

Abstract
Among the earliest and most important stages during tumorigenesis is the activation of the angiogenic process, an event that is termed the “switch to the angiogenic phenotype.” We have developed an in vivo system that can reliably recapitulate the stages in tumor development that represent this transition. Using this model, we have harvested and studied tumor nodules that can be distinguished from each other on the basis of their degree of vascularization. Angiogenic tumor nodules were characterized by the presence of capillary vessels as determined by factor VIII immunohistochemistry, and both angiogenic and proteolytic activities in vitro. In contrast, preangiogenic nodules were devoid of microvessels and showed little angiogenic or proteolytic activity in vitro. Addition of a specific metalloproteinase inhibitor resulted in the abrogation of both angiogenic and proteolytic activities of the angiogenic nodules in vitro. Comparative substrate gel electrophoresis detected the presence of a prominent matrix metalloproteinase (MMP-2) in the angiogenic nodules when compared with the preangiogenic ones. Suppression of MMP-2 activity by antisense oligonucleotides in the vascular nodules resulted in the loss of angiogenic potential both in vitro and in vivo in the chick chorioallantoic membrane assay. Moreover, this suppression of MMP-2 activity in angiogenic nodules inhibited tumor growth in vivo by approximately 70%. These results strongly implicate the activity of MMP-2 as a requirement for the switch to the angiogenic phenotype and validate this model as a reliable and reproducible tool by which to study other cellular and biochemical factors involved in the acquisition of the angiogenic phenotype.
One of the earliest events during the transition of a tumor from the preneoplastic to the tumorigenic phenotype is the acquisition of the angiogenic phenotype. This transition is marked by changes in the remodeling of both the pericapillary membrane and of its surrounding extracellular matrix (ECM), by endothelial cell proliferation, and by capillary tube formation (1). It is now widely appreciated that a major rate-limiting step in ECM remodeling is the activity of matrix metalloproteinases (MMPs). This multigene family of metal-dependent ectoenzymes includes more than 25 members to date, generally organized into five broad categories originally based on their substrate specificity (2).
Much research attention has been focused on the role of MMPs in tumor invasion and metastasis (3–5); however, relatively little is known about the functional consequences of MMP expression during the onset of the angiogenic phenotype during tumorigenesis. Our laboratory and those of others have shown that certain endogenous MMP inhibitors are potent inhibitors of angiogenesis both in vivo and in vitro (6–10). Synthetic MMP inhibitors recently have been shown to inhibit neovascularization as well (11–13), and currently, a number of these synthetic inhibitors are in various stages of clinical testing as inhibitors of angiogenesis and as anticancer agents.
There is a convincing body of evidence that demonstrates that the angiogenic switch is regulated by the net balance between positive and negative regulators of new capillary growth, formally termed the “balance hypothesis for the angiogenic switch” (1, 14). However, no specific molecule has been identified as a key modulator of the switch to the angiogenic phenotype during early tumorigenesis in vivo. Studies of this type have been limited by the lack of reproducible in vivo models that can reliably recapitulate the stages comprising the transition to the angiogenic phenotype. Given that MMP activity is one of the earliest and sustained activities required for successful neovascularization (15, 16), and because many inhibitors of MMP activity are themselves angiogenesis inhibitors, we hypothesized that a shift in the net proteolytic balance between MMPs and their inhibitors, in favor of MMP inhibition, would result in the suppression of the angiogenic phenotype and subsequent tumor growth. To test this hypothesis, we have developed an in vivo model of chondrosarcoma development that provided us with the opportunity to harvest and test chondrosarcoma nodules that represent both the preangiogenic and angiogenic phenotype. Based on this model, we report data that support a key role for the activity of a specific MMP, MMP-2, in the setting of the angiogenic switch.
Materials and Methods
Animal Model.
We have modified a previously reported method by Folkman and coworkers for the study of adenocarcinoma (17). Swarm rat chondrosarcoma was maintained s.c. on both hips of Sprague–Dawley male rats (100–125 g) (Charles River Breeding Laboratories) as described (18, 19). Chondrosarcoma cell suspensions were prepared by suspending finely minced tumor in Ringer's lactate solution, consecutively passing it through a steel sieve and through syringes containing 20- to 30-gauge needles. This preparation (6 × 105 cells in 1 ml) was then injected into an air sac created by s.c. injection of 25 ml of air into in the sacral region of the backs of male Sprague–Dawley rats (approximately 100 g). Chondrosarcoma nodules were harvested after approximately 10–12 days. Avascular and vascular nodules were selected based on visual and dissecting microscopic observations.
Histology and Immunohistochemistry.
For paraffin sections, tissues were fixed overnight in Carnoy's fixative and processed by standard histological procedures. Alternatively, tissues used for cryostat sections were fixed in 10% buffered formalin and sectioned. The presence of microvessels in tissue samples was detected by staining endothelial cells for factor VIII (Dako) as described (20).
Collagen Gel Assay.
Bovine capillary endothelial cells (EC) were the kind gift of Judah Folkman (Children's Hospital, Harvard Medical School). EC were maintained in DMEM (JRH Biosciences, Lenexa, KS) supplemented with 10% calf serum (HyClone), 1% glutamine penicillin streptomycin (Irvine Scientific, Santa Ana, CA) (DMEM/10), and 3 ng/ml basic fibroblast growth factor (Scios Nova, Mountain View, CA). This collagen gel assay is a modification of that of Folkman and coworkers (21). Briefly, capillary EC were treated with trypsin, and 4 × 105 cells were resuspended in 1 ml of DMEM/10 and mixed at a ratio of 1:1 with chilled Vitrogen 100 (Collagen Corp.), which had been neutralized with 0.1 M sodium hydroxide and 1/10 volume of 10× MEM (GIBCO/BRL). Aliquots (250 μl) of the vitrogen-EC mixture were loaded into each well of 48-well tissue culture plates (Costar). Single chondrosarcoma nodules were added to each well before the solution was allowed to polymerize at 37°C for 60 min, then 250 μl of complete DMEM was added to each well, and the plate was incubated in a 10% CO2 incubator. Where specified, EC were labeled with 4 μg/ml rhodamine-conjugated DiI-acetylated low density lipoprotein (Biomedical Technologies, Stoughton, MA) for 24 h before use in the collagen gel assay.
Capillary EC Proliferation Assay.
Capillary EC proliferation was measured as previously reported by us (6, 22) by using a modification of the method of Connolly and coworkers (23). Results were verified by cell counting assays using a coulter counter (6, 20, 22).
Substrate Gel Electrophoresis.
Tissue extracts were prepared as described (24). Protein concentration was determined by using a Bio-Rad protein assay. MMP activity was detected by using gelatin zymography (25, 26). To verify that the gelatinase activities detected were, in fact, MMP activities, samples were electrophoresed and the gels were then incubated in substrate buffer in the presence or absence of 1,10-phenanthroline (10 mM) (Sigma), an MMP inhibitor. To distinguish latent from active forms of MMPs, samples were exposed to 4-aminophenylmercuric acetate (1 mM) (Sigma), a mercurial agent that has been shown to process MMPs from their latent to their active forms (25).
Immunoblot Analysis.
Tissue extracts were separated by electrophoresis on a 12% SDS/PAGE gel and transferred electrophoretically to nitrocellulose membranes (24, 26). Blots were blocked with 5% nonfat dry milk and incubated with mouse anti-MMP-2 antibodies (Calbiochem) and horseradish peroxidase-conjugated goat anti-rabbit IgG. Protein bands were visualized by using the ECL system (Amersham Pharmacia).
Reverse Transcription–PCR (RT-PCR).
Total RNA was extracted from nodules by using a Qiagen RNA kit (Qiagen, Chatsworth, CA). RT-PCRs were performed and optimized, according to standard protocols. PCR primers for MMP-2 were: forward, 5′-ATCTGGTGTCTCCCTTACGG and reverse, 5′-GTGCAGTGATGTCCGACAAC.
Antisense Oligonucleotide Treatment.
Rat MMP-2 antisense, control, and FITC-labeled phosphorothioate DNA oligonucleotides were designed and synthesized by Biognostik, Göttingen, Germany. The sequence of the MMP-2 specific antisense oligonucleotides was 5′-CACACCTTGCCATCG and that of control oligonucleotides was 5′-CGTCCCTATACGACC. To verify uptake of oligonucleotides by tumor cells, nodules were incubated with FITC-labeled oligonucleotides in 10% FBS MEM with 1% glutamine penicillin streptomycin for 12 h. Nodules were then fixed in methanol, sectioned by using a cryostat, and examined under a fluorescent microscope. To neutralize MMP-2 expression, vascularized (red) tumor nodules were dissected and incubated in 10% FBS MEM containing either antisense or control oligonucleotides in polypropylene tubes for 3 days. Both culture media and oligonucleotides were replaced daily.
Chick Chorioallantoic Membrane (CAM) Assay.
The chick CAM assay for angiogenesis was conducted as reported (8, 22). On day 6, test tumor nodules were placed on the surface of CAM just above the subectodermal plexus. Embryos were examined for blood vessel growth after 48 and 72 h under a dissecting microscope.
Tumor Growth In Vivo.
Male Sprague–Dawley rats (100 g) were anesthetized, and an incision of 0.5 cm was made along the back midline. Tumor nodules were implanted s.c. into both sides at a site approximately 1.5 cm away from the midline incision. The incisions were sewn and animals were returned to cages. Animals were killed after 3 weeks and tumors were dissected and weighed.
Results
Animal Model for the Study of the Development of the Angiogenic Phenotype.
To study the biochemical and molecular mechanisms underlying the switch to the angiogenic phenotype, we have developed an in vivo model based on the development of Swarm rat chondrosarcoma. After approximately 10–12 days, injection of chondrosarcoma cell suspensions into air sacs in the sacral region of Sprague–Dawley rats resulted in the formation of distinct three-dimensional nodules that could be visually distinguished from each other on the basis of their degree of vascularization, i.e., avascular (white) from vascular (red) (Fig. 1 A and B). This model permitted the harvesting of multiple tumor nodules of each type for further study.
Figure 1.

Distinctive avascular and vascular phenotypes of chondrosarcoma tumor nodules. (A) Injection of tumor cell suspensions into air sacs in the sacral region of rats resulted in the formation of distinct tumor nodules, which could be visually distinguished from each other on the basis of their degree of vascularization i.e., vascular (white arrows) from avascular (black arrows). At day 10, approximately 50% of the tumor nodules were vascularized and by day 12, the majority of the nodules were vascularized. These results were consistently observed throughout more than 15 independent experiments. (B) Representative avascular and vascular nodules, respectively, are shown. Factor VIII immunostaining demonstrates the avascularity of representative control xiphoid cartilage sections (C) in which microvessels are not detected, in contrast to sections of chondrosarcoma in which a number of microvessels are easily seen (D). A lack of microvessels is observed in avascular chondrosarcoma nodules (E) when compared with vascular nodules in which positive microvessel staining is apparent (F). When avascular tumor nodules (G) were dissected from air sacs and implanted s.c. in rats, the majority of avascular nodules (11/13) became vascularized tumors (H). (Scale bars: A and B, 2 mm; C–F, 100 μm; G, 2 mm; H, 10 mm.)
To confirm that this visual distinction was in fact caused by bona fide differences in vascularization, we used a number of experimental approaches. First, factor VIII immunohistochemistry was conducted on the sections of normal rat cartilage tissue and whole chondrosarcoma tissue, as well as on avascular and vascular tumor nodules. As demonstrated in Fig. 1C, normal cartilage is avascular as evidenced by a striking lack of microvessels. In contrast, chondrosarcoma is vascularized as determined by the appearance of numerous microvessels after factor VIII staining (Fig. 1D). With respect to chondrosarcoma nodules, factor VIII staining showed that the “white” nodules were characterized by the absence of microvessels (Fig. 1E) and the “red” nodules were vascular, as evidenced by the appearance of microvessels (Fig. 1F).
When avascular nodules (n = 13) were transplanted s.c. into rats, the majority of them eventually became vascularized (n = 11), developing into tumors with an average weight of 4.4 g after 4 weeks (Fig. 1 G and H). When vascular nodules (n = 6) were transplanted, most of them (n = 5) gave rise to larger tumors with an average weight of 7.3 g after 4 weeks.
Different Angiogenic Phenotypes of Tumor Nodules in Vitro.
Having determined that these white and red nodules represent distinct vascular stages in the transition to the angiogenic phenotype, the angiogenic activity of these nodules was then determined in two different bioassays that represent different angiogenic processes. In the first series of studies, we used an in vitro three-dimensional collagen gel assay for angiogenic activity (21, 27). As depicted in Fig. 2A, the vascular nodules implanted in collagen gels in which EC were dispersed, induced an extensive capillary EC concentration around the red nodule, in contrast to the avascular ones (Fig. 2B). As the EC localized around the angiogenic nodule, the appearance of a gradient of EC became apparent. This difference in angiogenic potential between avascular and vascular chondrosarcoma nodules was consistently observed in the course of study of more than 100 nodules of each type, obtained from five separate experiments. These results are consistent with others in the literature that attribute this distinctive EC behavior to the angiogenic activity of the sample being tested (21).
Figure 2.

In vitro angiogenesis assay of vascular and avascular chondrosarcoma tumor nodules. Vascular tumor nodules (TN), implanted in collagen gels throughout which capillary EC were dispersed, induced the concentration and alignment of capillary EC around the nodule (A) by day 3 in culture, in contrast to the lack of angiogenic response to avascular tumor nodules (B). When capillary EC as in A are labeled with fluoresceinated low density lipoprotein, the corona of EC around the vascular nodules is highlighted (C). To show the position of the vascular tumor nodule that is not fluorescent, the nodule in C is shown as a superimposed image taken with bright-field illumination. (Scale bar: A–C, 150 μm.)
To verify that the cells surrounding the chondrosarcoma nodules were, in fact, the capillary EC that had been mixed into the collagen, we repeated these experiments after labeling the EC with fluoresceinated low density lipoprotein (LDL), a specific EC marker (28). As depicted in Fig. 2C, after approximately 6 days in culture, the capillary EC had concentrated around the vascular nodules such that one could detect an increase concentration of LDL-labeled EC surrounding the vascular nodule. Therefore, the avascular and vascular nodules from chondrosarcoma represent distinct, bona fide stages in the vascularization of this tumor (preangiogenic and angiogenic) as determined by visual appearance, factor VIII immunohistochemistry, and angiogenic activity in vitro.
In the second bioassay, we tested avascular and vascular tumor nodules for their effects on capillary EC proliferation. When equal amounts of protein extracted from these nodules were tested, the vascular nodules were nearly two times as stimulatory as the avascular nodules (Fig. 3).
Figure 3.

Comparison of EC mitogenic activity between avascular and vascular tumor nodules. Protein was extracted from nodules, dialyzed against PBS, and added (10 μg/well) to each well of capillary EC. Stimulation of EC proliferation by nodule extracts was expressed in comparison to maximal stimulation (100%) by basic fibroblast growth factor (1 ng/ml, 0.4 ng/well), which resulted in a 4.2-fold increase in proliferation. All assays were done in duplicates. This result is representative of three independent experiments.
Vascular Tumor Nodules Are Characterized by Increased Proteolysis.
In addition to the differences in vascularization described above, it was consistently observed that in the case of the vascular nodules, the surrounding collagen gels were significantly degraded, in contrast to the collagen gels surrounding their avascular counterparts (Fig. 4 A and B). This observation suggested that vascular nodules might contain greater proteolytic activity than avascular nodules and that perhaps one important characteristic of the angiogenic phenotype in this model was increased proteolytic activity. Given that MMPs can degrade collagen, we asked whether the collagen gel could be rescued from degradation by the addition of the MMP inhibitor, 1,10-phenanthroline. Degradation of the gel was completely inhibited at 5 μM (Fig. 4C). This finding suggested that vascular and avascular nodules might be characterized by significantly different MMP activities.
Figure 4.
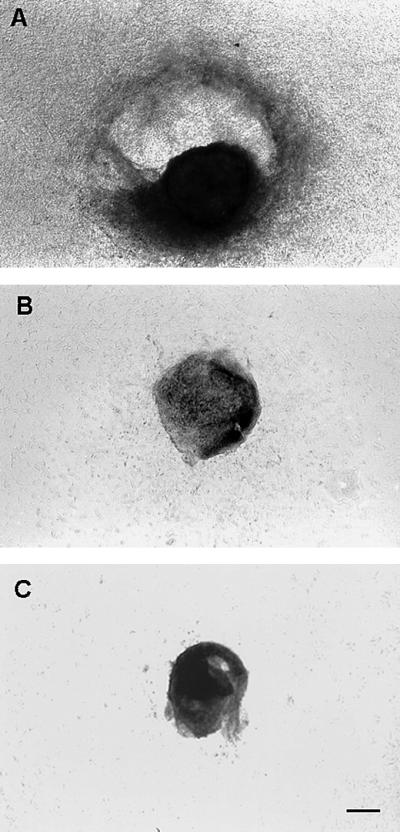
Comparison of proteolytic activity between vascular and avascular tumor nodules in vitro. By day 6 in culture, significant degradation of the collagen gel surrounding the vascular nodules was apparent (A), in contrast to gels surrounding the avascular nodules that remained intact (B). When 1,10-phenanthroline (5 μM) was added to the media overlaying the collagen gels in which vascular nodules were implanted, collagen gel degradation was completely suppressed (C). This result is representative of two independent assays (n = 7 for each treatment group). (Scale bar: A–C, 400 μm.)
To identify which MMPs might account for the differences between vascular and avascular nodules, we first compared MMP activity in vascular chondrosarcoma tumor versus avascular xiphoid cartilage extracts by zymography. Equivalent amounts of protein were tested in all cases. Results demonstrated that the predominant MMP activity in chondrosarcoma migrates at the relative molecular mass of approximately 65 kDa (Fig. 5A). Treatment of the samples with 4-aminophenylmercuric acetate did not result in a decrease in the molecular mass of either this 65-kDa proteolytic activity or that of a less intense 55-kDa species, suggesting that they represent the active form of MMP-2 (Fig. 5C) (25). The identity of these proteolytic activities as being MMP-2 was verified by immunoblot in which both bands were recognized by MMP-2-specific antibodies (Fig. 5D).
Figure 5.

MMP-2 activity is increased in chondrosarcoma tissue in comparison to cartilage control. Zymography revealed predominant proteolytic bands migrating at molecular weights consistent with their identification as MMP-2 species. (A) Levels of this proteolytic activity were greater in chondrosarcoma (CHSA) than in the xiphoid cartilage controls. (B) Densitometric analysis documents an approximate 3-fold increase in intensity in chondrosarcoma as compared with the cartilage control. (C) Treatment of tumor extracts with 4-aminophenylmercuric acetate (APMA) showed no difference in the molecular mass of the enzyme species, suggesting that they are both present in their active forms. (D) Identification of enzyme activities as being those of MMP-2 was verified by immunoblot analysis using monospecific MMP-2 antibodies. Human recombinant MMP-2 (rMMP2) was included as a positive control.
Scanning densitometric analysis demonstrated that the MMP-2 levels in chondrosarcoma were approximately three times greater than that in its avascular control, the xiphoid cartilage (Fig. 5B). Next, we compared MMP-2 activity levels in vascular versus avascular chondrosarcoma nodules. Consistent with the results obtained from analysis of vascular chondrosarcoma tumor and avascular xiphoid cartilage above, we detected the same two bands of proteolytic activity detected above, with the proteolytic levels in the vascular nodules being three times greater than those of their avascular nodule counterparts (Fig. 6 A and B). These results demonstrate that MMP-2 activity is significantly up-regulated in the vascular versus the avascular chondrosarcoma nodules.
Figure 6.

MMP-2 activity is up-regulated in vascular tumor nodules. (A and B) Zymographic analysis revealed that the MMP-2 levels were three times greater in vascular tumor nodules than in avascular nodules. RT-PCR analysis (C) demonstrated a significantly higher MMP-2 expression level in vascular nodules (V) when compared with avascular nodules (A). β-actin served as an internal control.
This finding was verified by using RT-PCR. When total RNA was extracted from white and red nodules and subjected to RT-PCR using β-actin as an internal control, it was shown that vascular chondrosarcoma nodules expressed more MMP-2 mRNA than their avascular counterparts (Fig. 6C).
Angiogenesis Is Inhibited by an MMP Inhibitor and MMP-2 Antisense in Vitro.
To determine whether an increase in the activity of MMP contributes to the process of angiogenesis during tumorigenesis in our model, we tested the effects of 1,10-phenanthroline on the interaction between the tumor nodules and the capillary EC in the collagen gel assay. The angiogenic response of capillary EC observed in the wells containing vascular tumor nodules was completely inhibited at a concentration of 5 μM 1,10-phenanthroline with no obvious cytotoxicity (data not shown).
Because zymography showed that MMP-2 was the major MMP activity detected in chondrosarcoma nodules, we next tested whether specifically blocking MMP-2 expression by antisense oligonucleotides could affect the interaction between EC and the tumor nodules in the collagen gel assay. We first demonstrated that uptake of FITC-labeled antisense-MMP-2 could be efficiently accomplished by chondrosarcoma cells in vascular tumor nodules within 12 h (Fig. 7A). To test whether specific antisense actually suppressed MMP-2 expression, the conditioned media of vascular nodules incubated in the presence of 10 μM MMP-2 antisense or control random oligonucleotides were analyzed by zymography. MMP-2 antisense treatment resulted in a significant decrease of the MMP-2 activity levels in conditioned medium compared with that of the controls (Fig. 7 B and C).
Figure 7.

Inhibition of MMP-2 expression and angiogenesis by antisense oligonucleotides in vitro. (A) Uptake of oligonucleotides by tumor cells in vitro was demonstrated by cellular localization of FITC-labeled oligonucleotides in vascular tumor nodules. (B and C) Treatment of vascular nodules with MMP-2 antisense oligonucleotides resulted in a 70% reduction in MMP-2 activity in the conditioned media in comparison to that of nodules treated with control oligonucleotides. Capillary EC responded to tumor nodules in the presence of control oligonucleotides (D), but did not in the presence of 10 μM MMP-2 antisense oligonucleotides (E). (Scale bars: A, 250 μm; D and E, 200 μm.)
When vascular nodules were cultured in the presence of 10 μM MMP-2 antisense in the collagen gel assay by using random oligonucleotides as the control, we detected a suppressed angiogenic response to the vascular nodule as characterized by a significantly reduced number of capillary EC around tumor nodules (Fig. 7 D and E).
MMP-2 Antisense Oligonucleotides Inhibit Angiogenesis in Vivo.
Given that MMP-2 antisense oligonucleotides were taken up efficiently by chondrosarcoma cells, resulting in both suppressed MMP-2 activity in tumor nodules and inhibition of angiogenesis in vitro, we next asked whether the inhibition of MMP-2 by antisense treatment could regulate the angiogenic process in vivo by using the chick CAM assay. When vascular nodules pretreated with MMP-2 antisense oligonucleotides (10 μM) were tested on the CAM, all nodules tested showed inhibition of angiogenesis in the area surrounding the nodules (Fig. 8A). In contrast, vascular nodules treated with random oligonucleotides stimulated capillary growth in this same assay (Fig. 8B) in comparison to control (untreated) CAMs (Fig. 8C).
Figure 8.

Suppression of angiogenesis and tumor growth by MMP-2 antisense in vivo. Vascular tumor nodules preincubated with 10 μM oligonucleotides inhibited the normal embryonic vasculature of the chick CAM (5/5) (A), in contrast to the control nodules that were pretreated with random oligonucleotides and that stimulated capillary growth (4/5) (B), and compared with untreated CAM control (C). Vascular tumor nodules that were preincubated with 10 μM MMP-2 antisense for 3 days and were implanted into animals (n = 6) resulted in 69% suppression of tumor growth in comparison to control nodules treated with random oligonucleotides (P < 0.05).
Suppression of Chondrosarcoma Growth by MMP-2 Antisense Oligonucleotides.
Given that the suppression of MMP-2 in vascular chondrosarcoma nodules resulted in a suppression of angiogenesis in vivo and in vitro, we next asked whether these nodules, in which MMP-2 was suppressed, exhibited any difference in their tumorigenic potential. When antisense-treated vascular nodules were implanted s.c. in rats and compared with nodules treated with random oligo controls implanted in the same animal on the opposite side, a suppression in tumor growth of 69% was detected in the antisense-treated group versus the control oligo group (Fig. 8D). Statistical analysis (Student's t test) showed a significant difference between two groups (P < 0.05).
Discussion
We report here the development of an in vivo model which reproducibly and predictably represents the switch to the angiogenic phenotype during tumorigenesis. This experimental tumor model was validated by comparative analyses of tumor nodules in four different in vitro and in vivo angiogenesis assays. The fact that each of these assays could independently distinguish between the preangiogenic and angiogenic phenotypes of these tumor nodules strongly supports this model as a useful one for the study of the development of neovascularization during tumorigenesis. In addition, this system may accurately mimic the transition to the angiogenic phenotype in human tumors because chondrosarcoma is a spontaneously occurring and naturally progressive cancer, as opposed to one that is the product of genetic or carcinogenic manipulation. Although a number of angiogenesis-related molecules have been suggested to play a role in the acquisition of the vascular phenotype (29, 30), the identification and characterization of the in vivo role of factors required for the initiation and subsequent development of the angiogenic phenotype has been limited by a lack of useful animal models through which one can reliably obtain tumor tissue that is representative of prevascular and vascular tumor stages.
The activity of MMPs has long been viewed as a rate-limiting step in the ECM degradation associated with normal and pathological events such as wound healing and tumor metastasis. Although the earliest description of the process of angiogenesis highlighted the importance of ECM degradation as being one of the earliest and sustained events in the process of new capillary formation (31), the important roles of MMPs and their endogenous inhibitors in the regulation of angiogenesis has only recently begun to be appreciated.
A number of studies have revealed that microvascular endothelial cells synthesize and secrete a variety of MMPs and tissue inhibitors of metalloproteinases (TIMPs) (15). Moreover, key angiogenic mitogens such as fibroblast growth factor and vascular endothelial growth factor have been shown to modulate the production and activity of these proteins (32–35). The finding that angiogenesis could be suppressed in vivo by using endogenous inhibitors of MMP activity provided some of the first convincing evidence of the importance of this enzyme family to new capillary formation (6, 9, 10). Recently, MMP-9 has been shown to be a key regulator of growth plate neovascularization during endochondral bone formation (36). In vitro studies of a variety of angiogenesis-related EC functions were also instrumental in implicating MMPs as important regulators of neovascularization (8, 27, 32). However, whether MMPs play a role in the switch to the angiogenic phenotype during tumorigenesis in vivo has not been studied.
MMPs may be modulating neovascularization in at least two other ways, both of which are related to the bioavailability of angiogenic modulators. As a function of their ability to degrade ECM, MMPs may be playing a role in the release of angiogenic mitogens that have been shown to be stored within the matrix (37). In addition, MMPs recently have been shown to process and release a variety of molecules which are regulators of vascular growth or function, including fibroblast growth factor receptor type 1 (38), tumor necrosis factor α (39), and heparin-binding-epidermal growth factor (40).
Our data demonstrate that MMP-2 is required for the switch to the angiogenic phenotype during the development of chondrosarcoma. We have demonstrated that metalloproteinases can play a key role in that switch. Given the redundancy of this enzyme family, it is particularly striking that the suppression of only one MMP could have such a profound effect on the angiogenic phenotype of these tumor nodules. These data do not preclude the possibility that other proteins, including other MMPs, also may be playing a role in the switch to the angiogenic phenotype in other systems. Although a number of studies have shown that MMP inhibitors can inhibit tumor growth (12, 13, 41), none of these inhibitors were specific to a particular MMP, all of them exhibiting broad MMP-inhibiting activity. Here, we show that the suppression of MMP-2 alone inhibits the transition from the prevascular to the vascular stage during tumor development and subsequently inhibits tumor growth.
The important role of this specific metalloproteinase, MMP-2, in the development of the angiogenic phenotype is supported by a number of important experimental observations. For example, EC have been shown to produce MMP-2 during differentiation into capillary tube-like structures and exogenous addition of MMP-2 was shown to enhance this process (42). In a recent study of human multiple myeloma, the progression of plasma cell tumors was accompanied by an increase of bone marrow neovascularization, which, in turn, was paralleled by an increase in the angiogenic and invasive potential of bone marrow plasma cells, functions shown to depend on basic fibroblast growth factor and MMP-2 production (43).
There may be significant clinical value in understanding the factors that regulate the switch to the angiogenic phenotype and in developing methods to intervene at this very early stage of tumor progression. For example, the induction of the angiogenic phenotype now has been correlated with the progression from ductal carcinoma in situ to invasive ductal carcinoma. Quantitative assessment of microvessels revealed that the occurrence of breast cancer metastases increases with increased microvessel count (44). This study also strongly supports the proposition that one strategy for controlling the inappropriate and dysregulated capillary growth characterizing angiogenic diseases may be one that is operative at the level of the control of MMP-2 activity. The model described here may be useful not only for studying the cellular and molecular mechanisms driving the angiogenic switch, but also may serve as an experimental template for the discovery of factors that may be useful in the suppression of the angiogenic phenotype.
Acknowledgments
We gratefully acknowledge Dr. Judah Folkman and Dr. Robert Langer for helpful discussions and advice and Evelyn Flynn for help with immunohistochemistry. This work was supported by a grant from the American Cancer Society (RPG 83821) to M.A.M.
Abbreviations
- MMP
matrix metalloproteinase
- ECM
extracellular matrix
- EC
endothelial cells
- CAM
chorioallantoic membrane
- RT-PCR
reverse transcription–PCR
References
- 1.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 2.Nagase H, Woessner J F., Jr J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 3.Cockett M I, Murphy G, Birch M L, O'Connell J P, Crabbe T, Millican A T, Hart I R, Docherty A J. Biochem Soc Symp. 1998;63:295–313. [PubMed] [Google Scholar]
- 4.Kahari V M, Saarialho-Kere U. Ann Med. 1999;31:34–45. doi: 10.3109/07853899909019260. [DOI] [PubMed] [Google Scholar]
- 5.Kleiner D E, Stetler-Stevenson W G. Cancer Chemother Pharmacol. 1999;43,Suppl.:42–51. doi: 10.1007/s002800051097. [DOI] [PubMed] [Google Scholar]
- 6.Moses M A, Sudhalter J, Langer R. Science. 1990;248:1408–1410. doi: 10.1126/science.1694043. [DOI] [PubMed] [Google Scholar]
- 7.Moses M A, Sudhalter J, Langer R. J Cell Biol. 1992;119:475–482. doi: 10.1083/jcb.119.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy A N, Unsworth E J, Stetler-Stevenson W G. J Cell Physiol. 1993;157:351–358. doi: 10.1002/jcp.1041570219. [DOI] [PubMed] [Google Scholar]
- 9.Johnson M D, Kim H R, Chesler L, Tsao-Wu G, Bouck N, Polverini P J. J Cell Physiol. 1994;160:194–202. doi: 10.1002/jcp.1041600122. [DOI] [PubMed] [Google Scholar]
- 10.Anand-Apte B, Pepper M S, Voest E, Montesano R, Olsen B, Murphy G, Apte S S, Zetter B. Invest Ophthalmol Vis Sci. 1997;38:817–823. [PubMed] [Google Scholar]
- 11.Hodgson J. Biotechnology. 1995;13:554–557. doi: 10.1038/nbt0695-554. [DOI] [PubMed] [Google Scholar]
- 12.Taraboletti G, Garofalo A, Belotti D, Drudis T, Borsotti P, Scanziani E, Brown P D, Giavazzi R. J Natl Cancer Inst. 1995;87:293–298. doi: 10.1093/jnci/87.4.293. [DOI] [PubMed] [Google Scholar]
- 13.Lozonschi L, Sunamura M, Kobari M, Egawa S, Ding L, Matsuno S. Cancer Res. 1999;59:1252–1258. [PubMed] [Google Scholar]
- 14.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 15.Moses M A. Stem Cells. 1997;15:180–189. doi: 10.1002/stem.150180. [DOI] [PubMed] [Google Scholar]
- 16.Stetler-Stevenson W G. J Clin Invest. 1999;103:1237–1241. doi: 10.1172/JCI6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Folkman J. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 18.Shing Y, Folkman J, Sullivan R, Butterfield C, Murray J, Klagsbrun M. Science. 1984;223:1296–1299. doi: 10.1126/science.6199844. [DOI] [PubMed] [Google Scholar]
- 19.Moses M A, Shing Y. Biochem Biophys Res Commun. 1994;199:418–424. doi: 10.1006/bbrc.1994.1245. [DOI] [PubMed] [Google Scholar]
- 20.O'Reilly M S, Holmgren L, Shing Y, Chen C, Rosenthal R A, Moses M, Lane W S, Cao Y, Sage E H, Folkman J. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 21.Folkman J, Watson K, Ingber D, Hanahan D. Nature (London) 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 22.Moses M A, Wiederschain D, Wu I, Fernandez C A, Ghazizadeh V, Lane W S, Flynn E, Sytkowski A, Tao T, Langer R. Proc Natl Acad Sci USA. 1999;96:2645–2650. doi: 10.1073/pnas.96.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connolly D T, Knight M B, Harakas N K, Wittwer A J, Feder J. Anal Biochem. 1986;152:136–140. doi: 10.1016/0003-2697(86)90131-4. [DOI] [PubMed] [Google Scholar]
- 24.Peters C A, Freeman M R, Fernandez C A, Shepard J, Wiederschain D G, Moses M A. Am J Physiol. 1997;272:R1960–R1965. doi: 10.1152/ajpregu.1997.272.6.R1960. [DOI] [PubMed] [Google Scholar]
- 25.Herron G S, Banda M J, Clark E J, Gavrilovic J, Werb Z. J Biol Chem. 1986;261:2814–2818. [PubMed] [Google Scholar]
- 26.Braunhut S J, Moses M A. J Biol Chem. 1994;269:13472–13479. [PubMed] [Google Scholar]
- 27.Montesano R, Orci L. Cell. 1985;42:469–477. doi: 10.1016/0092-8674(85)90104-7. [DOI] [PubMed] [Google Scholar]
- 28.Voyta J C, Via D P, Butterfield C E, Zetter B R. J Cell Biol. 1984;99:2034–2040. doi: 10.1083/jcb.99.6.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czubayko F, Liaudet-Coopman E D, Aigner A, Tuveson A T, Berchem G J, Wellstein A. Nat Med. 1997;3:1137–1140. doi: 10.1038/nm1097-1137. [DOI] [PubMed] [Google Scholar]
- 30.Lingen M W, DiPietro L A, Solt D B, Bouck N P, Polverini P J. Carcinogenesis. 1997;18:329–338. doi: 10.1093/carcin/18.2.329. [DOI] [PubMed] [Google Scholar]
- 31.Ausprunk D H, Folkman J. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- 32.Mignatti P, Tsuboi R, Robbins E, Rifkin D B. J Cell Biol. 1989;108:671–682. doi: 10.1083/jcb.108.2.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Unemori E N, Ferrara N, Bauer E A, Amento E P. J Cell Physiol. 1992;153:557–562. doi: 10.1002/jcp.1041530317. [DOI] [PubMed] [Google Scholar]
- 34.Lamoreaux W J, Fitzgerald M E, Reiner A, Hasty K A, Charles S T. Microvasc Res. 1998;55:29–42. doi: 10.1006/mvre.1997.2056. [DOI] [PubMed] [Google Scholar]
- 35.Zucker S, Mirza H, Conner C E, Lorenz A F, Drews M H, Bahou W F, Jesty J. Int J Cancer. 1998;75:780–786. doi: 10.1002/(sici)1097-0215(19980302)75:5<780::aid-ijc19>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 36.Vu T H, Shipley J M, Bergers G, Berger J E, Helms J A, Hanahan D, Shapiro S D, Senior R M, Werb Z. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlodavsky I, Folkman J, Sullivan R, Fridman R, Ishai-Michaeli R, Sasse J, Klagsbrun M. Proc Natl Acad Sci USA. 1987;84:2292–2296. doi: 10.1073/pnas.84.8.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levi E, Fridman R, Miao H Q, Ma Y S, Yayon A, Vlodavsky I. Proc Natl Acad Sci USA. 1996;93:7069–7074. doi: 10.1073/pnas.93.14.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gearing A J, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson A H, Drummond A H, Galloway W A, Gilbert R, Gordon J L, et al. Nature (London) 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki M, Raab G, Moses M A, Fernandez C A, Klagsbrun M. J Biol Chem. 1997;272:31730–31737. doi: 10.1074/jbc.272.50.31730. [DOI] [PubMed] [Google Scholar]
- 41.Bergers G, Javaherian K, Lo K M, Folkman J, Hanahan D. Science. 1999;30:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- 42.Schnaper H W, Grant D S, Stetler-Stevenson W G, Fridman R, D'Orazi G, Murphy A N, Bird R E, Hoythya M, Fuerst T R, French D L, et al. J Cell Physiol. 1993;156:235–246. doi: 10.1002/jcp.1041560204. [DOI] [PubMed] [Google Scholar]
- 43.Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, Dammacco F. Blood. 1999;93:3064–3073. [PubMed] [Google Scholar]
- 44.Weidner N, Semple J P, Welch W R, Folkman J. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]