

Abstract
The role of IL-13 in respiratory syncytial virus (RSV) immunopathogenesis is incompletely described. To assess the effect of IL-13 on primary RSV infection, transgenic mice which either overexpress IL-13 in the lung (IL-13 OE) or non-transgenic littermates (IL-13 NT) were challenged intranasally with RSV. IL-13 OE mice had significantly decreased peak viral titers four days after infection compared to non-transgenic littermates. In addition, IL-13 OE mice had significantly lower RSV-induced weight loss and reduced lung IFN-γ protein expression compared with IL-13 NT mice. In contrast, primary RSV challenge of IL-13 deficient mice resulted in a small, but statistically significant increase in viral titers on day four after infection, no difference in RSV-induced weight loss compared to wild type mice, and augmented IFN-γ production on day 6 after infection. In STAT1-deficient (STAT1 KO) mice, where primary RSV challenge produced high levels of IL-13 production in the lungs, treatment with an IL-13 neutralizing protein resulted in greater peak viral titers both four and six days after RSV and greater RSV-induced weight loss compared to mice treated with a control protein. These results suggest that IL-13 modulates illness from RSV-infection.
Keywords: Virus, IL-13, Interferon
Abbreviations: RSV, respiratory syncytial virus; sIL-13Ra, soluble IL-13 receptor; IFN, interferon; SP-A, surfactant protein-A
1. Introduction
Murine models suggest that IL-13 is an important mediator of enhanced pathology from RSV immunization strategies using either a recombinant vaccinia virus expressing RSV G glycoprotein or formalin-inactivated RSV [1], [2], [3]. Vaccination of mice with either the secreted RSV G glycoprotein or formalin-inactivated alum-precipitated RSV leads to Th2 immune responses characterized by augmented IL-5 and IL-13 expression with greater illness and pathology, decreased viral clearance, and heightened pulmonary eosinophilia with subsequent RSV challenge [1], [2]. Recent data from our group reveal that blocking IL-13 activity inhibits formalin-inactivated RSV-induced immune responses [2], and that IL-13 is sufficient for RSV G glycoprotein-induced eosinophilia after RSV challenge [4]. However, the role of IL-13 in the immune response to primary RSV challenge is not fully described.
IL-13 is produced by type 2 T lymphocytes, mast cells, basophils, and eosinophils [5]. IL-13 is a critical cytokine in allergic responses [6], and the interaction between allergic lung inflammation and RSV infection in causing airway dysfunction is of great importance in clinical medicine [7]. In addition, IL-13 is reported to be responsible for RSV-induced airway hyperresponsiveness [8], [9]. IL-13 has many of the same immunomodulatory properties as IL-4, partly because both cytokines signal through the IL-4 receptor α-chain [5]. Previously, our group found that IL-4 significantly delayed RSV clearance which was associated with suppression of the development of virus-specific cytotoxic lymphocytes [10], suggesting that IL-13 might have a similar effect. However, IL-13 plays a more critical role in immunity to the gastrointestinal nematode Nippostrongylus brasiliensis and the intracellular parasite Leishmania major than IL-4 [11], [12], highlighting divergent phenotypic effects of these cytokines in in vivo models of infection.
Given the functional similarities and differences between IL-4 and IL-13, we sought to determine the contribution of IL-13 to the immune response to primary RSV infection. In these experiments, we studied RSV infection in mice that overexpressed IL-13 (IL-13 OE) in airway epithelium [13] and in mice in which the IL-13 gene was deleted (IL-13 KO) [14], both from a C57BL/6 background. We also examined the role of IL-13 in RSV immunity by administering a soluble IL-13 receptor (sIL-13Ra) to RSV-infected STAT1 knock-out (STAT1 KO) mice of a BALB/c background. RSV infection in STAT1 KO mice of the BALB/c background elicits a mixed Th2/Th1 response with high levels of IL-13, while wild type BALB/c mice have a predominant Th1 immune response to RSV infection with high levels of interferon (IFN)-γ and extremely low or undetectable levels of IL-13 [15]. Comparison of the immune response of these three different models of RSV infection reveals that elevation of IL-13 levels in IL-13 OE mice results in decreased peak viral titers and protects against illness while decreasing lung IFN-γ levels. In contrast, RSV infection in IL-13 KO mice or in STAT1 KO mice in which the sIL-13Ra was administered results in higher peak viral tiers, delayed clearance, and augmented IFN-γ production in the lung.
2. Materials and methods
2.1. Mice
All mice were 8–12 weeks old and pathogen free. IL-13 OE mice were generated as previously described and had been bred back to a C57BL/6 background [13]. The IL-13 OE mice used in the experiments were heterozygous for the IL-13 OE transgene and non-transgenic littermates (IL-13 NT) were used as controls. Both the IL-13 OE and IL-13 NT were maintained on water until transgene activation was desired; at that time, doxycycline (0.5 mg/ml) was added to the animal's drinking water and sucrose (2%) was also added to mask the bitter taste of doxycycline [13]. After four days of treatment with doxycycline, the average IL-13 level was 357 ± 26 pg/ml in the ground lung homogenates of four IL-13 OE mice, while the average IL-13 level was 151 ± 13 pg/ml in four IL-13 OE mice treated with the vehicle. The IL-13 levels were undetectable in ground lung supernatants of the IL-13 NT, whether or not they were treated with doxycycline. Therefore, given that IL-13 OE produced some IL-13 when they were not treated with doxycycline, both the IL-13 OE and IL-13 NT mice were treated with doxycycline to avoid the confounding effect of doxycycline treatment in some groups and not in others.
IL-13 deficient (IL-13 KO) mice were generated as previously described and had been bred back to a C57BL/6 background [14]. Wild type, age-matched C57BL/6 mice were purchased from Jackson Laboratory as controls for the IL-13 KO mice.
STAT1 KO mice on a BALB/c background were generated as previously described [15]. Wild type, age-matched control BALB/c mice were purchased from Charles Rivers as controls for the STAT1 KO mice.
In caring for animals the investigators adhered to the Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (revised 1996).
2.2. Cells, virus, and soluble IL-13Ra
HEp-2 cells were maintained in Eagle's minimal essential media (EMEM) supplemented with glutamine, amphotericin, gentamicin, penicillin G, and 10% fetal bovine serum (10% EMEM). The A2 strain of RSV was provided by Dr. Robert Chanock, National Institutes of Health. Master stocks and working stocks of RSV were prepared as previously described [16]. sIL-13Ra was constructed by in-frame fusion of the extracellular domains of the murine IL-13R α-chain with domains 2 and 3 from the constant region of the human IgG1 heavy chain [17]. sIL-13Ra was shown to specifically bind and neutralize IL-13 with no effect on IL-4 activity [17]. The sIL-13Ra reagent and the appropriate isotype control (IgGc) were provided by Sandy Goldman (Wyeth, Cambridge, MA). In experiments utilizing the STAT1 KO mice, the mice were injected with 200 micrograms of either the sIL-13Ra or the control protein on days -2, -1, 0, 1, and 2.
2.3. Mouse infection
On day 0 mice were infected with RSV or given mock-infected culture media (MOCK) intranasally as previously described [16]. Briefly, the mice were anesthetized with intramuscular ketamine 40 μg/g and xylazine 6 μg/g. When held upright with the neck fully extended, the mice readily inhaled a 100 μl inoculum of stock virus placed over their nostrils with a micropipette. A titer of 107 PFU of RSV was used for the experiments in IL-13 OE and IL-13 KO with their appropriate control mice, and a titer of 105 PFU of RSV was used for the experiments in STAT1 KO and their appropriate control mice. A titer of 6 × 106 PFU was used in the IL-13 neutralization experiments using wild type BALB/c mice. RSV infection with this procedure causes bronchiolitis [16]. Pulmonary infection was confirmed in a subset of mice by plaque assay in HEp-2 cells as previously described [16].
2.4. Plaque assays
Lung tissue was removed, weighed, and immediately quick-frozen in EMEM. Briefly, one lung from each mouse was harvested in 2 ml of serum free media. The lung was snap frozen and then later thawed and ground using a mortar, pestle, and ground glass. The solution of the ground lung and the ground glass was then centrifuged at 2000 rpm for 15 min to obtain clarified lung supernatants. Clarified lung supernatants were diluted and inoculated onto subconfluent HEp2 cell monolayers in Costar 12-well plates (Costar, Cambridge, MA). After 1 h, plates were covered with 0.75% methylcellulose in 10% EMEM and incubated for 5 days at 37 °C. Monolayers were then fixed with 10% buffered formalin and stained with hematoxylin-eosin. Plaques were then counted with the aid of a dissecting microscope.
2.5. Quantitation of IFN-γ and IL-13 in lung tissues
Levels of IFN-γ and IL-13 in the ground lung supernatants (see paragraph above) were measured using commercially available ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer's protocols. The cytokine level from each lung was measured in duplicate.
2.6. Flow cytometry based measurement of RSV-specific IFN-γ producing CD8+ cells
Mice were sacrificed and lungs were harvested at days 6 and 8 post-infection. Lymphocytes were isolated manually by grinding lung tissue through a nylon strainer in RPMI 1640. Lymphocytes were isolated by centrifugation on a cushion of Ficoll/Hypaque at room temperature, washed, and resuspended in RPMI 1640 plus 10% FBS. Lymphocytes were incubated for 5–6 h with 2 μg of the appropriate peptide (H-2Db-restricted CTL epitope from the M protein, corresponding to amino acids 187–195 (NAITNAKII)) in 5 ml RPMI 1640 plus 10% FBS and 1 μg/ml costimulatory Abs against CD28 and CD49d [18]. One hour into the incubation, 0.75 μg/ml GolgiStop (BD Pharmingen, San Diego, CA) was added to retain newly synthesized proteins within the cell. Cells were stimulated with 200 ng/ml PMA and 1 μM ionomycin as a positive control or with 2 μg of ovalbumin peptide (corresponding to amino acid 323–329) in 5 ml media as a negative control. The incubation period was for 5 h at 37 °C. After the incubation, cells were fixed and permeabilized, according to the manufacturer's instructions (BD Pharmingen, San Diego, CA). Cells were stained with fluorochrome-conjugated Abs to CD8, IFN-γ, and TNF-α (BD Pharmingen) for 30 min at 4 °C and analyzed on a Becton Dickenson LSR II cytometer (BD Biosciences, San Jose, CA) equipped with a 488 nm Argon laser and 635 nm Helium Neon laser. Data were analyzed by using BD FACSDiva Software version 4.0 (BD Biosciences).
2.7. Immnuoblot analysis of surfactant protein-A (SP-A)
On the fifth day after infection, the mice were sacrificed and the lungs were lavaged with fixed aliquots of 0.7 ml of phosphate buffered saline (PBS). Equal volumes of fluid (20 μl) were solubilized in loading buffer (0.1 M Tris, pH 7.4, 50 μM dithiothreitol, 0.01% bromophenol blue, 2% sodium dodecyl sulfate (SDS), 10% glycerol) at 95 °C for 5 min. Proteins were resolved on pre-cast 4–20% tris-glycine polyacrylamide gels (#EC6025, Invitrogen Corp., Carlsbad, CA). After protein resolution, gels were transferred to nitrocellulose paper and incubated with either rabbit-anti-human SP-A antibody (1:1000 dilution) followed by peroxidase-conjugated goat anti-rabbit IgG (Sigma, 1:160,000 dilution). The immunodetected protein was detected with chemiluminescence ECL system (#RPN2135, Amersham Biosciences Corp, Piscataway, NJ) and recorded on high-performance chemiluminescence film. Quantization was performed using the imaging and quantitating abilities of the ChemiGenius2 imaging system (Syngene, Frederick, MD). The polyclonal IgG specific for surfactant protein-A was raised in rabbits injected with isolated human SP-A obtained from human lung lavage and was kindly provided by Dr. C.R. Mendelson (Southwestern University, Dallas, Texas).
2.8. Measurement of SP-D by ELISA
BAL fluid was obtained as described above on the fifth day after infection. Nunc-Immuno MaxiSorp plates were coated with 100 μl of monoclonal anti-mouse SP-D at 1 μg/ml diluted in 0.05 M sodium bicarbonate buffer overnight at 4 °C. The next morning, the wells were emptied and three hundred μl per well of blocking/dilution buffer was added to each well for 2 h. The blocking/dilution buffer consisted of 0.05 M Tris (pH = 7.5), 0.15 M Sodium Chloride, 0.001 M Calcium Chloride, with 0.05% (V/V) Tween-20 and 1.0% (W/V) Bovine Serum Albumin (BSA), fraction V. The well were again emptied and 50 μl per well of standards (125, 62.5, 31.25, 15.625, 7.8125 nanograms per ml) and samples were diluted in blocking/dilution buffer and added to the plate in duplicate. The plates were placed on a rocker and incubated at room temperature for 2 h. After incubation, the standards and samples were aspirated and the wells were washed three times with wash buffer which consisted of 0.05 M Tris (pH = 7.5), 0.15 M Sodium Chloride, 0.001 M Calcium Chloride, with 0.05% Tween-20. Next, 50 μl per well of a 1 to 700 dilution (in blocking/dilution buffer) of rabbit anti-SP-D was added to the plate and incubated for 1 to 2 h at room temperature with rocking. The contents of each well was aspirated and the wells were washed three times with wash buffer. Horseradish peroxidase conjugated donkey anti-rabbit IgG (H&L) (Jackson Laboratories, Bar Harbor, ME) was diluted 1:10,000 in blocking/dilution buffer and 100 μl was added to each well. After 2 h incubation, the wells were washed three times with wash buffer and then 100 μl TMB (BioFX Laboratories, Owings Mill, MD) was added to each well. The plates were stopped after color development with 100 μl of 2M H2SO4. The optical density of each well was read with a microtiter plate reader at a wavelength of 450 namometers.
2.9. RSV-specific antibody titers
Blood was drawn 30 days after infection, and after centrifugation, the serum was retained for measurement of RSV-specific antibodies. For measurement of anti-RSV-specific F antibody titers, purified F protein was diluted in bicarbonate buffer (pH 9.8), added to 96-well plates (Immulon II; Nunc, Roskilde, Denmark) at 10 μg/well, and incubated overnight at 4 °C. After coating, the plate was emptied and blocked with 150 μl of 1% bovine serum albumin (Sigma, St. Louis, Mo.). The plate was then washed four times with phosphate-buffered saline-0.05% Tween 20. The serum was then diluted to 1:500. Serum from an uninfected mouse was used as a negative control and blank. One hundred μl of the diluted serum was added to duplicate coated wells, and the plates were incubated at room temperature for 2 h. The plates were then washed five times with phosphate-buffered saline-0.05% Tween 20. After washing, 100 μl of horseradish peroxidase labeled anti-mouse immunoglobulin G1 (IgG1) and immunoglobulin G2a (IgG2a) (both from Southern Biotech, Birmingham, Ala.) were added to the appropriate wells at a dilution of 1:10,000 and incubated 2 h at room temperature. The plates were washed four times with phosphate-buffered saline-0.05% Tween 20. After washing, 100 μl of conjugate solution (R&D Systems, Minneapolis, MN) was added to each well and incubated 1 h at 37 °C. The plates were washed four times with phosphate-buffered saline without Tween 20. The reactions were stopped with 100 μl of 2.5 N H2SO4. Optical density was measured with a 450-nm filter, and values were calculated from a standard curve.
2.10. Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Measurements of weight loss curves were compared by ANOVA with Scheffe post-hoc analysis. Measurements of viral titers and cytokines were compared by t-test. Differences were considered to be significant if p < 0.05.
3. Results
3.1. IL-13 OE mice are protected against RSV-induced weight loss
Weight loss is a measure of RSV-induced disease and correlates with illness scores [16]. To determine the effect of IL-13 overexpression on RSV-induced weight loss, on day 0 we intranasally challenged two groups of mice with RSV (IL-13 OE-RSV and IL-13 NT-RSV) and two groups were mock-infected (IL-13 OE-MOCK and IL-13 NT-MOCK). As shown in Fig. 1, there was no difference in the weight loss curves of the IL-13 OE-RSV, IL-13 OE-MOCK, and IL-13 NT-MOCK; however, the non-transgenic littermate control IL-13 NT-RSV mice had significantly greater weight loss than any of the other three groups (p < 0.001). Therefore, the IL-13 OE mice were significantly protected against RSV-induced weight loss.
Fig. 1.
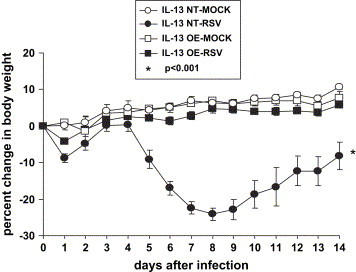
RSV-induced weight loss as a percentage of the pre-infection weight on day 0. (IL-13 NT-MOCK n = 5; IL-13 NT-RSV n = 10; IL-13 OE-MOCK n = 11; IL-13 OE-RSV n = 13). The data shown is representative of two separate experiments. *p < 0.001 WT-RSV compared to all other groups.
3.2. IL-13 OE mice have decreased peak RSV titers, enhanced viral clearance, and decreased lung IFN-γ production
In order to determine the effect of IL-13 overexpression on the kinetics of viral replication, we performed plaque assays on days 4 and 6 ( Fig. 2A). We found that on day 4 the IL-13 OE-RSV mice had significantly decreased peak viral titers compared to non-transgenic littermate control IL-13 NT-RSV mice (3.41 ± 0.1 vs. 5.39 ± 0.14 log10 PFU/g lung; p = 0.005). On day 6, the IL-13 OE-RSV mice again had significantly decreased viral titers compared to littermate control IL-13 NT-RSV mice (2.22 ± 0.46 vs. 3.14 ± 0.48 log10 PFU/g lung; p = 0.04). Therefore, the IL-13 overexpression resulted in decreased peak viral titers and enhanced viral clearance. In order to determine the effect of the IL-13 overexpression on the cellular immune cytokine response to RSV infection, we measured IFN-γ in the ground lung supernatants on day 6 after infection (Fig. 2B). We found that the IL-13 OE-RSV mice had significantly decreased IFN-γ compared to the non-transgenic littermate control IL-13 NT-RSV mice (151 ± 16 vs. 411 ± 35 pg/ml; p < 0.001). Thus, IL-13 overexpression in the lung has profound effects on RSV pathogenesis in decreasing peak RSV titers, enhancing viral clearance, and decreasing lung IFN-γ production.
Fig. 2.
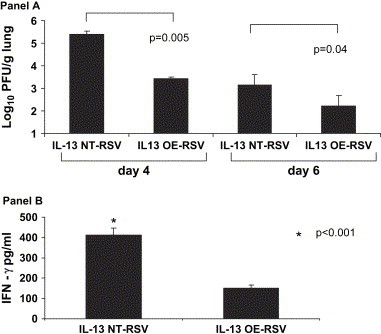
A. The effect of lung IL-13 overexpression on viral titers in IL-13 OE and non-transgenic littermate control (IL-13 NT) mice. Viral titers represented as log10 plaque forming units/gram of lung tissue on days 4 and 6 after infection (n = 4 in each group). The data shown is representative of two separate experiments. B. The effect of lung IL-13 overexpression on lung INF-γ levels in IL-13 OE and non-transgenic littermate control (IL-13 NT) mice. Levels of IFN-γ protein were measured in lung supernatants on day 6 after infection (n = 4 in each group). The data shown is representative of two separate experiments. *p < 0.001.
3.3. Deficiency of IL-13 has no effect on RSV-induced weight loss
In order to determine the effect of IL-13 on illness as a result of RSV infection, we measured the weights of IL-13 KO and WT C57BL/6 control mice that were either mock-infected or RSV-infected for 14 days ( Fig. 3). We found that the RSV-infected IL-13 KO and RSV-infected WT C57BL/6 mice had significantly greater weight loss compared to the mock-infected IL-13 KO mice (p < 0.001). We found that there was no difference in weight loss between the RSV-infected IL-13 KO and RSV-infected WT C57BL/6 mice. Thus, deficiency in IL-13 had no effect on the degree of RSV-induced weight loss.
Fig. 3.
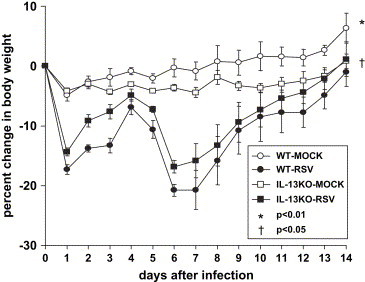
RSV-induced weight loss as a percentage of the pre-infection weight on day 0. (WT-MOCK n = 3; WT-RSV n = 6; IL-13KO-MOCK n = 4; IL-13KO-RSV n = 8). The data shown is representative of two separate experiments. *p < 0.01, WT-MOCK compared to either WT-RSV or IL-13KO-RSV; †p < 0.05, IL-13KO-MOCK compared to either WT-RSV or IL-13KO-RSV.
3.4. IL-13 KO mice have increased peak RSV titers and augmented lung IFN-γ production
In order to determine the effect of IL-13 deficiency on the kinetics on RSV replication, we performed plaque assays on days 4 and 6 ( Fig. 4A). We found that on day 4 the IL-13 KO-RSV mice had a small, but statistically significant increase in peak viral titers compared to WT-RSV mice (4.59 ± 0.16 vs 4.25 ± 0.16 log10 PFU/g lung; p = 0.05). On day 6, the IL-13 KO-RSV mice again had a trend toward increased viral titers compared to WT-RSV mice (3.86 ± 0.36 vs 3.44 ± 0.11 log10 PFU/g lung; p = 0.08). Although IL-13 deficiency resulted in an increase in peak viral titers that was statistically significant, the biologic relevance of a difference that is less than 0.5 log10 is uncertain. In order to determine the effect of the IL-13 deficiency on the cellular immune cytokine response to RSV infection, we measured IFN-γ in the ground lung supernatants on day 6 after infection (Fig. 4B). We found that the IL-13 KO-RSV mice had significantly increased IFN-γ compared to the littermate control WT-RSV mice (1540 ± 430 vs. 498 ± 75 pg/ml; p = 0.04). Thus, IL-13-deficiency results in increased peak RSV titers and augmented lung IFN-γ production.
Fig. 4.
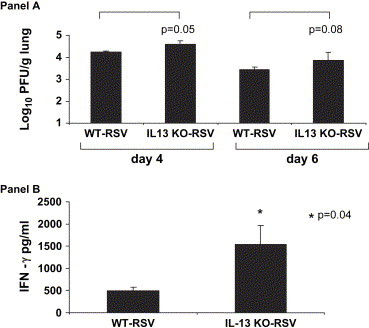
A. The effect of IL-13 deficiency on viral titers in IL-13 KO and WT mice. Viral titers represented as log10 plaque forming units/gram of lung tissue on days 4 and 6 after infection (n = 6–8 in each group). The data shown is representative of two separate experiments. B. The effect of IL-13 deficiency on lung IFN-γ protein expression in IL-13 KO and WT mice. Levels of IFN-γ protein were measured in lung supernatants on day 6 after infection (n = 3–4 in each group). The data shown is representative of two separate experiments. *p = 0.04.
3.5. Neither IL-13 overexpression nor IL-13 deficiency altered SP-A or SP-D expression in conjunction with RSV infection
IL-13 OE in which there is constitutive expression of the IL-13 transgene have been previously reported to exhibit a significant increase in SP-A and SP-D expression compared to non-transgenic littermates [19]. Since we saw a small, but statistically significant change in peak viral titers in the IL-13 KO-RSV mice compared to the WT-RSV mice, we sought to determine if these differences in viral titer might be related to IL-13 deficiency regulating lung SP-A or SP-D expression, as SP-A and SP-D both have antiviral properties. We measured SP-A and SP-D in bronchoalveolar lavage fluid and found no difference in the level of either SP-A or SP-D protein in the WT-MOCK and IL-13 KO-MOCK groups, nor in the WT-RSV and IL-13 KO-RSV groups. As mentioned in the results above, we found a significant reduction in viral titers in the IL-13 OE mice compared to IL-13 NT mice in which the IL-13 transgene is induced with doxycycline two days prior to RSV infection. However, we did not find differences in either SP-A or SP-D protein levels in BAL fluid from either IL-13 NT-MOCK or IL-13 OE-MOCK, nor between the IL-13 NT-RSV and IL-13 OE-RSV groups. Therefore the differences in viral titers between the WT and IL-13 KO groups, and the differences in viral titers between the IL-13 NT and IL-13 OE groups, are not likely to be related to differences in SP-A and SP-D expression in these mice.
3.6. IL-13 overexpression or deficiency has no effect on number of RSV-specific IFN-γ-expressing CD8+ cells
We assessed how IL-13 affected the number of RSV-specific IFN-γ-expressing CD8+ cells by measuring the percentage of CD8 cells that produced IFN-γ in response to a newly identified RSV-specific H-2Db-restricted CTL epitope [18]. The results listed are the percentages of IFN-γ-staining cells in the lymphocyte gate. There was no difference in the peptide-stimulated CD8 cells expressing IFN-γ from either the IL-13 OE or WT mice on either day 6 (0.6 ± 0.1 vs. 0.8 ± 0.1% respectively, n = 4 for each group) or day 8 (2.0 ± 0.5 vs. 2.2 ± 0.9% respectively, n = 4 for each group). Similarly, there was no difference in the peptide-stimulated CD8 cells expressing IFN-γ from either the IL-13 KO or WT mice on either day 6 (2.7 ± 0.7 vs. 1.6 ± 0.2% respectively, n = 4 for each group) or day 8 (5.6 ± 0.9 vs. 5.0 ± 1.0% respectively, n = 5 for each group). Therefore, neither overexpression of IL-13 nor deficiency of IL-13 appears to influence the numbers of RSV-specific IFN-γ-producing CD8 T lymphocytes.
3.7. Effect of IL-13 overexpression and IL-13 deficiency on RSV-specific IgG1 and IgG2a titers
We examined how IL-13 modulated RSV-specific antibody titers by measuring anti-RSV F IgG1 and IgG2a levels in serum 30 days after primary infection. There was no difference in either RSV-specific IgG1 or IgG2a between the IL-13 NT and IL-13 OE mice ( Fig. 5, panel A). There was a trend toward an increase (p = 0.07) in the RSV-specific IgG1 levels in the WT mice compared to the IL-13 KO mice, but no difference between these groups in RSV-specific IgG2a levels (Fig. 5, panel B). Therefore, neither overexpression of IL-13 nor deficiency of IL-13 appears to influence RSV-specific antibody titers that are measured 30 days after primary infection.
Fig. 5.
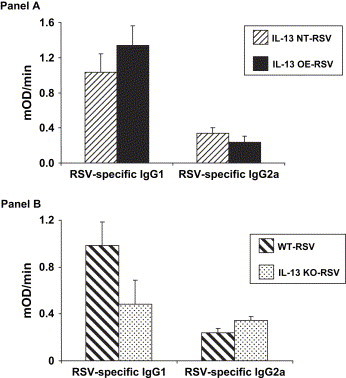
A. The effect of IL-13 overexpression on serum IgG1 and IgG2a antibody titers drawn 30 days after primary infection (n = 3 in each group). B. The effect of IL-13 deficiency on serum IgG1 and IgG2a antibody titers drawn 30 days after primary infection (n = 5 in each group).
3.8. IL-13 neutralization in RSV-infected STAT1 KO mice leads to greater weight loss
IL-13 expression is induced in RSV-infected STAT1 KO mice [15]. We have previously shown that STAT1 KO mice have greater weight loss compared to WT BALB/c mice when infected with 1 × 105 PFU [15]. In order to determine the effect of neutralizing IL-13 on RSV-induced illness in these mice, we administered sIL-13Ra or control protein (IgGc) intraperitoneally prior to and on the day of infection ( Fig. 6). We found that RSV-infected STAT1 KO mice treated with sIL-13Ra (STAT1 KO-RSV sIL-13Ra) had significantly greater weight loss (p = 0.008) compared to RSV-infected STAT1 KO mice treated with IgGc (STAT1 KO-RSV IgGc). This suggests that IL-13 has a protective effect against RSV-induced weight loss in the STAT1 KO mice.
Fig. 6.
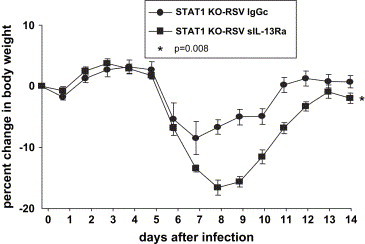
The effect of IL-13 neutralization on RSV-induced weight loss in STAT1 KO mice. RSV-induced weight loss is expressed as a percentage of the pre-infection weight on day 0. (n = 14 in each group). The data shown is representative of two separate experiments. *p = 0.008 STAT1 KO-RSV IgGc vs. STAT1 KO-RSV sIL-13Ra.
3.9. IL-13 neutralization in RSV-infected STAT1 KO mice increases viral titers and lung IFN-γ levels
Since STAT1 KO-RSV sIL-13Ra mice had greater weight loss than the STAT1 KO-RSV IgGc group, we determined the effect of IL-13 on viral titers in these two groups ( Fig. 7A). We found that the STAT1 KO-RSV sIL-13Ra mice had significantly greater viral titers than the STAT1 KO-RSV IgGc group on both day 4 (5.17 ± 0.10 vs 4.49 ± 0.33 log10 PFU/g lung; p = 0.02) and day 6 (5.64 ± 0.11 vs 4.21 ± 0.27 log10 PFU/g lung; p = 0.002) compared to the STAT1 KO-RSV IgGc mice. As the STAT1 KO-RSV sIL-13Ra mice had greater viral titers than the STAT1 KO-RSV IgGc group, we decided to measure lung levels of IFN-γ in these two groups (Fig. 7B). On day 6, the STAT1 KO-RSV sIL-13Ra mice had a 160% increase in lung IFN-γ levels than the STAT1 KO-RSV IgGc (5894 ± 1797 vs. 3512 ± 1672 pg/ml; p = 0.39), although this difference was not statistically significant. However, on day 8, the STAT1 KO-RSV IgGc had undetectable lung levels of IFN-γ, while the STAT1 KO-RSV sIL-13Ra mice had 2139 ± 562 pg/ml (p = 0.02). Therefore, these results indicate that IL-13 has a role in diminishing viral titers in RSV infection and decreasing lung expression of IFN-γ.
Fig. 7.
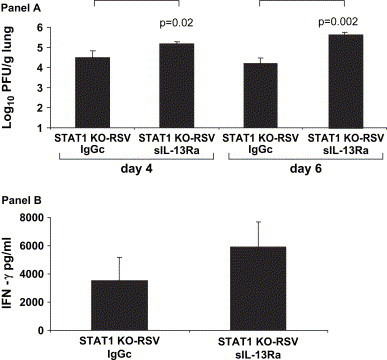
A. The effect of IL-13 neutralization on viral titers in STAT1 KO mice. Viral titers represented as log10 plaque forming units/gram of lung tissue on days 4 and 6 after infection (n = 4 in each group). The data shown is representative of two separate experiments. B. The effect of IL-13 neutralization on lung IFN-γ protein in STAT1 KO mice. Levels of IFN-γ protein measured in lung supernatants measured on day 6 after infection (n = 3–4 in each group). The data shown is representative of two separate experiments.
4. Discussion
In these studies, we investigated the effects of overexpression of IL-13, deficiency in IL-13 production, or neutralization of IL-13 using a soluble receptor on the immune response to primary RSV challenge. Inducible overexpression of IL-13 prior to RSV infection decreased viral titers, diminished lung IFN-γ production, and protected against RSV-induced weight loss. In contrast, mice which express no IL-13 have higher viral titers and increased IFN-γ production, while there is no effect of RSV-induced weight loss. Furthermore, administration of a sIL-13Ra to STAT1 KO mice that express IL-13 with primary RSV challenge leads to increased viral titers, prolonged IFN-γ expression and greater RSV-induced weight loss. In these experiments, we therefore found that IL-13 was inversely related to viral titers. Diminished viral antigen load probably abrogated the need for a strong adaptive immune response with resultant decreased IFN-γ production in the lung. The absence of IL-13 increased viral load by a yet unknown mechanism, resulting in a greater requirement for adaptive immunity to clear virus, hence the increase in lung IFN-γ levels. These results suggest that IL-13 modulates the immune response to RSV infection.
There are very few studies examining the effect of IL-13 in viral infection. In vitro studies reveal varying effects of IL-13 on anti-viral immunity depending on the virus studied. Primary blood-derived human macrophages that are exposed to IL-13 decrease the infection and replication of HIV-1 [20]. IL-13 blocks the completion of reverse transcription, thus decreasing virus production, and diminishes the infectivity of the progeny virus [21]. In addition, IL-13 induced a 50% reduction in the number of the CD4 and CCR5 binding sites and completely inhibited surface expression of CXCR4 [22]. CD4 is the HIV receptor, while CCR5 and CXCR4 are co-receptors [22]; these are not known to be receptors for RSV. IL-13 also protects bronchoalveolar macrophages from productive infection with HIV-1 [23]. While HIV-1 production is strongly inhibited in alveolar macrophages treated with IL-13, IL-13 treatment increases human cytomegalovirus expression in these same cells [24].
IL-13 has been examined in two other in vivo murine models of RSV infection. Tekkanat and Lukacs reported that primary RSV infection in BALB/c mice induced expression of IL-13 in the supernatants of lung homogenates [9]. In this study, a clinical isolate from the University of Michigan was used, not strain A2. Using this clinical isolate, RSV-induced IL-13 was associated with airway hyperresponsiveness and airway epithelial mucus production, features which were blocked by the administration of anti-IL-13 antibody [9]. These investigators also found that RSV-infected mice treated with anti-IL-13 antibodies had significantly reduced viral antigen levels in lung homogenates as measured by an ELISA specific for RSV proteins, but they did not measure PFU in this study [9]. They also did not measure the effect of anti-IL-13 on IFN-γ production.
In contrast, Park and colleagues found that RSV infection caused airway hyperresponsiveness that was independent of IL-13 [25]. These studies used human RSV long strain, type A that was purified in sucrose gradients [25]. In these experiments, they used two protocols, one examining IL-13 KO mice on a BALB/c background and another using wild type BALB/c mice with the same sIL-13Ra reagent used in our study [25]. These investigators also found that RSV induced airway epithelial mucus production that was absent in the IL-13 KO mice and inhibited in wild type mice treated with sIL-13Ra [25]. Neither viral titers nor IFN-γ were measured in these experiments. In our studies, we have been unable to detect IL-13 with primary RSV (A2 strain) infection in wild type BALB/c mice either by measuring protein in ground lung homogenates nor lung mRNA by RNAse protection assay, nor have we found airway hyperresponsiveness in RSV-infected mice [26]. The differences between the studies of Tekkanat and Lukacs, Park, and our group may be explained by the different RSV strain utilized, preparation of virus stocks, the dose, the route of administration, and the mechanism by which airway responsiveness was measured. In collaboration with the Lukacs group, we recently reported that there are differential immune responses and pulmonary pathophysiology induced by two different A strains of respiratory syncytial virus [27].
IL-13 and IL-4 induce similar biological functions. This stems from the fact that the IL-4Rα chain is shared by the receptors for both IL-4 and IL-13 [28], and both trigger the STAT-6 signaling pathway. However, T cells do not express IL-13 receptors [29], and effects mediated directly by the cytokines on T cell effectors may differ. Studies of tumor surveillance suggest that IL-13 may alter CD8 + T cell function through an intermediate CD1d-restricted cell type [30]. CD8+ cytolytic function was also increased when IL-13 was inhibited in the setting of recombinant vaccinia infection, but virus titers and illness were not reported [31]. IL-4 has been shown to increase the virulence of poxvirus infection and is associated with a reduction in CTL precursor frequency [32]. IL-4 has also been shown to influence the CTL response to RSV. Overexpression of IL-4 during RSV infection results in delayed virus clearance and enhanced illness, and is associated with abrogated CD8+ cytolytic T cell activity [10]. The effect on CD8 + T cell function in this setting is related to altered cytolytic function, and does not alter frequency or IFN-γ production in CD8 + T cells [33], [34]. Our current study clearly shows that IL-13 serves a protective role during RSV infection, and does not alter the frequency of IFN-γ producing CD8 + T cells. While RSV-specific cytolytic activity was not measured, the rapid virus clearance (Fig. 2A) suggests that CD8 + T cell function was intact. The differences in the effect of IL-4 and IL-13 on the biological function of CD8+ CTL and the consequent impact on virus pathogenesis is likely due to the difference in the specific receptor distribution and the different pattern of cells with STAT-6 activation.
The mechanism by which IL-13 protects against RSV-induced weight loss in both the IL-13 OE mice and the experiments using the sIL-13Ra in STAT1 KO mice is still unclear. While constitutive expression of the IL-13 transgene in IL-13 OE mice increased surfactant proteins (SP)-A, -B, and -C two-fold, and SP-D was increased 70-fold over levels found in non-transgenic littermates [19], we found no difference in these surfactant proteins between IL-13 OE mice in which the transgene was induced two days prior to RSV infection and IL-13 NT mice. This is relevant to our model because SP-A and SP-D are collectins that both enhance pulmonary clearance of RSV [35], [36]. We also found that deficiency of IL-13 did alter either SP-A or SP-D levels in BAL fluid; therefore, surfactant regulation of RSV replication does not seem to be the explanation for the IL-13 regulatory effect on RSV-induced immune responses.
Other possible mechanisms by which IL-13 may modulate RSV pathogenesis include IL-13 regulation of matrix metalloproteinase-9 (MMP-9) and/or cathepsin. MMP-9 lung expression is induced in IL-13 OE mice [37]. MMP-9 enhanced syncytial formation leading to RSV multiplication and spread, and these effects were blunted by MMP inhibitors [38]. Therefore, this would suggest that IL-13 induces MMP-9 which in turn would increase viral titers. However, in our study, we found that IL-13 overexpression reduced viral titers and IL-13 deficiency increased viral titers, thus it is not clear how the interaction of IL-13 regulation of MMP-9 might be playing a role in viral replication in our in vivo studies. IL-13 also induced expression of cathepsins [39] which are a major factor in endosomal and lysosomal system proteases and are critical in intracellular antigen processing and presentation [40]. Cathepsins have been shown to be essential for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry [41] and also enhanced entry mediated by the Ebola virus glycoprotein [42]. However, the role of cathepsins in RSV entry or pathogenesis has not been described.
In summary, we found that IL-13 overexpression protects against RSV-induced weight loss, and decreases both viral titers and lung IFN-γ production. On the other hand, IL-13 deficiency results in increased viral titers and lung IFN-γ production. Neutralization of IL-13 in the STAT1 KO mouse, where IL-13 is induced with primary RSV challenge, results in enhanced weight loss and increased viral titers and lung IFN-γ production. These results reveal that IL-13 significantly modulates RSV-induced disease.
Acknowledgement
This work was supported by The American Academy of Allergy, Asthma and Immunology ERT Award, R01-HL-069949, R01-AI-054660, and R01-AI-045512. The authors would like to thank Mr. William Hull and Dr. Jeffrey Whitsett for measurements of surfactant protein D and Dr. Hoyin Mok for providing the RSV F protein coated ELISA plate.
References
- 1.Johnson T.R., Johnson J.E., Roberts S.R., Wertz G.W., Parker R.A., Graham B.S. Priming with secreted glycoprotein G of respiratory syncytial virus (RSV) augments interleukin-5 production and tissue eosinophilia after RSV challenge. J. Virol. 1998;72:2871–2880. doi: 10.1128/jvi.72.4.2871-2880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson T.R., Graham B.S. Secreted respiratory syncytial virus G glycoprotein induces interleukin-5 (IL-5), IL-13, and eosinophilia by an IL-4-independent mechanism. J. Virol. 1999;73:8485–8495. doi: 10.1128/jvi.73.10.8485-8495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Openshaw P.J., Clarke S.L., Record F.M. Pulmonary eosinophilic response to respiratory syncytial virus infection in mice sensitized to the major surface glycoprotein G. Int. Immunol. 1992;4:493–500. doi: 10.1093/intimm/4.4.493. [DOI] [PubMed] [Google Scholar]
- 4.Johnson T.R., Parker R.A., Johnson J.E., Graham B.S. IL-13 is sufficient for respiratory syncytial virus G glycoprotein-induced eosinophilia after respiratory syncytial virus challenge. J. Immunol. 2003;170:2037–2045. doi: 10.4049/jimmunol.170.4.2037. [DOI] [PubMed] [Google Scholar]
- 5.Wynn T.A. IL-13 effector functions. Annu. Rev. Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 6.Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T.Y., Karp C.L., Donaldson D.D. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 7.Peebles R.S., Jr., Hashimoto K., Graham B.S. The complex relationship between respiratory syncytial virus and allergy in lung disease. Viral Immunol. 2003;16:25–34. doi: 10.1089/088282403763635429. [DOI] [PubMed] [Google Scholar]
- 8.Lukacs N.W., Tekkanat K.K., Berlin A., Hogaboam C.M., Miller A., Evanoff H., Lincoln P., Maassab H. Respiratory syncytial virus predisposes mice to augmented allergic airway responses via IL-13-mediated mechanisms. J. Immunol. 2001;167:1060–1065. doi: 10.4049/jimmunol.167.2.1060. [DOI] [PubMed] [Google Scholar]
- 9.Tekkanat K.K., Maassab H.F., Cho D.S., Lai J.J., John A., Berlin A., Kaplan M.H., Lukacs N.W. IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 2001;166:3542–3548. doi: 10.4049/jimmunol.166.5.3542. [DOI] [PubMed] [Google Scholar]
- 10.Fischer J.E., Johnson J.E., Kuli-Zade R.K., Johnson T.R., Aung S., Parker R.A., Graham B.S. Overexpression of interleukin-4 delays virus clearance in mice infected with respiratory syncytial virus. J. Virol. 1997;71:8672–8677. doi: 10.1128/jvi.71.11.8672-8677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barner M., Mohrs M., Brombacher F., Kopf M. Differences between IL-4R alpha-deficient and IL-4-deficient mice reveal a role for IL-13 in the regulation of Th2 responses. Curr. Biol. 1998;8:669–672. doi: 10.1016/s0960-9822(98)70256-8. [DOI] [PubMed] [Google Scholar]
- 12.Urban J.F., Jr., Noben-Trauth N., Donaldson D.D., Madden K.B., Morris S.C., Collins M., Finkelman F.D. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. 1998;8:255–264. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- 13.Zheng T., Zhu Z., Wang Z., Homer R.J., Ma B., Riese R.J., Jr., Chapman H.A., Jr., Shapiro S.D., Elias J.A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenzie G.J., Emson C.L., Bell S.E., Anderson S., Fallon P., Zurawski G., Murray R., Grencis R., McKenzie A.N. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9:423–432. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 15.Durbin J.E., Johnson T.R., Durbin R.K., Mertz S.E., Morotti R.A., Peebles R.S., Graham B.S. The role of IFN in respiratory syncytial virus pathogenesis. J. Immunol. 2002;168:2944–2952. doi: 10.4049/jimmunol.168.6.2944. [DOI] [PubMed] [Google Scholar]
- 16.Graham B.S., Perkins M.D., Wright P.F., Karzon D.T. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 1988;26:153–162. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 17.Donaldson D.D., Whitters M.J., Fitz L.J., Neben T.Y., Finnerty H., Henderson S.L., O'Hara R.M., Jr., Beier D.R., Turner K.J., Wood C.R., Collins M. The murine IL-13 receptor alpha 2: molecular cloning, characterization, and comparison with murine IL-13 receptor alpha 1. J. Immunol. 1998;161:2317–2324. [PubMed] [Google Scholar]
- 18.Rutigliano J.A., Rock M.T., Johnson A.K., Crowe J.E., Jr., Graham B.S. Identification of an H-2D(b)-restricted CD8+ cytotoxic T lymphocyte epitope in the matrix protein of respiratory syncytial virus. Virology. 2005;337:335–343. doi: 10.1016/j.virol.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 19.Homer R.J., Zheng T., Chupp G., He S., Zhu Z., Chen Q., Ma B., Hite R.D., Gobran L.I., Rooney S.A., Elias J.A. Pulmonary type II cell hypertrophy and pulmonary lipoproteinosis are features of chronic IL-13 exposure. Am. J. Physiol. Lung Cell Mol. Physiol. 2002;283:L52–L59. doi: 10.1152/ajplung.00438.2001. [DOI] [PubMed] [Google Scholar]
- 20.Montaner L.J., Doyle A.G., Collin M., Herbein G., Illei P., James W., Minty A., Caput D., Ferrara P., Gordon S. Interleukin 13 inhibits human immunodeficiency virus type 1 production in primary blood-derived human macrophages in vitro. J. Exp. Med. 1993;178:743–747. doi: 10.1084/jem.178.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montaner L.J., Bailer R.T., Gordon S. IL-13 acts on macrophages to block the completion of reverse transcription, inhibit virus production, and reduce virus infectivity. J. Leukoc. Biol. 1997;62:126–132. doi: 10.1002/jlb.62.1.126. [DOI] [PubMed] [Google Scholar]
- 22.Bailer R.T., Lee B., Montaner L.J. IL-13 and TNF-alpha inhibit dual-tropic HIV-1 in primary macrophages by reduction of surface expression of CD4, chemokine receptors CCR5, CXCR4 and post-entry viral gene expression. Eur. J. Immunol. 2000;30:1340–1349. doi: 10.1002/(SICI)1521-4141(200005)30:5<1340::AID-IMMU1340>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 23.Denis M., Ghadirian E. Interleukin 13 and interleukin 4 protect bronchoalveolar macrophages from productive infection with human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses. 1994;10:795–802. doi: 10.1089/aid.1994.10.795. [DOI] [PubMed] [Google Scholar]
- 24.Hatch W.C., Freedman A.R., Boldt-Houle D.M., Groopman J.E., Terwilliger E.F. Differential effects of interleukin-13 on cytomegalovirus and human immunodeficiency virus infection in human alveolar macrophages. Blood. 1997;89:3443–3450. [PubMed] [Google Scholar]
- 25.Park J.W., Taube C., Yang E.S., Joetham A., Balhorn A., Takeda K., Miyahara N., Dakhama A., Donaldson D.D., Gelfand E.W. Respiratory syncytial virus-induced airway hyperresponsiveness is independent of IL-13 compared with that induced by allergen. J. Allergy Clin. Immunol. 2003;112:1078–1087. doi: 10.1016/j.jaci.2003.08.046. [DOI] [PubMed] [Google Scholar]
- 26.Peebles R.S., Jr., Sheller J.R., Collins R.D., Jarzecka A.K., Mitchell D.B., Parker R.A., Graham B.S. Respiratory syncytial virus infection does not increase allergen- induced type 2 cytokine production, yet increases airway hyperresponsiveness in mice. J. Med. Virol. 2001;63:178–188. [PubMed] [Google Scholar]
- 27.Lukacs N.W., Moore M.L., Rudd B.D., Berlin A.A., Collins R.D., Olson S.J., Ho S.B., Peebles R.S., Jr. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 2006;169:977–986. doi: 10.2353/ajpath.2006.051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilton D.J., Zhang J.G., Metcalf D., Alexander W.S., Nicola N.A., Willson T.A. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc. Natl. Acad. Sci. U.S.A. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zurawski G., de Vries J.E. Interleukin 13, an interleukin 4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol. Today. 1994;15:19–26. doi: 10.1016/0167-5699(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 30.Terabe M., Matsui S., Park J.M., Mamura M., Noben-Trauth N., Donaldson D.D., Chen W., Wahl S.M., Ledbetter S., Pratt B., Letterio J.J., Paul W.E., Berzofsky J.A. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J. Exp. Med. 2003;198:1741–1752. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahlers J.D., Belyakov I.M., Terabe M., Koka R., Donaldson D.D., Thomas E.K., Berzofsky J.A. A push-pull approach to maximize vaccine efficacy: abrogating suppression with an IL-13 inhibitor while augmenting help with granulocyte/macrophage colony-stimulating factor and CD40L. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13020–13025. doi: 10.1073/pnas.192251199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma D.P., Ramsay A.J., Maguire D.J., Rolph M.S., Ramshaw I.A. Interleukin-4 mediates down regulation of antiviral cytokine expression and cytotoxic T-lymphocyte responses and exacerbates vaccinia virus infection in vivo. J. Virol. 1996;70:7103–7107. doi: 10.1128/jvi.70.10.7103-7107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aung S., Tang Y.W., Graham B.S. Interleukin-4 diminishes CD8(+) respiratory syncytial virus-specific cytotoxic T-lymphocyte activity in vivo. J. Virol. 1999;73:8944–8949. doi: 10.1128/jvi.73.11.8944-8949.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aung S., Graham B.S. IL-4 diminishes perforin-mediated and increases Fas ligand-mediated cytotoxicity In vivo. J. Immunol. 2000;164:3487–3493. doi: 10.4049/jimmunol.164.7.3487. [DOI] [PubMed] [Google Scholar]
- 35.LeVine A.M., Gwozdz J., Stark J., Bruno M., Whitsett J., Korfhagen T. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J. Clin. Invest. 1999;103:1015–1021. doi: 10.1172/JCI5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LeVine A.M., Elliott J., Whitsett J.A., Srikiatkhachorn A., Crouch E., DeSilva N., Korfhagen T. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004;31:193–199. doi: 10.1165/rcmb.2003-0107OC. [DOI] [PubMed] [Google Scholar]
- 37.Lanone S., Zheng T., Zhu Z., Liu W., Lee C.G., Ma B., Chen Q., Homer R.J., Wang J., Rabach L.A., Rabach M.E., Shipley J.M., Shapiro S.D., Senior R.M., Elias J.A. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J. Clin. Invest. 2002;110:463–474. doi: 10.1172/JCI14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeo S.J., Yun Y.J., Lyu M.A., Woo S.Y., Woo E.R., Kim S.J., Lee H.J., Park H.K., Kook Y.H. Respiratory syncytial virus infection induces matrix metalloproteinase-9 expression in epithelial cells. Arch. Virol. 2002;147:229–242. doi: 10.1007/s705-002-8316-1. [DOI] [PubMed] [Google Scholar]
- 39.Fulkerson P.C., Fischetti C.A., Hassman L.M., Nikolaidis N.M., Rothenberg M.E. Persistent effects induced by IL-13 in the lung. Am. J. Respir. Cell Mol. Biol. 2006;35:337–346. doi: 10.1165/rcmb.2005-0474OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zavasnik-Bergant T., Turk B. Cysteine cathepsins in the immune response. Tissue Antigens. 2006;67:349–355. doi: 10.1111/j.1399-0039.2006.00585.x. [DOI] [PubMed] [Google Scholar]
- 41.Qiu Z., Hingley S.T., Simmons G., Yu C., Das S.J., Bates P., Weiss S.R. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006;80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schornberg K., Matsuyama S., Kabsch K., Delos S., Bouton A., White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]