamic psychotherapy of borderline patients. New York: Basic Books; 1989:210 p.
- 47.Gillespie CF, Ressler KJ. Emotional learning and glutamate: translational perspectives. CNS Spectr 2005;10:831-9. [DOI] [PMC free article] [PubMed]
- 48.Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 2004;61:1136-44. [DOI] [PubMed]
- 49.Hofmann SG, Meuret AE, Smits JA, et al. Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gen Psychiatry 2006;63:298-304. [DOI] [PubMed]
- 50.Goodman M, New A, Siever L. Trauma, genes, and the neurobiology of personality disorders. Ann N Y Acad Sci 2004;1032:104-16. [DOI] [PubMed]
- 51.Battle CL, Shea MT, Johnson DM, et al. Childhood maltreatment associated with adult personality disorders: findings from the Collaborative Longitudinal Personality Disorders Study. J Personal Disord 2004;18:193-211. [DOI] [PubMed]
- 52.Johnson JG, Cohen P, Brown J, et al. Childhood maltreatment increases risk for personality disorders during early adulthood. Arch Gen Psychiatry 1999;56:600-6. [DOI] [PubMed]
- 53.Bandelow B, Krause J, Wedekind D, et al. Early traumatic life events, parental attitudes, family history, and birth risk factors in patients with borderline personality disorder and healthy controls. Psychiatry Res 2005;134:169-79. [DOI] [PubMed]
- 54.Fonagy P, Target M, Gergely G. Attachment and borderline personality disorder. A theory and some evidence. [vii-viii.]. Psychiatr Clin North Am 2000;23:103-22. [DOI] [PubMed]
- 55.Fertuck EA, Lenzenweger MF, Clarkin JF. The association between attentional and executive controls in the expression of borderline personality disorder features: a preliminary study. Psychopathology 2005;38:75-81. [DOI] [PubMed]
- 56.Wagner AW, Linehan MM. Facial expression recognition ability among women with borderline personality disorder: implications for emotion regulation? J Personal Disord 1999;13:329-44. [DOI] [PubMed]
- 57.Camras L, Roibordy S, Hill J, et al. Maternal facial behavior and the recognition and production of emotional expressions by maltreated and nonmaltreated children. Dev Psychol 1990;26:304-12.
- 58.Blair RJ, Cipolotti L. Impaired social response reversal. A case of ‚acquired sociopathy'. Brain 2000;123:1122-41. [DOI] [PubMed]
- 59.Fonagy P. Attachment and borderline personality disorder. [discussion 1175-87]. J Am Psychoanal Assoc 2000;48:1129-46. [DOI] [PubMed]
- 60.Fonagy P, Target M. Attachment and reflective function: their role in self-organization. Dev Psychopathol 1997;9:679-700. [DOI] [PubMed]
- 61.Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 2000;57:925-35. [DOI] [PubMed]
- 62.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci 1999;22:105-22. [DOI] [PubMed]
- 63.Bredy TW, Zhang TY, Grant RJ, et al. Peripubertal environmental enrichment reverses the effects of maternal care on hippocampal development and glutamate receptor subunit expression. Eur J Neurosci 2004;20:1355-62. [DOI] [PubMed]
- 64.Burgess N, Maguire EA, O'Keefe J. The human hippocampus and spatial and episodic memory. Neuron 2002;35:625-41. [DOI] [PubMed]
- 65.Rosen JB, Donley MP. Animal studies of amygdala function in fear and uncertainty: Relevance to human research. Biol Psychol. 2006. [DOI] [PubMed]
- 66.Debiec J, Doyere V, Nader K, et al. Directly reactivated, but not indirectly reactivated, memories undergo reconsolidation in the amygdala. Proc Natl Acad Sci U S A 2006;103:3428-33. [DOI] [PMC free article] [PubMed]
- 67.Johnson LR, Farb C, Morrison JH, et al. Localization of glucocorticoid receptors at postsynaptic membranes in the lateral amygdala. Neuroscience 2005;136:289-99. [DOI] [PubMed]
- 68.Herpertz SC, Dietrich TM, Wenning B, et al. Evidence of abnormal amygdala functioning in borderline personality disorder: a functional MRI study. Biol Psychiatry 2001;50:292-8. [DOI] [PubMed]
- 69.Gallagher HL, Frith CD. Functional imaging of ‚theory of mind'. Trends Cogn Sci 2003;7:77-83. [DOI] [PubMed]
- 70.Brune M. “Theory of mind” in schizophrenia: a review of the literature. Schizophr Bull 2005;31:21-42. [DOI] [PubMed]
- 71.Bora E, Vahip S, Gonul AS, et al. A. Evidence for theory of mind deficits in euthymic patients with bipolar disorder. Acta Psychiatr Scand 2005;112:110-6. [DOI] [PubMed]
- 72.Frith U. Mind blindness and the brain in autism. Neuron 2001;32:969-79. [DOI] [PubMed]
- 73.Rowe AD, Bullock PR, Polkey CE, et al. “Theory of mind” impairments and their relationship to executive functioning following frontal lobe excisions. Brain 2001;124:600-16. [DOI] [PubMed]
- 74.Teicher MH, Dumont NL, Ito Y, et al. Childhood neglect is associated with reduced corpus callosum area. Biol Psychiatry 2004;56:80-5. [DOI] [PubMed]
- 75.Elberger AJ, Deng J. Corpus callosum and visual cortex of mice with deletion of the NMDA-NR1 receptor: I. Accelerated development of callosal projection neurons. Brain Res Dev Brain Res 2003;144:121-33. [DOI] [PubMed]
- 76.Dapretto M, Davies MS, Pfeifer JH, et al. Understanding emotions in others: mirror neuron dysfunction in children with autism spectrum disorders. Nat Neurosci 2006;9:28-30. [DOI] [PMC free article] [PubMed]
- 77.Hebb DO. The organization of behavior; a neuropsychological theory. New York: Wiley; 1949:xix, 335 p.
- 78.Bliss TV, Lomo T. Plasticity in a monosynaptic cortical pathway. J Physiol 1970;207:61P. [PubMed]
- 79.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol 1983;334:33-46. [DOI] [PMC free article] [PubMed]
- 80.LeVay S, Wiesel TN, Hubel DH. The development of ocular dominance columns in normal and visually deprived monkeys. J Comp Neurol 1980;191:1-51. [DOI] [PubMed]
- 81.Wiesel TN, Hubel DH. Single-Cell Responses in Striate Cortex of Kittens Deprived of Vision in One Eye. J Neurophysiol 1963;26:1003-17. [DOI] [PubMed]
- 82.Fox K, Daw N, Sato H, et al. Dark-rearing delays the loss of NMDA-receptor function in kitten visual cortex. Nature 1991;350:342-4. [DOI] [PubMed]
- 83.Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9:984-97, 979. [DOI] [PubMed]
- 84.Rowland LM, Astur RS, Jung RE, et al. Selective cognitive impairments associated with NMDA receptor blockade in humans. Neuropsychopharmacology 2005;30:633-9. [DOI] [PubMed]
- 85.Flohr H. An Information Processing Theory of Anesthesia. In: Baars BJ, Banks WP, Newman JB. Essential sources in the scientific study of consciousness. Cambridge, Mass.: MIT Press; 2003:901-912.
- 86.Pawlak V, Schupp BJ, Single FN, et al. Impaired synaptic scaling in mouse hippocampal neurones expressing NMDA receptors with reduced calcium permeability. J Physiol 2005;562:771-83. [DOI] [PMC free article] [PubMed]
- 87.Philpot BD, Espinosa JS, Bear MF. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci 2003;23:5583-8. [DOI] [PMC free article] [PubMed]
- 88.Kawahara H, Kawahara Y, Westerink BH. The role of afferents to the locus coeruleus in the handling stress-induced increase in the release of noradrenaline in the medial prefrontal cortex: a dual-probe microdialysis study in the rat brain. Eur J Pharmacol 2000;387:279-86. [DOI] [PubMed]
- 89.Nihei MK, Desmond NL, McGlothan JL, et al. N-methyl-D-aspartate receptor subunit changes are associated with lead-induced deficits of long-term potentiation and spatial learning. Neuroscience 2000;99:233-42. [DOI] [PubMed]
- 90.Coyle JT, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl) 2004;174:32-8. [DOI] [PubMed]
- 91.Lee FJ, Liu F. Direct interactions between NMDA and D1 receptors: a tale of tails. Biochem Soc Trans 2004;32:1032-6. [DOI] [PubMed]
- 92.Ladd CO, Huot RL, Thrivikraman KV, et al. Long-term adaptations in glucocorticoid receptor and mineralocorticoid receptor mRNA and negative feedback on the hypothalamo-pituitary-adrenal axis following neonatal maternal separation. Biol Psychiatry 2004;55:367-75. [DOI] [PubMed]
- 93.Mirescu C, Gould E. Stress and adult neurogenesis. Hippocampus 2006;16:233-8. [DOI] [PubMed]
- 94.Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 1992;267:1244-52. [PubMed]
- 95.Bonne O, Grillon C, Vythilingam M, et al. Adaptive and maladaptive psychobiological responses to severe psychological stress: implications for the discovery of novel pharmacotherapy. Neurosci Biobehav Rev 2004;28:65-94. [DOI] [PubMed]
- 96.McMahon SD, Grant KE, Compas BE, et al. Stress and psychopathology in children and adolescents: is there evidence of specificity? J Child Psychol Psychiatry 2003;44:107-33. [DOI] [PubMed]
- 97.Lee HJ, Choi JS, Brown TH, et al. Amygdalar nmda receptors are critical for the expression of multiple conditioned fear responses. J Neurosci 2001;21:4116-24. [DOI] [PMC free article] [PubMed]
- 98.Farb C, Aoki C, Milner T, et al. Glutamate immunoreactive terminals in the lateral amygdaloid nucleus: a possible substrate for emotional memory. Brain Res 1992;593:145-58. [DOI] [PubMed]
- 99.Minor TR, Hunter AM. Stressor controllability and learned helplessness research in the United States: sensitization and fatigue processes. Integr Physiol Behav Sci 2002;37:44-58. [DOI] [PubMed]
- 100.Bredy TW, Grant RJ, Champagne DL, et al. MJ. Maternal care influences neuronal survival in the hippocampus of the rat. Eur J Neurosci 2003;18:2903-9. [DOI] [PubMed]
- 101.De Bellis MD, Hall J, Boring AM, et al. A pilot longitudinal study of hippocampal volumes in pediatric maltreatment-related posttraumatic stress disorder. Biol Psychiatry 2001;50:305-9. [DOI] [PubMed]
- 102.Lehmann K, Butz M, Teuchert-Noodt G. Offer and demand: proliferation and survival of neurons in the dentate gyrus. Eur J Neurosci 2005;21:3205-16. [DOI] [PubMed]
- 103.Schore AN. Affect dysregulation & disorders of the self. 1st ed. New York: W.W. Norton; 2003:266-306.
- 104.Kling A, Steklis HD. A neural substrate for affiliative behavior in nonhuman primates. Brain Behav Evol 1976;13:216-38. [DOI] [PubMed]
- 105.Baron-Cohen S. Mindblindness: an essay on autism and theory of mind. Cambridge, Mass.: MIT Press; 1995:171.
- 106.Barbas H. Anatomic basis of cognitive-emotional interactions in the primate prefrontal cortex. Neurosci Biobehav Rev 1995;19:499-510. [DOI] [PubMed]
- 107.Bohn I, Giertler C, Hauber W. NMDA receptors in the rat orbital prefrontal cortex are involved in guidance of instrumental behaviour under reversal conditions. Cereb Cortex 2003;13:968-76. [DOI] [PubMed]
- 108.Palencia CA, Ragozzino ME. The influence of NMDA receptors in the dorsomedial striatum on response reversal learning. Neurobiol Learn Mem 2004;82:81-9. [DOI] [PubMed]
- 109.Brunner R, Parzer P, Schmitt R, et al. Dissociative symptoms in schizophrenia: a comparative analysis of patients with borderline personality disorder and healthy controls. Psychopathology 2004;37:281-4. [DOI] [PubMed]
- 110.Paris J. Understanding self-mutilation in borderline personality disorder. Harv Rev Psychiatry 2005;13:179-85. [DOI] [PubMed]
- 111.Schmahl C, Greffrath W, Baumgartner U, et al. Differential nociceptive deficits in patients with borderline personality disorder and self-injurious behavior: laser-evoked potentials, spatial discrimination of noxious stimuli, and pain ratings. Pain 2004;110:470-9. [DOI] [PubMed]
- 112.Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology 2002;23:659-68. [DOI] [PubMed]
- 113.Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 1994;51:199-214. [DOI] [PubMed]
- 114.Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol 2002;42:165-79. [DOI] [PubMed]
- 115.Holcomb HH, Lahti AC, Medoff DR, et al. Sequential regional cerebral blood flow brain scans using PET with H2(15)O demonstrate ketamine actions in CNS dynamically. Neuropsychopharmacology 2001;25:165-72. [DOI] [PubMed]
- 116.Carlsson ML. Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate - serotonin interactions for pharmacotherapy. J Neural Transm 1998;105:525-35. [DOI] [PubMed]
- 117.Waterhouse L, Fein D, Modahl C. Neurofunctional mechanisms in autism. Psychol Rev 1996;103:457-89. [DOI] [PubMed]
- 118.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex traits: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 2003;60:1187-92. [DOI] [PubMed]
- 119.Sawamoto N, Honda M, Okada T, et al. Expectation of pain enhances responses to nonpainful somatosensory stimulation in the anterior cingulate cortex and parietal operculum/posterior insula: an event-related functional magnetic resonance imaging study. J Neurosci 2000;20:7438-45. [DOI] [PMC free article] [PubMed]
- 120.Wu MF, Pang ZP, Zhuo M, et al. Prolonged membrane potential depolarization in cingulate pyramidal cells after digit amputation in adult rats. Mol Pain 2005;1:23. [DOI] [PMC free article] [PubMed]
- 121.Inturrisi CE. The role of N-methyl-D-aspartate (NMDA) receptors in pain and morphine tolerance. Minerva Anestesiol 2005;71:401-3. [PubMed]
- 122.Etkin A, Pittenger C, Polan HJ, et al. Toward a neurobiology of psychotherapy: basic science and clinical applications. J Neuropsychiatry Clin Neurosci 2005;17:145-58. [DOI] [PubMed]
- 123.Grosjean B. From synapse to psychotherapy: the fascinating evolution of neuroscience. Am J Psychother 2005;59:181-97. [DOI] [PubMed]
- 124.Pascual-Leone A, Amedi A, Fregni F, et al. The plastic human brain cortex. Annu Rev Neurosci 2005;28:377-401. [DOI] [PubMed]
- 125.Matsuoka N, Aigner TG. D-cycloserine, a partial agonist at the glycine site coupled to N-methyl-D-aspartate receptors, improves visual recognition memory in rhesus monkeys. J Pharmacol Exp Ther 1996;278:891-7. [PubMed]
- 126.Monahan JB, Handelmann GE, Hood WF, et al. D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol Biochem Behav 1989;34:649-53. [DOI] [PubMed]
- 127.Heresco-Levy U, Javitt DC, Ermilov M, et al. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry 1999;56:29-36. [DOI] [PubMed]
- 128.Goff DC, Herz L, Posever T, et al. A six-month, placebo-controlled trial of D-cycloserine co-administered with conventional antipsychotics in schizophrenia patients. Psychopharmacology (Berl) 2005;179:144-50. [DOI] [PubMed]
- 129.Tsai G, Yang P, Chung LC, et al. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 1998;44:1081-9. [DOI] [PubMed]
- 130.Tsai G, Lane HY, Yang P, et al. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 2004;55:452-6. [DOI] [PubMed]
- 131.Tsai GE, Falk WE, Gunther J, et al. Improved cognition in Alzheimer's disease with short-term D-cycloserine treatment. Am J Psychiatry 1999;156:467-9. [DOI] [PubMed]
- 132.Heresco-Levy U, Kremer I, Javitt DC, et al. Pilot-controlled trial of D-cycloserine for the treatment of post-traumatic stress disorder. Int J Neuropsychopharmacol 2002;5:301-7. [DOI] [PubMed]