

Abstract
A full set of SP6 promoter variants with all possible single substitutions at positions −17 to +5 was constructed. Transcription activities of these variants were individually measured in vivo and in vitro to determine the contribution of each base pair to the promoter activity. The in vivo activity was measured indirectly by transcriptional interference of the replication of promoter-bearing plasmids. This activity depends most highly on residues −11, −9, −8, −7, and +1 (initiation site). All substitutions at −11, −9, −8, and −7 abolished formation of closed complexes, except for A−8C. These residues are involved in base-specific interactions with the polymerase, and the substitutions exhibit the same strong inhibition in vitro. In contrast, the in vitro activities of some other variants, measured on linearized templates, were different from those in vivo. Some variants at −13, −4, and −2, among others, showed exceptionally higher activities in vivo than in vitro, supporting the possibility that these residues are involved in postbinding steps, including template melting and bending. The A−3T variant showed much lower activity in vivo than in vitro, but it bound to the polymerase 2-fold more than the consensus sequence and is possibly involved in polymerase binding. A quantitative hierarchy of all the base pairs is graphically displayed by activity logos, revealing the energetic contribution of each base pair to the activity.
The bacteriophage SP6 genome encodes a single-subunit RNA polymerase that is very similar to the T7, T3, and K11 RNA polymerases. SP6 promoters consist of a highly conserved 20-bp sequence that extends from positions −17 to +3 and exhibits a strong homology to the T7, T3, and K11 promoter sequences (1). Despite their homology, each polymerase shows highly stringent specificity for its own promoter sequence. Mutational studies with phage T7 promoters have yielded detailed information about the relationship of structure to function and the recognition of them by RNA polymerase. The promoter consists of two domains: an initiation domain downstream of −4 and a binding domain upstream of −5 (2, 3). Single base changes in the initiation domain have little effect on promoter binding but reduce the rate of transcription initiation. In contrast, changes in the binding domain reduce the efficiency of promoter binding but have little effect on the initiation of transcription. Base pairs at positions −11 through −7 of the T7 promoter are essential to the binding (4–7).
The interactions of phage RNA polymerases with their promoters have also been studied by a variety of biochemical methods. Footprinting studies with methidiumpropyl-EDTA-Fe(II) indicate that the T7 polymerase protects the region from −17 to −4 (8, 9). Methylation and ethylation interference studies indicate that the major groove of the promoter between −5 and −12 is important for polymerase binding (10). More recent studies with base analog substitutions reveal that, at positions −11 and −10 of the T3 and T7 promoters, their RNA polymerases recognize functional groups along the nontemplate strand wall of the major groove (11, 12). Li et al. (13) identified specific functional groups of residues −9 to −5 as primary contacts with T7 RNA polymerase.
The SP6 promoter has not been studied as thoroughly as the T7 promoter. Although the two promoters share a high degree of homology, there are some differences, especially in the promoter–RNA polymerase interaction region. Mutational studies with the SP6 promoter suggest that the SP6 polymerase binding domain could extend approximately to position −3 and that the polymerase-DNA contacts could be distributed in a broader region than in the T7 promoter (1, 14). In vitro transcription results with −9 and −8 mutants at various salt conditions suggest that SP6 RNA polymerase uses more nonionic forces for promoter interaction than T7 polymerase does (1).
In this study, we have constructed a library of SP6 promoter variants with all possible single base pair substitutions at positions −17 to +5. The strengths of these promoter variants were measured in vivo and in vitro to determine the contribution of each base pair to the promoter activity. Such a full saturation mutagenesis has not been applied even to the T7 promoter. A quantitative view of the base pair contributions is displayed by a method called activity logos, a term analogous to sequence logos (15).
Materials and Methods
Saturation Mutagenesis of the Phage SP6 Promoter.
A full set of single residue substitutions from −17 to +5 of the SP6 promoter was constructed by chemical synthesis. Oligonucleotides MutAC (5′-CTGGATCCaTTTaGGTGacacTaTaGaaGaAGTGATCA-GTCTAGATGCG-3′) and MutGT (5′-CTGGATCCAtttAggtgACACtAtAgAAgAAGTGATCAGTCTAGATGCG-3′) were synthesized on a 0.2-μmol scale with a Gene Assembler Plus DNA synthesizer (Amersham Pharmacia). They carried mixtures of bases in the positions designated by lower case letters; in these positions, the indicated base was 91%, and the other three bases were 3% each. The sequences are of the upper, nontemplate strands from −25 to +24. A 16-nt primer was synthesized, annealed to the 3′ ends of MutAC and MutGT, and extended by the Klenow fragment of Escherichia coli DNA polymerase I to make double-stranded DNA. The resulting 49-bp duplexes were then cleaved with XbaI and inserted into the XbaI–SmaI site of pSV2. The promoter assay vector pSV2 was derived from pGEM4Z (Promega). The 2.5-kilobase NdeI–EcoRI fragment of pGEM4Z was self-ligated after the ends were filled in. The 1-kilobase BamHI–BglII fragment of pGEMEX-2 (Promega) was inserted at the BamHI site of the resulting promoterless plasmid, resulting in pSV2. Individual promoter variants cloned in pSV2 (pSV-no.) were sequenced, and 40 of the 66 possible mutants were isolated from these random pools. The remaining 26 point mutants were obtained from the other nine oligonucleotides in the same way.
Determination of Plasmid Copy Numbers for in Vivo Promoter Strength Assays.
When E. coli cells harboring each pSV-no. were transformed with pACSP6R containing the SP6 RNA polymerase gene (16), each pSV copy number depended on the strength of its promoter (17). The copy number of pACSP6R was invariantly 220 per cell in the presence of strong or weak promoters, measured against the chromosome copy number as previously described by Lin-Chao and Bremer (18). Thus, the variable copy number of each pSV was determined by comparison to pACSP6R. JM109 cells grown in LB medium containing 100 μg/ml ampicillin and 25 μg/ml tetracycline were harvested, and plasmids were prepared from 3-ml aliquots by the alkaline lysis method (19). After electrophoresis in 0.8% agarose, gels were stained with ethidium bromide and destained in water. DNA bands were photographed by using Polaroid film type 667, and negative scan images were subjected to density analysis by imagequant version 3.3 (Molecular Dynamics). The copy number of each pSV, Ni, was calculated as follows:
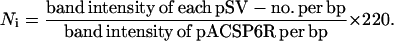 |
Measurement of Promoter Strength in Vitro.
The strengths of SP6 promoter variants were individually determined in vitro by measuring the production of RNA from a linearized plasmid containing each variant. Transcription reactions were carried out in 20 μl of mixture containing 40 mM Tris⋅HCl (pH 7.5), 6 mM MgCl2, 2 mM spermidine, 10 mM DTT, 0.5 mM each ribonucleotide, 0.2 units/μl RNasin, 1 μg of linear plasmid DNA, 0.5 units/μl of SP6 RNA polymerase (Promega), and 0.3 μM [α-32P]CTP (400 Ci/mmol; 1 Ci = 37 GBq). The reactions were terminated after a 30-min incubation at 37°C by adding 20 μl of the gel loading buffer containing 80% (vol/vol) deionized formamide, 10 mM EDTA (pH 8.0), 0.025% xylene cyanole, and 0.025% bromophenol blue. After 8 M urea/6% polyacrylamide gel electrophoresis, the gels were exposed to Agfa x-ray film Curix at −70°C. By using the autoradiogram as a guide, the radioactive bands were excised from the gel, and the radioactivity was measured by liquid scintillation counting. Alternatively, it was measured by PhosphorImager analysis with a Storm 860 scanner (Molecular Dynamics).
Results
Relative Promoter Strength in Vivo.
To determine the contribution of each residue of the SP6 promoter to the transcription initiation efficiency, all single residue substitutions were introduced in every position from −17 to +5. (The initiation site is numbered +1.) Transcription activity of each cloned variant was measured in vivo by measuring the copy number of an SP6 promoter-bearing plasmid, rather than by measuring a reporter gene expression. When SP6 RNA polymerase is present in E. coli JM109 cells, the stability of SP6 promoter-bearing plasmids depends on the orientation of the promoter relative to the replication origin and on the sequence of the origin (17). When the SP6 promoter is directed toward the replication origin of pGEM4Z, the copy number is quantitatively correlated with the efficiency of phage transcription (17). The promoter variants were cloned in pSV2, a derivative of pGEM4Z, and the resulting recombinants, pSV-nos., were introduced into JM109/pACSP6R cells producing SP6 RNA polymerase under the control of lac promoter, as shown in Fig. 1. When a promoter variant is active, an extended form of RNA I is synthesized that acts as an inhibitor of the replication primer, RNA II, resulting in reduction in the pSV copy number.
Figure 1.

In vivo transcription assay based on interference of the replication of pSV-no. plasmids by SP6 transcription. SP6 RNA polymerase is produced from the gene under the control of the lac promoter in plasmid pACSP6R. It recognizes an SP6 promoter variant on a pSV-no. plasmid and produces a transcript that contains a 3′ RNA I sequence. This RNA interacts with RNA II and reduces the pSV-no. copy number. Thus, the copy number depends on strength of the promoter variant.
A kinetic model for replication control of ColE1-type plasmids was previously developed by Brendel and Perelson (20). The plasmid copy number depends most strongly on the rate of RNA I synthesis. It also increases linearly with RNA II synthesis rate and with cell doubling time, but in our studies, these two parameters were constant. Minor modifications were made in our studies to relate the in vivo promoter activity (rate of RNA I synthesis) to the copy number of pSV (Fig. 2). In our system, Rom protein is not involved, and the interaction between RNA I and RNA II is weakened, resulting in the high copy number of pGEM4Z (19). All the kinetic parameters were adopted from the original model, except for those involving the RNA I–RNA II complex (k1, k−1, and k2). The parameter k−2 was not changed, because its value was too small compared with k2. Values of the three parameters were adjusted to set the copy number (sum of all the forms in Fig. 2) at 400 per cell (for pGEM4Z derivatives). The copy number of each pSV-no. variant will depend on its rate of RNA I synthesis, kI, that is controlled by an SP6 promoter variant. The kI for each promoter variant, kIi, was calculated from the measured plasmid copy number, Ni, of that variant.
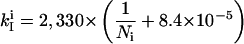 |
The relative activity of each promoter variant, ai, compared with the consensus promoter, was calculated as follows:
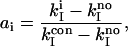 |
where kIcon is for the plasmid containing the consensus SP6 promoter and kIno is for pSV2 that does not contain an SP6 promoter sequence.
Figure 2.

Kinetic scheme of the ColE1 replication control mechanism slightly modified from the model of Brendel and Perelson (20). The parent plasmid of pSV-no., pGEM4Z, does not produce the Rom protein. The values of parameters k1, k−1, and k2 were modified to 4.2 × 107 M−1⋅min−1, 202 min−1, and 16.5 min−1, respectively, to fix the copy number of pGEM4Z at 400 per cell. Other parameters are the same as described (20). Plasmid DNA occurs either free (D) or in association with RNA II transcripts (DIIs for short transcripts or DIIl for longer transcripts), with RNA II primer (Dp), or with bound complex (Dc* and Dc). Short-length plasmid-bound RNA II (DIIs) forms an unstable complex with RNA I (Dc*). Replication converts primed DNA (Dp) to free DNA (D). The conversions between the different states occur at the rates indicated.
The relative in vivo activities of the 66 SP6 promoter variants determined are shown in Fig. 3. None showed significantly higher activity than the consensus promoter. The effects of the substitutions varied greatly: 6 substitutions did not affect the activity at all, and a third (n = 22) of the 66 substitutions reduced the activity to less than 10% of that of the consensus promoter.
Figure 3.

Relative in vivo transcription efficiencies of SP6 promoter variants compared with the consensus SP6 promoter. Each position was classified as described in the text, and the classifications (classes A to D) are shown at the bottom. An average of more than three measurements was taken for each variant.
The 22 positions from −17 to +5 were divided into four classes depending on the substitution effects. All three substitutions reduced the activity to below one-third the consensus level in positions −11, −9, −8, −7, and +1 (class A). Two of the three substitutions showed such reduction in positions −15, −14, −6, −5, −4, −2, and −1 (class B), whereas only one substitution did so in positions −13, −3, and +2 (class C). In the remaining seven positions, no substitutions showed this much reduction (class D). This classification qualitatively reflects the relative importance of the position to the promoter activity (see below), class A being the highest.
Relative Promoter Strength in Vitro.
Transcription activities of SP6 promoter variants on linearized plasmid templates were measured individually under standard conditions (at 6 mM MgCl2) and compared with the consensus promoter activity (Fig. 4). Again, the effects of the substitutions greatly varied. The activity range was greater in vitro (from 0.01 to 120% of the consensus promoter activity) than in vivo (from 5 to 100%). Interestingly, T-to-A substitution at the −10 position (designated T−10A here) increased the activity by 20%, and T−10G, A−3T, and G+4T substitutions did not affect it (98–105 ± 13%) within experimental error. About a half of the variants (n = 35) had less than one-third of the consensus promoter activity. When the 22 positions were divided into four classes based on in vitro data, 6 positions changed class compared with the in vivo results. Three more positions, −5, −4, and −2, joined the class A in vitro. Position −13 joined class B, and positions −1 and +3 joined class C. All these changes represent an increase in importance in vitro, except for −1.
Figure 4.

Relative in vitro transcription efficiencies of SP6 promoter variants compared with consensus SP6 promoter. Each position was classified as described in the text, and the classifications (classes A to D) are shown at the bottom. An average of more than three measurements was taken for each variant.
The differences between the in vivo and in vitro activities of each variant are shown in Fig. 5. Some are significantly outside the maximum standard deviation for the difference values (about ± 20%). The most dramatic difference was shown by the T−4C variant: 100 ± 10% in vivo but 2 ± 1% in vitro. Several other variants showed much higher activities in vivo than in vitro, including A−13C. Variant A−3T was the only one that showed the opposite effect.
Figure 5.

Difference between in vivo and in vitro activities of the promoter variants. Each bar results from subtracting the in vitro activity from the in vivo activity, separately expressed as the percentage of the consensus promoter activity. Differences shown in the shaded area are within experimental error. Residues possibly involved in polymerase binding (filled boxes) and those displaying high in vivo:in vitro activity ratios (open boxes) are highlighted at the bottom.
Some of the variants were assayed for polymerase binding or formation of closed complex by gel retardation (data not shown), as previously described (21). All substitutions at −11, −9, −8, and −7 abolished polymerase binding, except for the variant A−8C that retained a third of the binding affinity of the consensus promoter. Thus, loss or reduction of transcription activity by these substitutions is attributed by loss or reduction of polymerase binding. The binding affinity of T−10A, the only variant that was significantly more active in vitro than the consensus promoter, was similar to that of the consensus, suggesting that the mutation enhanced a kinetic step after polymerase binding. Variant A−3T bound to the polymerase over 2-fold more than the consensus, suggesting that a subsequent step or steps were inhibited both in vitro and in vivo.
Abortive initiation products from variants carrying substitutions at −1 through +5, except for +1, were closely examined. The largest products of abortive initiation cycling were 6 nt long as previously reported (22). Only the G+4T variant produced greatly increased amounts of 6-mers, suggesting that this residue plays an important role in abortive initiation cycling.
Activity Logos: A Method for the Display of Saturation Mutagenesis Data.
All possible base pairs at each position were individually tested in this study for their effects on promoter activity in in vivo and in vitro assays. A graphic method to display all these data is desirable. Sequence logos (15) display consensus sequence information from a set of comparative sequence data and provide a quantitative view of the amount of information at each point in the functional site, based on information theory. In this study, a method displaying any saturation mutagenesis data was developed based on energetics; the resulting displays are called activity logos, a term analogous to sequence logos.
Activity is not simply a sum of individual residue contributions, but as a first approximation, it can be treated this way (23). In a binding assay, the binding constant, K, can be considered to reflect the free energies, ΔGi, contributed by each residue in the binding site.
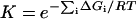 |
If a pure rate is measured, the rate constant k2 reflects an activation energy, with contribution from each residue, Ei:
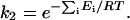 |
When the assay depends on the product of the rate and a preequilibrium binding, the activity, V, will depend on:
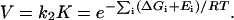 |
For example, transcription assays in this study depend on the product of the equilibrium of polymerase-promoter binding and the rates of isomerization and promoter clearance.
Because all of these equations have the same form, we can describe the activity a, for any assay as:
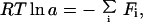 |
where Fi indicates the relative energetic contribution of the ith position. If we change the identity of the base at position i from the normal base to base b, an altered activity abi may result. The energy change at that position will be Fbi, if we treat the positions as independent.
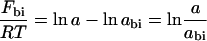 |
The relative energetic importance of the ith position, Hio can be estimated by adding the effects of each possible base on activity.
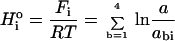 |
The normal base should refer to the base yielding the maximum activity, which is not necessarily a consensus or wild-type base, such that ln(a/abi) is always positive. This formalism weights equally any depression in energetics that affects activity. Thus, a position with relative activities A = 1, C = G = T = 0.1 and a position with A = C = G = 1, T = 0.001 would contribute equally.
At each position, an activity logo, the equivalent of a sequence logo, can be constructed by weighting Hio by the relative activities seen with each of the four bases.
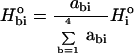 |
A correction must be made for the limited dynamic range and signal to noise of activity measurements. To prevent abi from ever assuming an unrealistically small value, all observed activities can be incremented by a small constant ɛ, say 0.01 a. Thus, the final form of the activity logos becomes
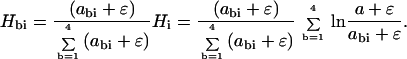 |
Hi is the height of the logo for position i, and Hbi is the portion for base b.
Our extensive set of in vivo and in vitro activity data on all the point mutations of the SP6 promoter is displayed by activity logos in Fig. 6. The qualitative classification of base pair positions, described earlier, is fairly consistent with this quantitative view of the energetic contribution of every position, except for a few positions in classes B and C. The activity logos in Fig. 6 are also compared with sequence logos constructed from 11 SP6 promoter sequences (1). Sequence logos appear to be high-contrast, low-resolution versions of the activity logos. They may resemble in vivo logos more than in vitro logos, as reflected by the results for position −3.
Figure 6.

Activity and sequence logos of the phage SP6 promoter. The two activity logos were constructed based on energetic contribution of each base pair in every position to the promoter activity as described in the text. Maximum heights of the activity logos simply reflect lower limits of measurements. The maximum information content in each residue is 2 in sequence logos. The sequence logos were generated from 11 known SP6 promoter sequences (1) by http://www.bio.cam.ac.uk/seqlogo/logo.cgi (15).
Discussion
Although extensive information on phage T7 promoters has accumulated through many independent approaches, none of them covered the entire length of the promoter. In this study, all possible substitutions were introduced into every position of the phage SP6 promoter, and their effects on the promoter activity were measured in vivo and in vitro, presenting the first example of full saturation promoter mutagenesis. Analysis of 11 known SP6 promoter sequences showed that only positions −17 to +3 contain significant sequence information (1), as shown in the sequence logos of Fig. 6. An in vitro evolution study showed no sequence requirements at positions −22 to −18 (24). Escape from abortive initiation cycling occurs after formation of 6-nt RNA in SP6 transcription (22). Thus, our coverage from −17 to +5 includes all the residues of the SP6 promoter.
The activity logos shown in Fig. 6 reveal quantitative hierarchies of all the possible base pairs in vivo and in vitro. Requirement for GTP as an initiating residue seems to be much more stringent in SP6 transcription than in T7 (5). Phage promoters have been thought to consist of two parts, largely based on studies on the T7 promoter. The region from −17 to −5 is for polymerase binding, and that from −4 to +3 is for melting of template duplex and initiation (2, 3). Within the presumably polymerase-binding region, the residues at −17, −16, −12, and −10 of the SP6 promoter show no evidence of base-specific interactions. The most important positions are −11, −9, −8, and −7, and these are involved in polymerase binding. We previously reported that the two base pairs at positions −9 and −8 distinguish between the SP6 and T7 promoters (1). The two consensus promoters are identical at −11 and −7. Although they differ also at −17, −16, −15, −12, and −10 (1), these positions do not contribute to the activity, except for −15. Substitution of T7-specific A for the SP6 T at −15 resulted in no activity loss at all in vivo and only half activity in vitro (Figs. 3 and 4).
Jorgensen et al. (10) reported that methylation of the G residues at positions −12, −11, −9, −7, and −5 interfered with binding of the polymerase and suggested that the major groove between −5 and −12 is a contact region. These results are consistent with ours, except for the role of the −12 residue. Any mutations in this position retained 56–100% activity, and a substitution of T (having a methyl group) resulted in 67–69% activity both in vivo and in vitro (Figs. 3 and 4).
The effects of mutation at the four most important positions on transcription activity did not show any significant differences in vivo and in vitro. These results probably reflect that the base-specific interactions with the polymerase are not significantly different in vivo and in vitro.
On the other hand, positions −17, −16, −15, −13, −12, −10, −4, −3, −2, and +3 showed great differences in vivo and in vitro (Fig. 5). Among these, −13C, −4C, −3T, −2A, and +3C showed especially meaningful differences (higher than two-thirds activity under one condition but lower than one-third activity under the other), whereas the others exhibited always higher than 50% activity. These differences in activity may be due to the difference in DNA template structure. The templates presumably formed negatively supercoiled structures in vivo but were linear in vitro. In fact, all the variants showing significant differential effects favored in vivo conditions, except for the A−3T (Fig. 5). The T7 RNA polymerase indirectly recognizes the A+T-rich sequence in the minor groove at −17 to −13 through its inherent flexibility, and these distortions cause a slight bend (25). If the SP6 polymerase recognizes the A+T-rich region in the same way, our results suggest that supercoiled structure might help the structural modulation.
The T7 promoter residues −4 to +3 are involved in postbinding steps, including melting of duplex in the complex (2, 3). The SP6 variant A−3T that showed higher activity in vitro than in vivo was found to bind the polymerase much more than the consensus promoter, according to the gel shift assay. Thus, it is possible that the SP6 −3 residue is involved in polymerase binding in a relatively nonspecific manner (14).
The relative in vivo activity of each promoter variant was measured in this study by the interference in plasmid replication by transcription. This indirect assay for promoter strength has an advantage over a direct assay of RNA measurement (like in our in vitro assay) or an indirect assay for activity of a reporter protein. In this in vivo assay, strong mutational effects result in high, rather than low, copy numbers of the plasmid, which can be measured very accurately. This advantage is significant especially in construction of activity logos, because strong mutational effects are weighted logarithmically more than weak ones.
The exploding availability of DNA sequence data creates a challenge to devise ways of displaying those data in a manner that conveys comprehensible clues about function. The kind of data most frequently available is comparative sequence information such as which sequences function naturally as promoters for a particular RNA polymerase or which mutations are known to inactivate a promoter. Molecular biologists have most often dealt with such data by constructing a consensus sequence. However, such sequences convey only part of the information available from a set of comparative sequence data. Sequence logos, soundly based on information theory, provide a quantitative view of the amount of information at each point in the functional site. They do not concern the different activities of individual sequences. Activity logos, based on energetics, provide a quantitative view of the energetic contribution of each residue to the function, from measurements of individual activities.
Acknowledgments
We thank Dr. John Rush and Prof. Charles C. Richardson at Harvard Medical School for sharing unpublished sequences of phage SP6 genomic promoters and Prof. Peter von Hippel at University of Oregon for very helpful comments on the manuscript. This work was partially supported by grants from the Ministry of Education Genetic Engineering Program and from the Korea Advanced Institute of Science and Technology.
References
- 1.Lee S S, Kang C. J Biol Chem. 1992;268:19299–19304. [PubMed] [Google Scholar]
- 2.Chapman K A, Burgess R R. Nucleic Acids Res. 1987;15:5413–5432. doi: 10.1093/nar/15.13.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman K A, Gunderson S I, Anello M, Wells R D, Burgess R R. Nucleic Acids Res. 1988;16:4511–4524. doi: 10.1093/nar/16.10.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider T D, Stormo G D. Nucleic Acids Res. 1989;17:659–674. doi: 10.1093/nar/17.2.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ikeda R A, Ligman C M, Warshamana S. Nucleic Acids Res. 1992;20:2517–2524. doi: 10.1093/nar/20.10.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda R A, Warshamana G S, Chang L L. Biochemistry. 1992;31:9073–9080. doi: 10.1021/bi00152a051. [DOI] [PubMed] [Google Scholar]
- 7.Diaz G A, Raskin C A, McAllister W T. J Mol Biol. 1993;229:805–811. doi: 10.1006/jmbi.1993.1086. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda R A, Richardson C C. Proc Natl Acad Sci USA. 1986;83:3614–3618. doi: 10.1073/pnas.83.11.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunderson S I, Chapman K A, Burgess R R. Biochemistry. 1987;26:1539–1546. doi: 10.1021/bi00380a007. [DOI] [PubMed] [Google Scholar]
- 10.Jorgensen E D, Durbin R K, Risman S S, McAllister W T. J Biol Chem. 1991;266:645–651. [PubMed] [Google Scholar]
- 11.Schick C, Martin C T. Biochemistry. 1993;32:4275–4280. doi: 10.1021/bi00067a016. [DOI] [PubMed] [Google Scholar]
- 12.Schick C, Martin C T. Biochemistry. 1995;34:666–672. doi: 10.1021/bi00002a034. [DOI] [PubMed] [Google Scholar]
- 13.Li T, Ho H H, Maslak M, Schick C, Martin C T. Biochemistry. 1996;35:3722–3727. doi: 10.1021/bi9524373. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Hong W J, Kang C. Biochem Mol Biol Int. 1993;31:153–159. [PubMed] [Google Scholar]
- 15.Schneider T D, Stephens R M. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong W J, Kang C. Biochem Mol Biol Int. 1997;42:711–716. doi: 10.1080/15216549700203131. [DOI] [PubMed] [Google Scholar]
- 17.Kwon Y, Kim J, Kang C. Genet Anal Biomol Eng. 1998;14:133–139. doi: 10.1016/s1050-3862(98)00007-2. [DOI] [PubMed] [Google Scholar]
- 18.Lin-Chao S, Bremer H. Mol Gen Genet. 1986;203:143–149. doi: 10.1007/BF00330395. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 20.Brendel V, Perelson A S. J Mol Biol. 1993;229:860–872. doi: 10.1006/jmbi.1993.1092. [DOI] [PubMed] [Google Scholar]
- 21.Muller D K, Martin C T, Coleman J E. Biochemistry. 1988;27:5763–5771. doi: 10.1021/bi00415a055. [DOI] [PubMed] [Google Scholar]
- 22.Nam S C, Kang C. J Biol Chem. 1988;263:18123–18127. [PubMed] [Google Scholar]
- 23.Berg O G, von Hippel P H. Trends Biochem Sci. 1988;13:207–211. doi: 10.1016/0968-0004(88)90085-0. [DOI] [PubMed] [Google Scholar]
- 24.Breakder R R, Banerji A, Joyce G F. Biochemistry. 1994;33:11980–11986. doi: 10.1021/bi00205a037. [DOI] [PubMed] [Google Scholar]
- 25.Cheetham G M, Jeruzalmi D, Steitz T. Nature (London) 1999;399:80–83. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]