

Abstract
The three-dimensional fold of a new protein sequence can often be inferred directly from sequence homology to a protein of known structure. The function of a new protein sequence is more difficult to predict, however, since homologues can have different molecular and cellular functions. To develop and automate computational methods for determining molecular function, we have analyzed ligand-binding specificity in two related families of binding proteins. One of these families includes Escherichia coli lactose repressor and ribose-binding protein, and the other includes E. coli sulfate- and phosphate-binding proteins. These proteins have similar folds but varying specificity, binding many different small molecules, including mono- and disaccharides, purines, oxyanions, ferric iron, and polyamines. Starting from template structural alignments, alignments of over 90 sequences per family were generated by iterative database searches with hidden Markov models. Phylogenetic trees were made of full-length sequences and of subsets of residues lining the binding cleft, to determine whether subbranches of the trees correlate with ligand-binding preference. Automated analyses of residues in the binding pocket were also used to predict ligand-binding function for many uncharacterized database sequences and to identify specific side chain–ligand contacts in proteins without solved structures. Our results demonstrate the utility of anchoring functional annotation within a protein family context.
The need for computational assignment of gene function is becoming more pressing as the number of new sequences far surpasses our ability to perform experiments to determine their functions. Initiatives for targeted three-dimensional structure determination for proteins of unknown fold (1, 2), coupled with advances in threading and other fold prediction methods, will soon lead to correct automated fold assignment for most new protein sequences. These advances will not as readily lead to correct functional annotation, however, since many different molecular and cellular functions may be associated with the same protein fold.
The functional annotation of a new protein is often transferred from the homologous protein with the highest blast score. Because of the modularity of protein domains and divergence of function in paralogous proteins, this practice has led to many incorrect annotations and the propagation of annotation errors in sequence databases (3–5). Annotation accuracy may be improved by considering the evolutionary history of each protein family (6) or by requiring a match to particular functional residues in a sequence motif, in addition to overall homology, in order for functional annotation to be transferred. These methods generally require careful analysis of a particular family of sequences and have found the most success in recognizing conserved catalytic residues in the active sites of enzymes (7–10).
Many proteins, however, have molecular functions that are defined by noncatalytic interactions with ligands or other proteins. Function is more difficult to derive from sequence in these families, because there may not be strictly conserved residues responsible for the common binding function. This is the case for several related families of periplasmic binding proteins (PBPs) in bacteria, which serve as receptors for many different ligands. One family of these proteins includes Escherichia coli ribose-binding protein (RbsB) and also the effector-binding domains of E. coli lactose repressor (LacI), purine repressor (PurR), and trehalose repressor (TreR). A second family, with distant sequence homology to the first and a slightly different three-dimensional fold, includes E. coli sulfate-binding protein (SubI) and molybdate-binding protein (ModA). Altogether there are more than a dozen different protein–ligand complexes in these two families whose structures have been solved crystallographically. Despite the diversity of ligands, these proteins have virtually identical backbone structures surrounding the ligand-binding cleft, implying that residue side chains within the binding pocket are critical determinants of specificity.
We have analyzed ligand specificity for the RbsB/LacI and SubI families to develop methods of structure–function classification from sequence that are generally applicable to families of receptors and binding proteins as well as to enzymes. Starting with multiple structural alignments, we constructed multiple sequence alignments of diverse family members with an iterated approach using profile hidden Markov models (HMMs). We have previously developed programs to automate the merger of structural information with multiple sequence alignments (11) and continue this work here with software that automates the integration of ligand-binding information from known structures with multiple alignments. These programs allowed direct comparison of binding-site residues across the two families, facilitating the prediction of ligand-binding function for several unannotated sequences and prediction of specific residue–ligand contacts in proteins without solved structures. Phylogenetic classifications of the families and of subalignments of binding-site residues were also used to identify clusters of protein sequences with similar binding pockets. Our results also offer to functional and structural genomics efforts an example of how densely the space of protein folds must be sampled with experimental results to predict molecular function for families of receptors.
Several other computational methods have been used to relate sequence and function for proteins of known structure, including hierarchical analyses of residue conservation patterns (12, 13) and multivariate analysis (14, 15). Two of these methods, evolutionary trace (13) and sequence space (15), have been used recently to address the important problem of identifying interaction surfaces and other functional residues in proteins of known structure. While these methods use prior knowledge of protein functional classes to predict the location of binding sites, the methods we apply here use prior knowledge of the binding-site residues to predict protein function.
Methods
Multiple Alignments.
Multiple alignments were constructed by iterative HMM searches of Swiss-Prot (16) and GenBank (17) nonredundant databases by using hmmer 2.1 (18), starting from seed multiple structural alignments. For the LacI/RbsB family, the initial structural alignment was generated using the Homology module of insightii (Molecular Simulations, San Diego). Only the effector-binding domains were used from the repressors. For the SubI family, the seed multiple structural alignment was generated by dali/fssp (19), with a few corrections made by hand in the loop regions. Automated HMM searches and multiple sequence alignment were performed for the SubI family by using alignmentbuilder, which was written in Perl to use hmmer 2, with starting hmmsearch E-value of e−130, step factor e20, and final E-value of e−10. The alignment model was allowed to converge at each E-value step before decreasing the stringency of the search, and sequences more than 90% identical to model sequences were not included. alignmentbuilder was run first with Swiss-Prot, then the alignment was checked by hand for concurrent alignment of close homologues and to eliminate a few gaps in loop regions. A new HMM was then constructed from this alignment, and alignmentbuilder was run on the nonredundant database with the same parameters. One additional alignmentbuilder step was then performed to a final convergence at E = 1.0e−9. The “withali” option of hmmer 2 was used for all hmmalign steps to avoid model drift as new sequences were added. Sequence edition was performed by using seaview (20).
Ligand-Binding-Site Analysis.
Binding-site residues for each structure were defined as those with a side-chain heavy atom < 4.5 Å from the ligand. We included α-carbon atoms in the side-chain definition so that glycine residues that form part of the binding pocket were not excluded. Residues with side chains making a crystallographically defined water-mediated hydrogen bond were also considered part of the binding pocket. Contacts to both sugar anomers were included for 1byk (21) and 1abe (22), and contacts from 2gbp (23) were included in the analysis of the nearly identical structure 1gca (24). Contacts to trehalose and trehalose-6-phosphate were included for 1byk, and contacts to the bound phosphate moiety that coordinates the iron atom in 1mrp (25) were also included. Hydrogen bonds from backbone atoms were ignored.
A series of Perl programs was written to automate the comparison of alignment sequences to each known binding site. A protein sequence was added to the list of possible matches for a given structure if it had binding-site identity > 50% or similarity > 65%. The output list was ordered by using a score based on the following measures: percentage identity and similarity to binding-site residues, the number of standard deviations of these from the mean over all sequences in the alignment, and whole-domain percentage identity and similarity.
Binding-site residue matches were determined by the presence of appropriate interacting chemical groups at each alignment position. Hydrophobic and van der Waals contacts were evaluated by size and hydrophobicity in the context of the structure. Binding-site residues with side chains projecting away from the binding site or out into solvent, or residues whose only contact is a water-mediated hydrogen bond, were not included in the determination of binding-site matches (the nonshaded columns in Figs. 4 and 5).
Figure 4.

High-scoring sequence matches to the ligand-binding residue set of LacI, showing that residues in contact with IPTG are largely preserved for the sequence that has been annotated as “LacI” in Klebsiella pneumoniae and for ORF 3367755. A simple scoring scheme was used to order the output on the basis of subset and whole-domain sequence similarity. Numbers below the alignment indicate the interacting hydroxyl groups of the IPTG structure (below). Columns that are not highlighted correspond to residues that have a side-chain atom ≤4.5 Å from IPTG, but which do not appear to make direct contact. Asterisks indicate less plausible residue substitutions in the binding site (see text).
Figure 5.

High-scoring sequence matches to the 4.5-Å ligand-binding residue set of E. coli ribose-binding protein. Residues that cannot duplicate the chemical interaction of RBSB_ECOLI with ribose at a particular alignment position are circled. Sequences with circled residues are not considered to have binding-site matches. Two lower-scoring sequences that qualify as binding-site matches are DEGA_BACSU and 2612909 (below the dotted line).
Phylogenetic trees for whole-domain alignments and binding-site subalignments were created with phylip Ver. 3.5c (J. Felsenstein and Department of Genetics, University of Washington, Seattle) with 300–500 bootstrap replicates using the neighbor-joining method and the Pam–Dayhoff distance matrix. Multiple alignments, dendrograms, and tables of binding-site matches are available at http://winslow.med.harvard.edu/johnson/.
Results and Discussion
We analyzed two evolutionarily related classes of PBP-like proteins with slightly different topological arrangements of a central β-sheet core (26). Both Type I and Type II PBPs are bilobate α/β structures with a central ligand-binding cleft (Fig. 1). The residues lining the binding cleft are distributed throughout the primary amino acid structure, such that there is no local sequence motif that may be associated with a particular binding function. PBP sequence alignments were created with an iterative HMM-based approach, starting from initial alignments of closely related structures (see Methods). Two structural alignments were used as starting sequence alignment models, one of Type I PBP structures most similar to E. coli ribose-binding protein and the effector domain of LacI, and one of Type II PBP structures similar to E. coli sulfate-binding protein. We used structural alignments as seeds for larger HMM-based sequence alignments to increase sequence diversity in the alignments without compromising alignment accuracy.
Figure 1.
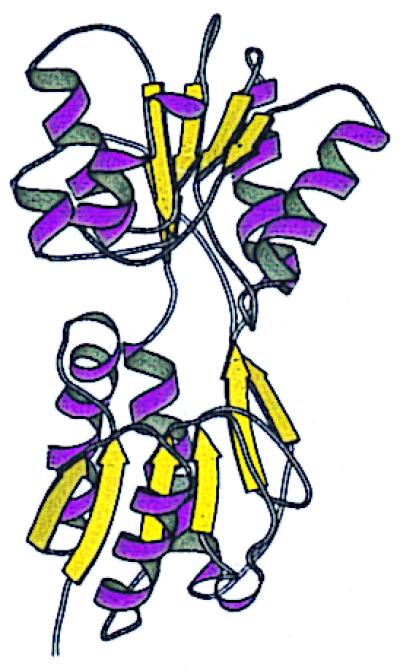
Ribbon structure of the effector-binding domain of trehalose repressor, a member of the LacI/RbsB family, with β-strands in yellow and α-helices in violet (21). Trehalose (not shown) binds in the central cleft between the two lobes. The figure was created with molscript (45).
LacI/RbsB Family Alignment.
The Protein Data Bank (PDB; ref. 27) structures 1tlf, 2dri, 1abe, 1byk, 1gca, 1rpj, and 1wet were used for the starting LacI/RbsB family HMM; each has less than 3.0 Å rms deviation (Cα) relative to the structure of E. coli RbsB (2dri) over a minimum of 240 residues. These structures were solved with bound isopropyl-d-thiogalactoside (IPTG), ribose, arabinose, trehalose-6-phosphate, galactose, allose, and guanine, respectively (21, 22, 24, 28–31). Sequences were added gradually to the alignment model, which was checked manually after every iteration to ensure that the seed structures maintained alignment across structurally conserved regions (SCRs), that closely related sequences aligned to the model with concurrent gaps, and that new alignment gaps made structural sense (e.g., to avoid unnecessary insertions in SCRs). The final sequence alignment for this family of Type I PBPs comprised 102 sequences with 20% average sequence identity.
LacI/RbsB Family Phylogenetic Tree.
A phylogenetic tree was generated from this alignment to identify clusters of similar protein domains (Fig. 2), and statistically significant branches were compared with previously recognized functional subdivisions of the family. Although we refer to these dendrograms as phylogenetic trees, we are using this technique only to cluster similar sets of residues rather than to make inferences about the evolutionary history of these proteins. Known periplasmic receptors segregate on one significant branch, apart from known DNA-binding proteins, even though the DNA-binding domains of the transcriptional regulators were not included in the multiple alignment. Thus, for example, ribose PBPs such as RBSB_ECOLI do not cluster with ribose-binding repressors (RBSR_ECOLI, RBSR_HAEIN, and RBSR_BACSU), and xylose PBPs (e.g., XYLF_ECOLI) do not cluster with xylose operon repressors (e.g., XYLR_ECOLI). However, within each of the two broad functional categories, several statistically significant smaller clusters appear to correlate with known or hypothesized ligand preferences (see Fig. 2). Several exceptions to this are also apparent. For example, Gram-negative ribose repressors (RBSR_ECOLI and RBSR_HAIEN) are more similar to Gram-negative purine repressors (e.g., PURR_HAEIN) than they are to the Gram-positive ribose repressor RBSR_BACSU. Further, some proteins annotated as sucrose operon repressors (e.g., SCRR_SALTY) cluster with fructose repressors, others cluster with the Bacillus subtilis ribose repressor (e.g., SCRR_PEDPE), and yet another, SCRR_STAXY, clusters with trehalose-binding regulatory proteins. Thus, the clusters of the whole-domain phylogenetic tree are generally not predictive of common ligand-binding function, except in the outermost branches, where sequence identity approaches 50% or more. In addition, proteins that bind the same ligand are frequently found in different whole-domain phylogenetic clusters.
Figure 2.

Three methods for predicting groups of functionally related sequences, applied to the LacI/RbsB family. Significant clusters from a phylogenetic tree of the LacI/RbsB family, created from the final whole-domain multiple alignment by using phylip (see Methods), are indicated with black solid lines. Tree roots not present in ≥30% of the bootstrap replicates were removed. Known PBPs fall into one significant phylogenetic cluster (Left). The remaining sequences are likely to be effector-binding domains of DNA-binding proteins. Proteins are labeled with Swiss-Prot or GenBank accession numbers and with boldface type for solved PDB structures. The bound ligands are the following: RBSB, ribose; YJCX, allose; DGAL, glucose/galactose; ARAF, arabinose; PURR, guanine; LACI, IPTG; TRER, trehalose. The second clustering method is indicated by yellow boxes. These sequence groups were present in ≥30% of bootstrap replicates for the subalignment of ligand-binding residues. A third method of function prediction is indicated by blue boxes; these are sequences whose aligned binding-site residues are capable of matching the ligand-binding interactions of a known structure. Green shading indicates overlap between yellow and blue groups. Only one cluster, the binding-site matches to E. coli RBSB_ECOLI, includes both DNA-binding proteins and PBPs, indicated by a connecting dotted line. Asterisks identify sequences that did not fall into a significant cluster in the whole-domain phylogeny but that belong to a significant cluster using one of the other two methods.
LacI/RbsB Family Binding-Site Phylogenetic Tree.
We hypothesized that the residues lining the binding cleft of this family might be better predictors of ligand-binding function than whole-domain sequences. For each solved structure in the alignment, residues with side chains within 4.5 Å of their respective ligands were identified, and the columns of the multiple alignment that contained these binding-site residues were extracted. This composite set of 32 contact alignment positions compiled from seven structures, which we will refer to as the binding-site subalignment, was then used for phylogenetic clustering.
With only 32 alignment positions, the binding-site subalignment produces fewer statistically significant clusters than the whole-domain alignment, which has over 400 alignment positions. However, clusters present in the whole-domain phylogeny that were also significant in the binding-site phylogeny (Fig. 2, yellow and green boxes) correlate well with known ligand preferences. Only the cluster that includes E. coli ribose and purine repressors is obviously representative of multiple ligand types. The other clusters from the whole-domain phylogeny that appear to represent multiple ligands, including all of the sucrose repressor clusters, have subdivided into smaller clusters in the binding-site subalignment that are more likely to represent common binding functions. One group of two sequences, CYTR_ECOLI and MALR_CLOBU, was significant at the binding-site level but was not in the whole-domain phylogeny. CytR is known to be regulated by cytidine (32), and MalR in Clostridium butyricum is involved in regulation of 4-α-glucanotransferase (33). The significance of binding-site similarity between these two proteins is unclear.
Matches to Ligand-Binding Sites of Known LacI/RbsB Family Structures.
The set of contact residues for each known structure was also compared with each sequence in the multiple alignment to look for matches. Fig. 3 shows the results of this sequence comparison for E. coli LacI, highlighting two proteins that match the LacI ligand-binding residues with the highest similarity. The first, a protein that has been annotated as LacI from Klebsiella pneumoniae, is 40% identical to E. coli LacI over the whole domain, but is almost 90% identical over the set of ligand-binding residues, strongly supporting its functional annotation. The second, an ORF from Streptomyces coelicolor (GenBank accession no. 3367755), is less than 30% identical to LacI over the whole domain, but has a very similar binding site (Fig. 3, arrows). The alignment of the LacI binding-site residues with LACI_KLEPN and 3367755 is shown in Fig. 4. This level of analysis adds support to the hypothesis that these two proteins bind ligands similar to lactose, because the hydrophobic and hydrogen-bonding interactions made by the side chains of E. coli LacI to IPTG could be replicated by the aligned residues from both homologues. Molecular modeling of 3367755 based on the LacI binding site indicates that an arginine residue can be accommodated at position 76 with small conformational changes to neighboring residues. However, the incorporation of a tryptophan residue at position 191 would require either a ligand smaller than IPTG or major conformational changes to neighboring residues, suggesting that lactose may not be the natural ligand for 3367755.
Figure 3.

Scatter plots of sequence identity (a) and similarity (b) of each sequence in the multiple alignment to the LacI effector-binding domain. The vertical axes show identity/similarity over the whole domain, whereas the horizontal axes show identity/similarity over the ligand contact subset of 17 residues (see text). Gray circles highlight proteins previously predicted or known to bind lactose. Arrows indicate positions of the ORF with GenBank accession no. 3367755.
Given an input multiple alignment and list of protein–ligand contacts (generated from PDB files), our ligand analysis software creates plots such as Figs. 3 and 4 for each known structure. The sets of binding-site matches to proteins of known structure for the LacI/RbsB family are diagrammed relative to the phylogenetic grouping methods in Fig. 2. No binding-site matches were found for allose-binding protein or arabinose-binding protein within the 102 alignment sequences.
Sequences with binding-site residues similar to E. coli RbsB are shown in Fig. 5 as a second example. We will refer to a sequence that has residues capable of duplicating the RbsB–ribose interaction at every contact position as having a “binding-site match” to RbsB. The sequences in Fig. 5 that cannot match the RbsB–ribose interaction at a particular alignment position either do not bind ribose or do not bind ribose with the same set of binding interactions as RbsB. Interestingly, the ribose operon repressor (RbsR) from B. subtilis has a binding-site match to RbsB, despite appearing in a different phylogenetic cluster when the full domains are used as input (Fig. 2). We thus predict that this Gram-positive repressor binds ribose with the same set of protein–ligand contacts as the periplasmic ribose-binding protein in E. coli. Oddly, the ribose repressors from E. coli and Haemophilus influenzae (not shown) do not have the same set of binding interactions with ribose, since they have mismatches at RbsB positions 90 and 137. Two other B. subtilis DNA-binding proteins, DegA, which may regulate the degradation of phosphoribosylpyrophosphate amidotransferase (34), and the sequence with GenBank accession no. 2612909, a regulatory protein of (otherwise) unknown function, may also be hypothesized to bind molecules similar to ribose as a result of this analysis. These two proteins also did not cluster with RbsB in the whole-domain phylogeny. Finally, it is worthy of note that this level of functional sequence analysis also succeeds in separating the ligand-binding functions of the purine and ribose repressors. Although these sequences clustered together in both phylo-genetic analyses, neither can match the other's binding-site residues.
SubI Family Alignment.
The six structures used for the PBP Type II structural alignment were 1sbp, 1ixh, 1mrp, 1pot, 1wod, and 1atg. These are the most similar structures in the PDB to 1atg (rms deviation ≤ 4.0 over a minimum of 209 residues), and bind sulfate, phosphate, ferric iron, polyamines, molybdate, and tungstate (25, 35–39). A program was written to increase the automation level of iterated HMM-based alignment building, using parameters and strategies that proved useful in the LacI/RbsB family alignment and in previous alignments of divergent protein families (11). alignmentbuilder uses hmmer algorithms to find and incorporate all database sequences with E-values below a given threshold into the alignment model, repeating until convergence. It then gradually decreases the search stringency, converging at each new E-value until it reaches a user-defined threshold (see Methods). Some manual adjustments were made to the alignment to ensure concurrent alignment of close homologues. The final alignment for the SubI family, which converged at E = 1e−9, contained 94 sequences with 17% average pairwise sequence identity.
SubI Family Domain and Binding-Site Phylogenetic Trees.
Fig. 6 shows the significant clusters derived from whole-domain phylogenetic analysis of the SubI family multiple alignment. Groups correlate fairly well with known functions. For example, one cluster contains several proteins previously annotated as iron binding (including FBP_NEIGO, HITA_HAEIN, 1651916, and 3978164), and another contains known sulfate and thiosulfate receptors (e.g., SUBI_SALTY and CYSP_ECOLI). A phylogenetic tree of the SubI family binding-site subalignment was also constructed. This analysis identified five significant clusters of sequences that were subsets of significant whole-domain phylogenetic groups and one new cluster of phosphate-binding proteins (2182813 and 541315; see Fig. 6). Further, one sequence of unknown ligand-binding preference, Y131_HAEIN, may be hypothesized to bind iron because of its phylogenetic linkage at the whole-domain and binding-site levels to AfuA of Actinobacillus pleropneumoniae (1469286), which is an iron-binding PBP (40).
Figure 6.
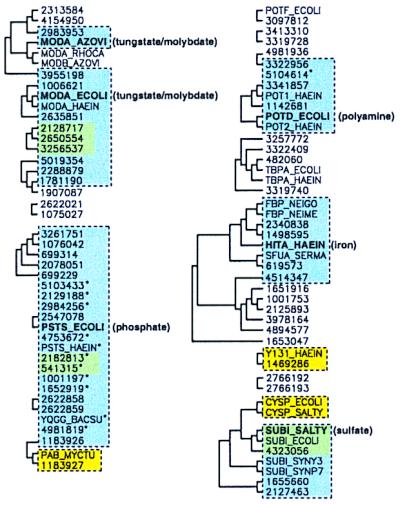
Comparison of three functional clustering methods for the SubI family of periplasmic binding proteins. Solid black lines designate clusters from the whole-domain phylogenetic tree present in ≥30% of the bootstrap replicates. Phylogenetic clusters from the binding-site subphylogeny (yellow boxes) were considered significant if present in ≥25% of the replicates. A lower value was used for the SubI family because there are fewer contact positions in the alignment. Binding-site matches to the six known structures are shown with blue rectangles (see text), and green shading represents overlap between blue and yellow clusters. Asterisks identify sequences that did not fall into a significant cluster in the whole-domain phylogeny but that match the binding-site residues of a known structure.
Matches to Ligand-Binding Sites of Known SubI Family Structures.
As with the LacI/RbsB family, the binding-site residues of each known structure were compared with all aligned sequences. Proteins with binding-site matches to known SubI family structures, that is, those with reasonable residue matches at all contact positions, are shown in Fig. 6 inside blue boxes. These clusters represent predictions both of ligand-binding specificity and of side chain–ligand interactions. Eight of the sequences with binding-site matches do not currently have ligand-binding annotation in GenBank, and four of these eight are not members of significant phylogenetic clusters. Thus, the ligand-binding functions of these proteins are not likely to have been predicted correctly from whole-domain sequence similarity alone. It was somewhat surprising to observe that proteins binding the similar ligands sulfate, molybdate, and phosphate, using the same fold and backbone conformation (2.7 Å average pairwise Cα rms deviation), can be classified readily into functional groups on the basis of sequence. Each of these functional groups, however, uses a different and conserved constellation of amino acids to accomplish the binding interaction. Full lists of binding-site matches and new function predictions for the SubI and LacI/RbsB families are available at http://winslow.med.harvard.edu/johnson/.
As a final example of our results, binding-site matches to E. coli molybdate-binding protein ModA, a SubI family member, are depicted in Fig. 7. Many of these binding-site matches are already known to bind molybdate, but four of the matching sequences are from putative or hypothetical proteins of unknown binding function (GenBank accession nos. 5019354, 2650554, 2128717, and 3256537). Further, all four proteins have whole-domain sequence identity to ModA between 18% and 30%, in the range where transfer of functional information between homologues is highly error prone. The effector domains of E. coli purine and ribose repressors, for example, are 42% identical but bind structurally unrelated ligands.
Figure 7.
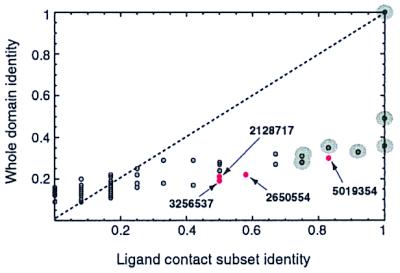
Scatter plot of sequence identity of each protein in the SubI family multiple alignment to ModA of E. coli, over the whole domain and over the 12 binding-site residues of ModA. Sequences with a binding-site match to ModA are highlighted with gray circles for previously annotated molybdate receptors or in red for sequences without a molybdate-binding annotation.
Evaluation of Methods for Classifying Sequences into Ligand-Specific Groups.
In summary, three classification methods were used to separate the sequences of these families into ligand-specific groups. The first method, domain-level sequence similarity or identity, was useful in identifying new family members, mapping the sequence space of the family, and generating hypotheses about functional relatedness among phylogenetic branches. Whole-domain sequence similarity was not a particularly reliable indicator of ligand-binding function, however. We observed significant clusters with very different ligand-binding functions, as well as many proteins with the same ligand-binding preference that were not clustered together at a significant level.
Information about the location of the binding site for a group of sequences can clearly improve ligand-prediction methods. This is demonstrated in Fig. 7, which shows that molybdate receptors have higher sequence identity in the ligand-binding pocket than in the binding domain as a whole. The second method, binding-site phylogenetic analysis, was thus useful in eliminating false positives from whole-domain functional groupings, because proteins that do not bind the same ligand did not co-cluster at the binding-site level. This method also served to group proteins of similar ligand-binding sites but unknown binding function.
The third method, matching sequences to the binding sites of known structures across a multiple alignment, is likely to be the most reliable way to identify ligand-binding function, because it requires chemical similarity for all protein–ligand contact residues. One of the most striking examples is the RbsR sequence from B. subtilis, RBSR_BACSU, which is only 22% identical to E. coli RbsB over the whole domain but is easily identified through automated methods as a match to the ribose-binding site of RbsB by this approach (Fig. 5). This technique is limited only by the number of known structures within a family and the ligand-binding diversity of those structures. Some true matches will be missed by this method, however, because one nonconserved contact residue does not necessarily signify a different function. Another potential pitfall of any homology-based functional annotation method is that conserved residues do not always have the same function, even when binding similar ligands. The chemically similar ligands galactose and glucose interact with conserved residues of arabinose-binding protein and glucose-/galactose-binding protein, for instance, but the sugars are bound in completely different orientations (41). Finally, all of the methods described here are limited by the availability of accurate multiple alignments.
Implications for Functional and Structural Genomics.
Our analysis of these two protein families demonstrates the challenge of extending predictions of three-dimensional structure to predictions of molecular function. For each protein of known structure in the LacI/RbsB and SubI families, an average of only five sequences from the alignment could be associated with confidence to the same ligand-binding function. Using the information from these structures in combination with multiple alignments, we were able to assign 55% of sequences in the two families to a group of common ligand-binding function, either by a binding-site match to a known structure or through the phylogenetic analysis of binding-site residues. However, several of these groups do not have a representative with a solved structure or known ligand-binding function. These groups of unknown ligand preference are good targets for future experiments, because the ligand-binding function of several proteins could be derived from the results of a single experiment.
The application of two of the methods we have described requires prior knowledge of the binding site or active site of a protein family to predict molecular function. With the growth of structure and sequence databases, we will soon be able to associate most sequences with solved structures, but for how many of these folds will binding sites be known? Russell et al. (42) have estimated that it is currently possible to predict the binding sites of 51% of newly solved structures, either by homology to proteins with known binding sites or by classifying them within a “superfold” of analogous structures that have the same binding-site locations. Thus, our methods should be applicable to most enzyme or receptor families of known structure. Overall, however, our results imply that for three-dimensional folds associated with many different functions, sequence space must be densely sampled with structures and experiments to assign molecular function to all members of the fold family.
One way to extend the methods presented here would be to construct homology models for each sequence in the alignment and then search for three-dimensional binding-site similarity to known structures or other homology models. Fetrow and Skolnick have recently demonstrated the utility of these low-resolution homology models for detecting enzyme active sites (43). Methods that transfer functional assignments between nonhomologous proteins (see ref. 44) may also be used to complement the homology-based methods described here and are particularly valuable when structural information is not available.
In conclusion, we have shown that many new ligand-binding annotations can be made computationally by combining binding-site structural information with multiple sequence alignments. These methods are applicable to receptors as well as enzymes and do not require localized sequence motifs. Moreover, the techniques described here will be straightforward to add to large-scale automated annotation algorithms used for new genomic data and will be particularly useful in assigning residue-specific function within other families of receptors. At a minimum, automated methods of functional annotation should check for residue conservation in the binding sites of receptors and the active sites of enzymes when assigning molecular function within families of known structure.
Acknowledgments
We are grateful to T. Smith, T. Springer, J. Hogle, A. McGuire, M. Bulyk, and F. Lam for helpful discussions and comments on the manuscript. J.M.J. and G.M.C. were supported by a grant from Hoechst Marion Roussel.
Abbreviations
- PBP
periplasmic binding protein
- RbsB
ribose-binding protein
- LacI
lactose receptor
- SubI
sulfate-binding protein
- ModA
molybdate-binding protein
- IPTG
isopropyl-d-thiogalactoside
- HMM
hidden Markov model
- PDB
Protein Data Bank
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050580897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050580897
References
- 1.Gaasterland T. Trends Genet. 1998;14:135. doi: 10.1016/s0168-9525(98)01430-9. [DOI] [PubMed] [Google Scholar]
- 2.Terwilliger T C, Waldo G, Peat T S, Newman J M, Chu K, Berendzen J. Protein Sci. 1998;7:1851–1856. doi: 10.1002/pro.5560070901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith T, Zhang X. Nat Biotechnol. 1997;15:1222–1223. doi: 10.1038/nbt1197-1222. [DOI] [PubMed] [Google Scholar]
- 4.Bork P, Gibson T J. Methods Enzymol. 1996;266:162–184. doi: 10.1016/s0076-6879(96)66013-3. [DOI] [PubMed] [Google Scholar]
- 5.Brenner S E. Trends Genet. 1999;15:132–133. doi: 10.1016/s0168-9525(99)01706-0. [DOI] [PubMed] [Google Scholar]
- 6.Eisen J A. Genome Res. 1998;8:163–167. doi: 10.1101/gr.8.3.163. [DOI] [PubMed] [Google Scholar]
- 7.Koonin E V, Tatusov R L. J Mol Biol. 1994;244:125–132. doi: 10.1006/jmbi.1994.1711. [DOI] [PubMed] [Google Scholar]
- 8.Babbitt P C, Hasson M S, Wedekind J E, Palmer D R, Barrett W C, Reed G H, Rayment I, Ringe D, Kenyon G L, Gerlt J A. Biochemistry. 1996;35:16489–16501. doi: 10.1021/bi9616413. [DOI] [PubMed] [Google Scholar]
- 9.Holm L, Sander C. Proteins Struct Funct Genet. 1997;28:72–82. [PubMed] [Google Scholar]
- 10.Zhang B, Rychlewski L, Pawlowski K, Fetrow J S, Skolnick J, Godzik A. Protein Sci. 1999;8:1104–1115. doi: 10.1110/ps.8.5.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson J M, Church G M. J Mol Biol. 1999;287:695–715. doi: 10.1006/jmbi.1999.2630. [DOI] [PubMed] [Google Scholar]
- 12.Livingstone C D, Barton G J. Comput Appl Biosci. 1993;9:745–756. doi: 10.1093/bioinformatics/9.6.745. [DOI] [PubMed] [Google Scholar]
- 13.Lichtarge O, Bourne H R, Cohen F E. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 14.Higgins D G. Comput Appl Biosci. 1992;8:15–22. doi: 10.1093/bioinformatics/8.1.15. [DOI] [PubMed] [Google Scholar]
- 15.Casari G, Sander C, Valencia A. Nat Struct Biol. 1995;2:171–178. doi: 10.1038/nsb0295-171. [DOI] [PubMed] [Google Scholar]
- 16.Bairoch A, Apweiler R. Nucleic Acids Res. 1997;25:31–36. doi: 10.1093/nar/25.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benson D, Boguski M S, Lipman D J, Ostell J, Oulette B F, Rapp B A, Wheeler D L. Nucleic Acids Res. 1999;27:12–17. doi: 10.1093/nar/27.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eddy S R. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 19.Holm L, Sander C. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 20.Galtier N, Gouy M, Gautier C. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 21.Hars U, Horlacher R, Boos W, Welte W, Diederichs K. Protein Sci. 1998;7:2511–2521. doi: 10.1002/pro.5560071204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quiocho F A, Vyas N K. Nature (London) 1984;310:381–386. doi: 10.1038/310381a0. [DOI] [PubMed] [Google Scholar]
- 23.Vyas N K, Vyas M N, Quiocho F A. Science. 1988;242:1290–1295. doi: 10.1126/science.3057628. [DOI] [PubMed] [Google Scholar]
- 24.Zou J-Y, Flocco M M, Mowbray S L. J Mol Biol. 1993;233:739–752. doi: 10.1006/jmbi.1993.1549. [DOI] [PubMed] [Google Scholar]
- 25.Bruns C M, Nowalk A J, Arvai A S, McTigue M A, Vaughan K G, Mietzner T A, McRee D E. Nat Struct Biol. 1997;4:919–924. doi: 10.1038/nsb1197-919. [DOI] [PubMed] [Google Scholar]
- 26.Fukami-Kobayashi K, Tateno Y, Nishikawa K. J Mol Biol. 1998;286:279–290. doi: 10.1006/jmbi.1998.2454. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein F C, Koetzle T F, Williams G J, Meyer E F J, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. Eur J Biochem. 1977;80:319–324. doi: 10.1111/j.1432-1033.1977.tb11885.x. [DOI] [PubMed] [Google Scholar]
- 28.Friedman A M, Fischmann T O, Steitz T A. Science. 1995;268:1721–1727. doi: 10.1126/science.7792597. [DOI] [PubMed] [Google Scholar]
- 29.Bjorkman A J, Binnie R A, Zhang H, Cole L B, Hermodson M A, Mowbray S L. J Biol Chem. 1994;269:30206–30211. [PubMed] [Google Scholar]
- 30.Chaudhuri B N, Ko J, Park C, Jones T A, Mowbray S L. J Mol Biol. 1999;286:1519–1531. doi: 10.1006/jmbi.1999.2571. [DOI] [PubMed] [Google Scholar]
- 31.Schumacher M A, Glasfeld A, Zalkin H, Brennan R G. J Biol Chem. 1997;272:22648–22653. doi: 10.1074/jbc.272.36.22648. [DOI] [PubMed] [Google Scholar]
- 32.Thomsen L E, Pedersen M, Nørregaard-Madsen M, Valentin-Hansen P, Kallipolitis B H. J Mol Biol. 1999;288:165–175. doi: 10.1006/jmbi.1999.2668. [DOI] [PubMed] [Google Scholar]
- 33.Goda S K, Eisa O, Akhter M, Minton N P. FEMS Microbiol Lett. 1998;165:193–200. doi: 10.1111/j.1574-6968.1998.tb13146.x. [DOI] [PubMed] [Google Scholar]
- 34.Bussey L B, Switzer R L. J Bacteriol. 1993;175:6348–6353. doi: 10.1128/jb.175.19.6348-6353.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pflugrath J W, Quiocho F A. J Mol Biol. 1988;200:163–180. doi: 10.1016/0022-2836(88)90341-5. [DOI] [PubMed] [Google Scholar]
- 36.Sugiyama S, Vassylyev D G, Matsushima M, Kashiwagi K, Igarashi K, Morikawa K. J Biol Chem. 1996;271:9519–9525. doi: 10.1074/jbc.271.16.9519. [DOI] [PubMed] [Google Scholar]
- 37.Hu Y, Rech S, Gunsalus R P, Rees D C. Nat Struct Biol. 1997;4:703–707. doi: 10.1038/nsb0997-703. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Luecke H, Yao N, Quiocho F A. Nat Struct Biol. 1997;4:519–522. doi: 10.1038/nsb0797-519. [DOI] [PubMed] [Google Scholar]
- 39.Lawson D M, Williams C E, Mitchenall L A, Pau R N. Structure (London) 1998;6:1529–1539. doi: 10.1016/s0969-2126(98)00151-8. [DOI] [PubMed] [Google Scholar]
- 40.Chin N, Frey J, Chang C F, Chang Y F. FEMS Microbiol Lett. 1996;143:1–6. doi: 10.1111/j.1574-6968.1996.tb08452.x. [DOI] [PubMed] [Google Scholar]
- 41.Vyas N K, Vyas M N, Quiocho F A. J Biol Chem. 1991;266:5226–5237. [PubMed] [Google Scholar]
- 42.Russell R B, Sasieni P D, Sternberg M J E. J Mol Biol. 1998;282:903–918. doi: 10.1006/jmbi.1998.2043. [DOI] [PubMed] [Google Scholar]
- 43.Fetrow J S, Skolnick J. J Mol Biol. 1998;281:949–968. doi: 10.1006/jmbi.1998.1993. [DOI] [PubMed] [Google Scholar]
- 44.Marcotte E M, Pellegrini M, Thompson M J, Yeates T O, Eisenberg D. Nature (London) 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- 45.Kraulis P J. J Appl Crystallogr. 1991;26:283–291. [Google Scholar]