

Abstract
Specificity for double-stranded DNA can arise due to somatic mutations within one of the branches of an autoreactive B cell clone. However, it is not known whether a different autospecificity predates anti-dsDNA and whether separate offshoots of an expanding B cell clone retain or evolve alternative specificities. We compared 3H9, an anti-dsDNA IgG, to 4H8 and 1A11, antibodies produced by hybridomas representing an alternative branch of the 3H9 B cell clone. All three IgG bound chromatin in ELISA and apoptotic cells in confocal microscopy, yet only 3H9 bound dsDNA, as measured by plasmon resonance. Moreover, we demonstrate that despite the unique specificity of 3H9 for dsDNA, all three clone members exhibited indistinguishable binding to chromatin. The binding to chromatin and apoptotic cells was unaffected by N-linked glycosylation in L chain CDR1, a modification that results from a replacement of serine 26 with asparagine in 4H8 and 1A11. These data provide the first evidence that specificity for nucleosome epitopes on apoptotic cells provides the initial positive stimulus for somatic variants that comprise a B cell clone, including those that subsequently acquire specificity for dsDNA. Conversely, selection of autoreactive B cells for binding to apoptotic cells leads to clonal expansion, antibody diversification, and the development of linked sets of anti-nuclear autoantibodies.
Keywords: Anti-DNA, Autoantibody, Autoimmunity, Apoptosis, B lymphocytes, Somatic Mutations, Systemic Lupus Erythematosus
1. Introduction
Nucleosomes, the complexes of DNA and histones that package genomic DNA into the repeating structure of chromatin, are the predominant target of autoantibodies in systemic lupus erythematosus (SLE) and murine models for SLE (Monestier and Kotzin, 1992; Burlingame et al., 1993; Jovelin et al., 1998). Autoantibodies to DNA, histones, or the native nucleosome particle have been identified and characterized at the molecular level (Shlomchik et al., 1990; Losman et al., 1992; Tillman et al., 1992). Nevertheless, a clear insight into the mechanisms that lead to the production of autoantibodies to these abundant nuclear antigens remains elusive.
The genetic analysis of anti-DNA and anti-histone autoantibodies from murine models for SLE argues that B cells derive a selective advantage from improvements in binding to nuclear antigens (Radic and Weigert, 1994; Stollar, 1994; Monestier and Novick, 1996; Rahman, 2004). The binding to DNA, histones, and their complexes benefits from the use of specific H and L chain combinations, a suitable arrangement of contact residues within the combining site, and the introduction of cationic residues (mostly arginines) within the gene junctions that code for complementarity-determining region 3 (CDR 3). The consistent choice of particular H and L chain V genes, recurrent motifs within the H and L chain junctions, and independent parallel mutations yielding the same amino acid replacements in different autoantibodies strongly suggest antigen-specific selection of autoreactive B cells. In addition, the positive selection of B cells with Ig receptors that recognize the native nucleosome particle (Losman et al., 1993; Seal et al., 2000) implies direct contacts between B cells and nuclear antigens.
An opportunity for nuclear autoantigens to exit the confines of the nucleus and gain access to the extracellular space arises during the energy-dependent modification and redistribution of cellular contents that characterize apoptosis, or programmed cell death (Casciola-Rosen et al., 1994). Nucleosomes are released from the apoptotic nucleus by the caspase-dependent activation of nucleases that cleave DNA between adjacent nucleosomes (Widlak and Garrard, 2005). Thereafter, nucleosome core particles associate with the outer membrane of the fragmenting nucleus (Radic et al., 2004). As nuclear fragments emerge from the plasma membrane, nucleosomes become exposed at the cell surface (Radic et al., 2004). The fact that surface blebs containing nuclear fragments constitute a site of enhanced autoantibody reactivity, suggests that apoptotic cells are the source of nuclear autoantigens (Cocca et al., 2002). Consistent with this view, genetic defects leading to increased abundance of apoptotic cells are among the most potent risk factors for the development of systemic autoimmunity (Botto et al., 1998; Bickerstaff et al., 1999; Lu and Lemke, 2001; Scott et al., 2001; Cohen et al., 2002).
We have examined interactions between autoantibodies and apoptotic cells in order to establish relevant criteria for the activation of autoreactive B cells (Cocca et al., 2001; Cocca et al., 2002; Cline and Radic, 2004b; Radic et al., 2004; Radic et al., 2006). Autoantibodies to DNA, individual histones, and epitopes of the native nucleosome core particle, showed consistent and characteristic binding to apoptotic blebs (Radic et al., 2004). One autoantibody in particular, 3H9, can be viewed as a typical representative of these anti-nuclear autoantibodies (ANA). This antibody binds DNA (Shlomchik et al., 1990), chromatin (Radic et al., 1991), and intact nucleosomes (Seal et al., 2000) and reacts with large apoptotic blebs containing nuclear fragments (Radic et al., 2004). The precise epitope of 3H9 on the nucleosome is the H2A/H2B/DNA complex (DN and MM, unpublished data). The H2A/H2B dimer occupies opposite, exposed sides of the nucleosome core particle that are available for interactions with adjacent nucleosomes in chromatin (Davey et al., 2002) or with viral proteins that specifically recognize the nucleosome (Barbera et al., 2006). These studies lead to the conclusion that nucleosomes arrayed on the surface of apoptotic blebs are accessible to ANA or anti-nuclear Ig receptors and may thus constitute a direct antigenic stimulus for B cells expressing ANA (Radic et al., 2004).
The B cell producing 3H9 arose as a member of an expanded B cell clone. This was concluded from the genetic analysis of 3H9 and its clonal relatives, 4H8 and 1A11, that were captured along with 3H9 as hybridomas in the same B cell fusion (Shlomchik et al., 1987). These B cells express the same Ig gene segments and VDJ and VJ rearrangement junctions. Because of their shared and unique mutations, the 4H8, 1A11, and 3H9 hybridomas could be linked in a genealogical dendrogram (Figure 1) that approximates the evolution of the expanded B cell clone.
Figure 1.
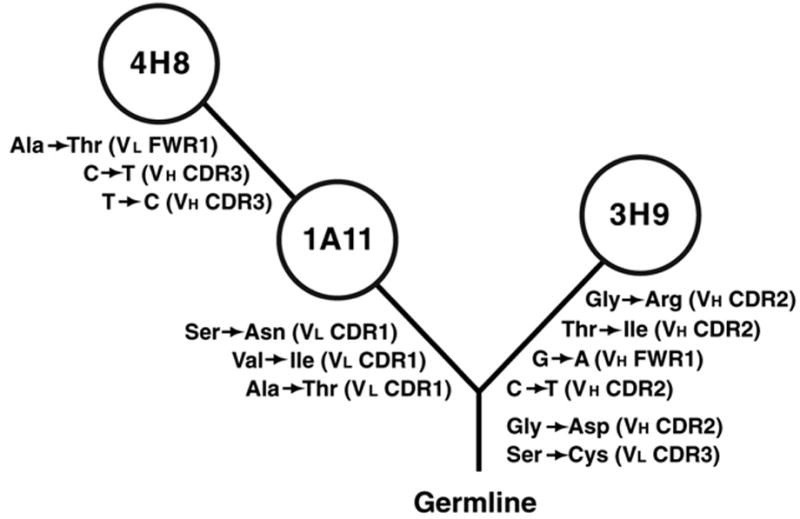
Dendrogram illustrating clonal relation between 3H9, 1A11, and 4H8. The diagram was constructed using shared and unique mutations in each antibody, assuming the minimum number of mutations (Shlomchik et al., 1990). Mutations were identified by comparison to the germline sequences of the closest VL (Thiebe et al., 1999) and VH genes (Haines et al., 2001). Silent mutations are listed as nucleotide changes, replacements as amino acid substitutions. The location of mutations is indicated in parentheses.
The comparison of binding between 3H9, 4H8 and 1A11, revealed that somatic mutations in 3H9 are responsible for its unique specificity for dsDNA (Shlomchik et al., 1990). The key role in DNA binding of the glycine to arginine mutation in VH3H9 was tested by reverting arginine 53 back to glycine: The R53G mutant lost specificity for dsDNA (Radic et al., 1993b). This result established that somatic mutations give rise to dsDNA specificity and that selection for anti-dsDNA B cells accounts for the expression of anti-dsDNA antibodies in murine models of SLE. However, this result also suggested that additional autospecificities must mediate positive selection of other members of this autoreactive B cell clone.
In continuation, we characterize the similarities and differences between 3H9 and its relatives and examine their specificity for DNA, chromatin, and apoptotic cells. Using surface plasmon resonance (SPR), we found that only 3H9 shows significant binding to dsDNA, whereas all three antibodies bind chromatin in ELISA and apoptotic cells in confocal microscopy. Somatic mutations within the other clone relatives did not alter binding to chromatin or apoptotic cells. Remarkably, despite N-glycosylation at asparagine 26 in L chain CDR1, binding was unaffected, indicating that the combining site in all three antibodies is asymmetric, with the H chain playing the dominant role in binding to nuclear antigens. Based on our data, we propose that B cells are stimulated to proliferate following Ig receptor engagement with autoantigens on apoptotic cells. As a result of positive selection, B cells in the expanding clone may evolve independent or overlapping autospecificities for DNA, histones, and nucleosomes.
2. Materials and Methods
2.1. Monoclonal antibody isolation
All monoclonal antibodies were isolated from a single MRL/lpr mouse, as described previously (Shlomchik et al., 1987). The IgG antibodies were purified from culture supernatants by binding to protein G sepharose (Pharmacia), extensive washing with PBS and elution with glycine HCl buffer (pH = 2.8). The eluted antibodies were dialyzed into PBS over night.
2.2. Gel electrophoresis and Western blotting
Two and 10 μg of 3H9, 4H8, or 1A11 were separated by reducing 10% SDS-PAGE. Gels were stained with Comassie blue for 1 h. Destaining was performed over night. Two μg of 3H9, 4H8, or 1A11 were separated by SDS-PAGE, proteins were transferred from the gels to nitrocellulose in a semi-dry blotter (Owl Separation Systems, Portsmouth, NH) with transfer buffer (48 mM Tris, 39 mM glycine, 0.04% SDS, and 20% methanol) at 0.8mA/cm2 for 90 min. Membranes were air-dried and blocked in PBS buffer containing 2% BSA, 3% FBS, 2.5 mM EDTA, and 0.25% Tween-20. All subsequent steps and washes were in 0.15 M NaCl, 50 mM Tris (pH=7.4), 0.2% Tween-20. Alkaline phosphatase (AP)-labeled secondary reagents were used according to manufacturers’ recommendations. Immunoreactive bands were visualized by using the chromogenic substrate in the AP color development kit (BioRad).
2.3. Biacore analysis
All SPR experiments were performed using a Biacore 2000 instrument (Biacore, Uppsala, Sweden). Biotinylated dsDNA of 500 bp average size was prepared by photobiotinylation, as previously described (Radic and Seal, 1997). A dilution to 40 ug/ml was prepared and 25uL was used to coat the SA sensor chip (Biacore, Uppsala, Sweden). A 0.05% SDS solution in HBSS was injected over the sensor chip surface to remove any loosely bound material from the surface and to allow the baseline to stabilize. The binding of the monoclonal antibodies to DNA was measured using antibodies diluted in HBSS to a final concentration of 75nM, 37.5nM, 18nM, and 9nM. The sensor chip surface was completely regenerated between runs using a solution of 0.05% SDS. All affinity measurements were performed using the BIAevalution v4.1 software.
2.4. Deglycosylation
Two μg of 3H9, 4H8, or 1A1 were treated with Protein: N-glycosidase F (PNGase F; New England Biolabs, Ipswich, MA), according to the manufacturers’ instructions. Briefly, 10 μg of 3H9, 4H8, or 1A11 were treated in a volume of 55 μl of 1x G7 Reaction Buffer (New England Biolabs), containing 1% NP-40 and 9 μl of the enzyme. Samples were incubated at 37° over night.
2.5. Chromatin ELISA
Chromatin was prepared from one bovine thymus by using a modification of a previously published procedure (Burlingame et al., 1993). Briefly, the thymus homogenate was prepared in 8–10 volumes of 0.25 M sucrose, 2 mM MgCl2, 20 mM Tris pH 7.4, filtered, and centrifuged at 2,000 g for 10 min. The nuclear pellet was washed in PBS containing 0.1% Triton X-100 and 1 mM EDTA, followed by centrifugation as in the preceding step. The pellet was washed with 50 mM Tris, pH 7.4 and 1 mM EDTA, centrifuged, and washed again in 10 mM Tris, pH 7.4 and 1 mM EDTA. The pellet was suspended in 100 ml of 1 mM EDTA, 10 mM Tris pH 7.4, containing 1 mM PMSF. Aliquots were frozen at −80oC.
For ELISA, Immulon 96 well plates were coated over night with 10 μg/ml poly-l-lysine, using 50 μl/well (in PBS pH-7.2), to provide a capture molecule for the binding of chromatin. Poly-L-lysine-coated plates were washed once with PBST and then 5 μg/ml (O.D. 260) of chromatin was adsorbed using 50 μl/well in PBS with 1.0 mM EDTA and 0.1 mM PMSF and incubating for 1.5 h. Following incubation, wells were washed 3 times with PBST, then blocked with 1% BSA in PBST for 1 h at RT. To control for non-specific binding, additional wells were blocked with 1% BSA in PBST. Serial 1:3 dilutions, starting at 20ug/ml of antibody (3H9, 4H8, or 1A11), were allowed to bind for 1.5 h at RT. Wells were washed 3 times with PBST and incubated with anti-mouse kappa AP conjugate (1:1000 dil.), for 1.0 h at RT. After the incubation, wells were washed 3 times with PBST and once with TBS and the bound antibodies were detected with 1.0 mg/ml PNPP substrate in 50 mM sodium bicarbonate with 10 mM MgCl2 and the readings were taken at 405 nm.
2.6. Binding to apoptotic cells
Jurkat cells (clone E6-1 from ATCC) were treated, as previously described (Cocca et al., 2001) with 2.0 μM camptothecin (Sigma Chemical Co., St. Louis, MO) for 3 to 6 h. Apoptosis was assessed by light microscopy and cells were harvested after at least 25% of cells in the population exhibited surface blebs. Antibody binding to apoptotic or viable cells was evaluated as described (Cocca et al., 2001; Cocca et al., 2002; Radic et al., 2004). Briefly, cells were washed in HBSS (supplemented to 3 mM CaCl2) and fixed for 15 min in ice-cold 6 % paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, PA). Fixed cells were washed and blocked in wash buffer (HBSS containing 3 mM CaCl2, 3% FBS, and 0.02% azide) for 5 min. Cells were suspended in buffer containing purified antibodies at 20 μg/ml. Following incubation with antibodies, cells were washed in wash buffer, pelleted as above, and incubated in a mixture of Alexa Fluor 647 rabbit anti-mouse IgG antisera (1:100 dilution), SYTOX Orange DNA stain (1:10,000 dilution), and Alexa Fluor 488 Annexin V (1:70 dilution). All secondary reagents and stains were obtained from Invitrogen.
Samples were viewed on a Zeiss LSM 510 laser scanning microscope (Carl Zeiss Inc., Thorwood, NY), by using a 100X Plan-Apochromat oil-immersion lens, and laser excitation at 488, 543, and 633 nm. Detection channels were set to record fluorescence emission above 650 nm for Alexa Fluor 647, between 560 and 615 nm for SYTOX Orange, and between 505 and 530 nm for Alexa Fluor 488. Consecutive images were collected at optimized intervals of between 0.4 and 0.8 μm to assemble complete three-dimensional representations of treated and untreated cells.
3. Results
3.1. Mutations that Distinguish 3H9 Give Rise to dsDNA Binding
Previously, reversion mutagenesis of VH3H9 identified the R53 mutation as the main determinant for dsDNA binding in 3H9 (Radic et al., 1993b). However, our previous work did not explore differences between 3H9 and its clonal relatives. To examine the differences in dsDNA binding between the clonal relatives, we constructed a binding substrate, consisting of biotinylated DNA attached to immobilized streptavidin, and examined antibody-DNA interactions using SPR. We established that the purified IgG were free of histones derived from hybridoma cultures (Figure 2), to limit the possibility that contaminating histones could give rise to apparent DNA binding (Guth et al., 2003). Moreover, we digested and removed all proteins from the substrate DNA prior to biotinylation.
Figure 2.
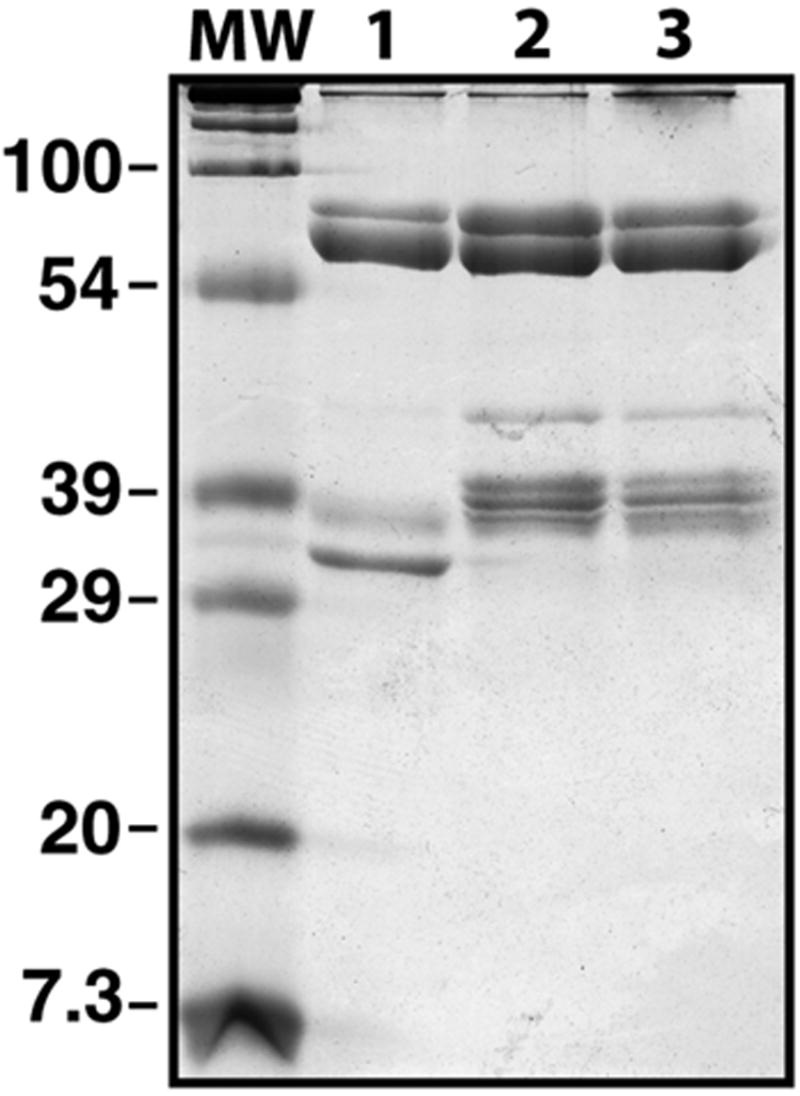
Reducing and denaturing SDS-PAGE of purified antibodies. The 3H9 (lane 1), 1A11 (lane 2), and 4H8 (lane 3) IgG were purified on Protein G-sepharose and examined on 12% PAGE. The mobilities of the IgG H and L chains were determined relative to molecular weight markers (MW). Note absence of contaminating proteins and difference in migration between the L chains of the clonally related IgG.
In our assays, different concentrations of antibodies were added to observe the extent of binding to dsDNA, and washing with buffer was used to determine the dissociation rate. Under these conditions, we determined that 3H9 was the only member of the B cell clone that showed affinity for dsDNA (Figure 3A). The method chosen, SPR, allowed us to estimate the KD of 3H9 binding to dsDNA at 2.1 x 10−7 M (Ka = 22,400/Ms and Kd = 0.0476/s). Because the association rate between 4H8 or 1A11 and dsDNA was negligible (Ka < 300), no reliable KD for their DNA binding could be derived.
Figure 3.
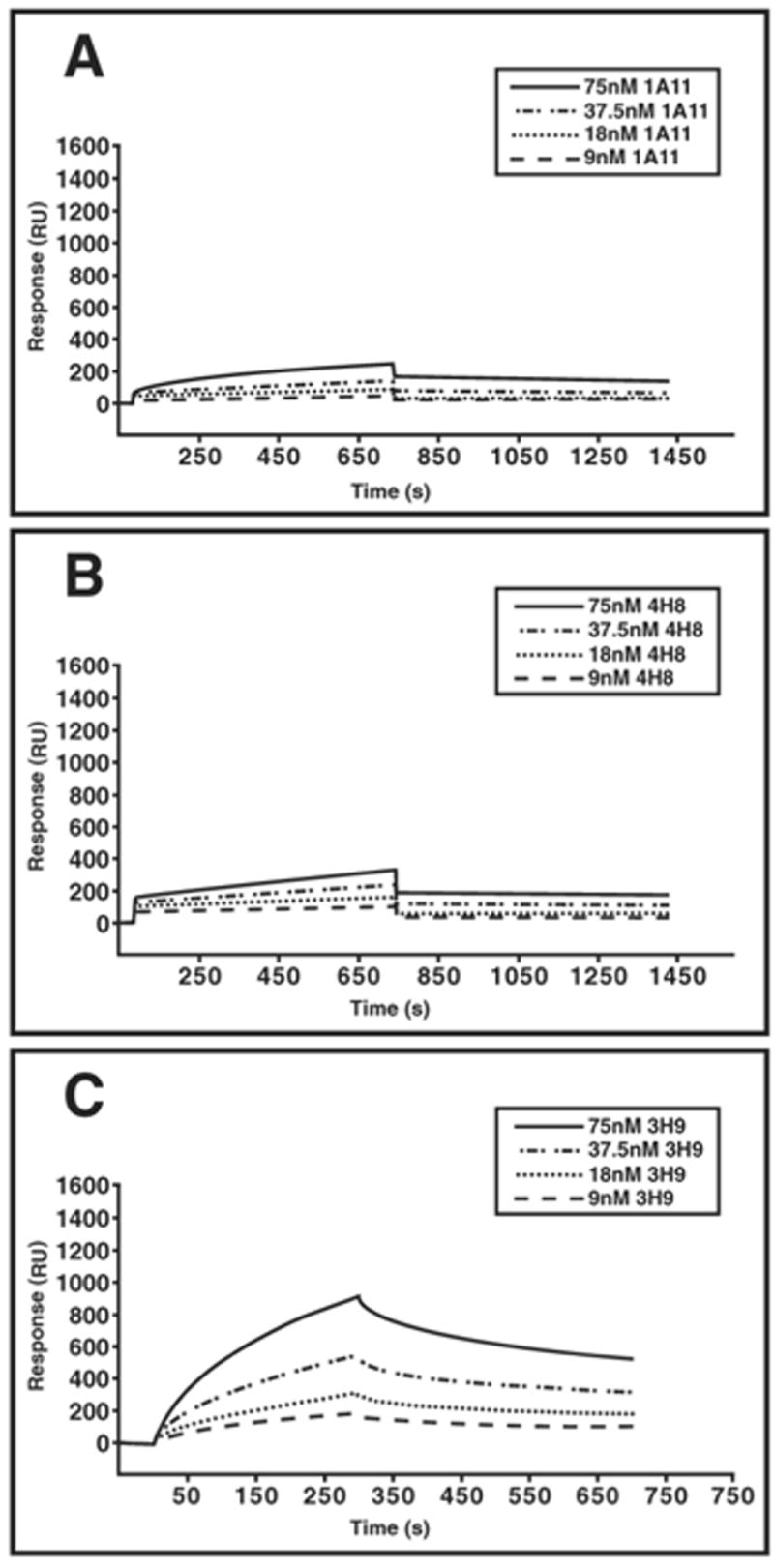
Binding of purified antibodies to dsDNA in SPR. The binding of all antibodies was measured using IgG concentrations of 75 nM, 37.5 nM, 18 nM, and 9 nM, as indicated in the legend. Binding of 1A11 (A), 4H8 (B), and 3H9 (C) is displayed in separate panels. Only 3H9 binds dsDNA.
3.2. Somatic Mutation Shared by 4H8 and 1A11 L Chains Allows N-Linked Glycosylation
The 4H8 and 1A11 members of the 3H9 clone share mutations that distinguish them from 3H9, as well as from the putative precursor B cell (Figure 1). One mutation in particular attracted our attention. The CDR1 of the 4H8 and 1A11 L chains acquired a serine 26 to asparagine replacement that forms a suitable acceptor site within a consensus sequence for N-linked glycosylation (Figure 4A). Consistent with N-glycosylation, the electrophoretic mobility of the 4H8 and 1A11 kappa chains was reduced in SDS-PAGE as compared to the 3H9 L chain (Figure 2), and treatment with protein N-glycosidase F (PNGase F) equalized the migration of all three L chains (Figure 4B). The removal of the N-glycosyl moiety could be accomplished using either denatured (data not shown) or native IgG. We estimate that, under the conditions used here, at least 95% of the N-linked carbohydrate was removed from the 4H8 and 1A11 L chains (Figure 4B).
Figure 4.
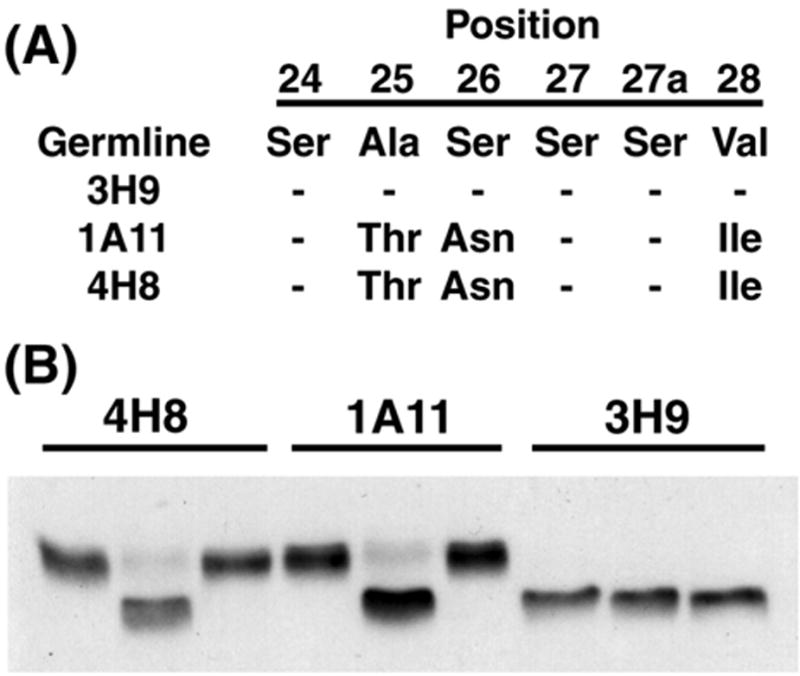
L chain mutations include serine 26 to asparagine (S26N) that directs N-linked glycosylation in CDR1 of the 1A11 and 4H8 L chains. (A) Amino acid sequences for portion of CDR1 are listed for germline, 3H9, 1A11, and 4H8, to illustrate substitutions. (B) Western blot of 4H8, 1A11, and 3H9 incubated in buffer alone (left lane in each set), treated with PNGase F under non-denaturing conditions (center lane), or untreated (right lane). L chains were detected using AP-labeled goat antibodies to mouse kappa (see Materials and Methods). Following PNGase F treatment, mobility of 4H8 and 1A11 L chains increases and differences in migration with 3H9 are eliminated.
3.3. Glycosylation of 4H8 and 1A11 L Chains has No Effect on Chromatin Binding
Glycosylation within the L chain combining site may alter binding to antigen (Tachibana et al., 1992). To test whether a carbohydrate moiety within CDR1 of the 4H8 and 1A11 L chains affects binding to chromatin, we used a chromatin ELISA to compare the binding of purified IgG, PNGase F-treated IgG, or IgG incubated in reaction buffer (Figure 5). Regardless of treatment, each of the antibody preparations showed superimposable binding curves (Figure 5), suggesting that N-glycosylation in L chain CDR1 of 4H8 and 1A11 has little or no effect on chromatin binding. In addition, each of the original antibodies exhibited nearly identical binding to chromatin (Figure 5), suggesting the mutations that distinguish the clonal relatives make no contribution to chromatin binding.
Figure 5.
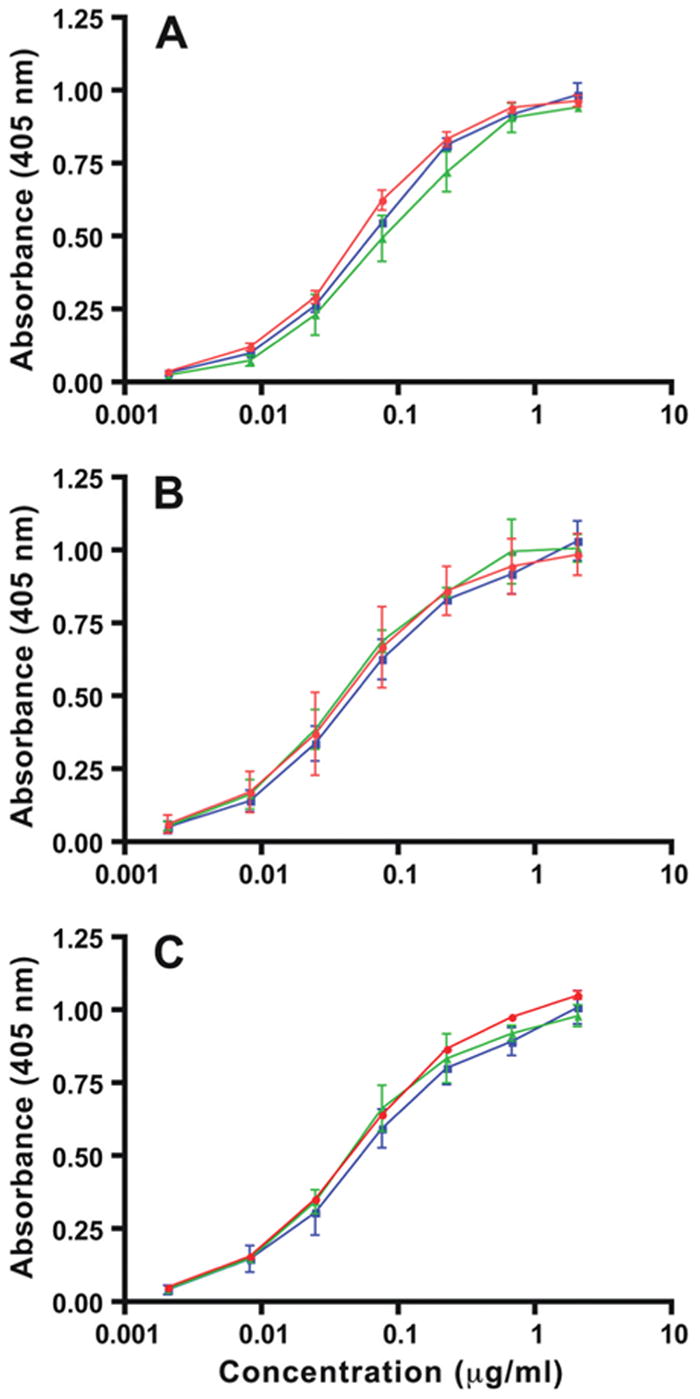
Binding of 4H8, 1A11, and 3H9 to chromatin is unaffected by PNGase F. Relative affinities for chromatin in solid phase ELISA were measured as described in Materials and Methods, and absorbance readings at 405 nm were plotted. Binding of 3H9 (A), 4H8 (B), and 1A11 (C) is shown in red, along with the binding of PNGase F- treated IgG (green), and with IgG incubated in buffer alone (blue). Triplicate values from two separate experiments were averaged, standard errors were computed, and displayed as vertical bars.
Because N-glycosylation at asparagine 26 in L chain CDR1 had no effect on chromatin binding (Figure 5), we propose that conservative mutations nearby, namely the replacements of alanine 25 by threonine and valine 28 by isoleucine (Figure 4A), are unlikely to affect chromatin binding. In turn, it follows that H chain mutations, including the glycine 53 to arginine replacement that gives 3H9 its ability to bind dsDNA (Figure 3A), also do not contribute in a significant way to chromatin binding.
3.4. Each Member of the 3H9 Clone Binds to Apoptotic Blebs
3H9 binds to the periphery of nuclear fragments that are generated during the execution phase of apoptosis (Radic et al., 2004). To explore the possibility that 4H8 and 1A11 share this specificity with 3H9, we used confocal microscopy to examine antibody binding to apoptotic cells. Twenty-five to 40% of apoptotic Jurkat cells exhibit large surface blebs as their most striking morphological feature, following 5 h of treatment with camptothecin (Cocca et al., 2002). The blebs are largely composed of separate fragments of the nucleus, as staining with a DNA binding fluorochrome demonstrates (Figure 6). Blebs form due to nuclear fragment movement and their protrusion from the cell surface that are mediated, at least in part, by the action of the Rho-associated kinase ROCK I (Cocca et al., 2002). At that time, the plasma membrane loses its phospholipid asymmetry, as visualized by the binding of annexin V (Figure 6). At that time, the membrane becomes porous, and allows the influx of macromolecules from outside the cell.
Figure 6.
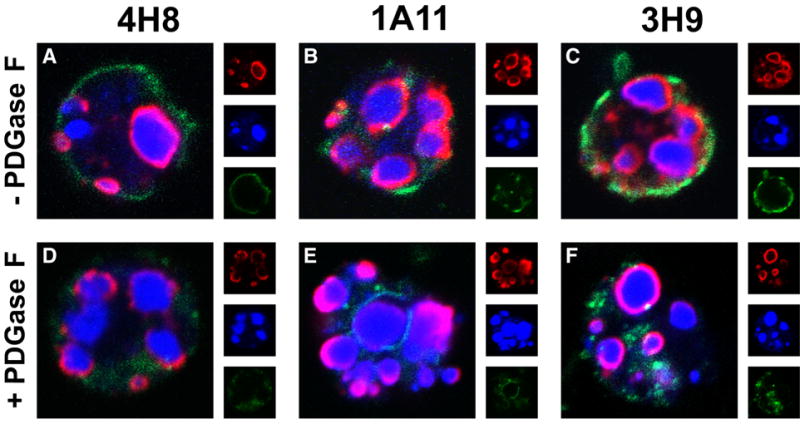
Binding of 4H8, 1A11, and 3H9 to apoptotic blebs and nuclear fragments is unaffected by PNGase F. Jurkat cells, treated with camptothecin to induce apoptosis, were fixed and incubated with 4H8 (A), 1A11 (B), and 3H9 (C). Equivalent reactions were incubated with 4H8 (D), 1A11 (E), and 3H9 (F) treated with PNGase F. Bound antibodies were detected with anti-mouse antibodies (displayed in red). Each of the antibodies, regardless of PNGase F treatment, bound at or near the surface of nuclear fragments. DNA, bound by Sytox Orange, is displayed in blue and annexin V in green. In addition to the composite images, separate wavelength channels are displayed at 1/3 size. Images represent optical cross sections. Controls included cells incubated in the absence of mouse IgG and cells incubated in the presence of z-VAD-fmk, neither of which showed antibody binding (data not shown).
4H8 and 1A11, regardless of L chain N-glycosylation, bound to apoptotic Jurkat cells in a pattern that was indistinguishable from 3H9 (Figure 6). Each of the antibodies bound to the periphery of nuclear fragments generated in apoptosis. The binding coincided with the release of a subset of nucleosomes from the nucleus and their association with the outer membrane of the nuclear envelope (Radic et al., 2004). The antibodies remained associated with the outside of nuclear fragments after they protruded from the cell membrane and formed apoptotic blebs. In fact, antibody binding, as a rule, was more intense along the face of the fragments that protruded from the cell (Figure 6B-E).
The binding of 4H8 and 1A11 to apoptotic blebs may be mediated by chromatin epitopes that are externalized during the execution phase of apoptosis because nucleosomes that are arrayed along the outside of nuclear fragments become exposed at the cell surface (Radic et al., 2004). The common precursor for 3H9, 4H8, and 1A11 itself must have been reactive with apoptotic blebs because binding to blebs is unlikely to have arisen independently within the two branches of the 3H9 B cell clone. Indeed, previously reported experiments using the 3H9 germline revertant established that even the earliest unmutated precursor of this B cell clone bound to apoptotic cells (Cocca et al., 2001).
4. Discussion
Over a decade ago, Casciola-Rosen and colleagues discovered the reactivity of SLE autoantibodies with blebs on apoptotic cells (Casciola-Rosen et al., 1994). In subsequent years, the importance of cell death in providing autoantigens that stimulate autoimmunity has become widely accepted (Cline and Radic, 2004a; Hall et al., 2004; Navratil et al., 2004). Using a panel of antigen-selected and affinity-matured murine autoantibodies, we identified the target of autoantibodies on the surface of apoptotic blebs as a histone-DNA complex whose composition is identical with the nucleosome core particle (Radic et al., 2004). Autoantibodies to DNA, individual core histones, or the intact core particle bind bleb antigens after they become externalized at the cell surface, suggesting the nucleosome core particle is at a pivotal point for maintaining tolerance or stimulating autoimmunity (Radic et al., 2004). Our current analysis of 3H9 and its clonal relatives indicates that divergent members of the 3H9 B cell clone share specificity for apoptotic blebs and that binding to blebs predates the development of anti-dsDNA specificity. Therefore, we propose that specificity for apoptotic blebs initiates clonal expansion.
Examination of 1A11 supports the idea that specificity for chromatin and apoptotic blebs arose early during the development of the 3H9 B cell clone. Following deglycosylation, 1A11 resembles the common branch point of the 3H9 clone, as it lacks H chain mutations unique to 3H9 and sheds the bulky carbohydrate moiety from its L chain. The PNGase F-treated 1A11 only retains three conservative replacements that distinguish it from the common branch point. The analysis of the deglycosylated 1A11 suggests that the branch point of the 3H9 clone bound chromatin and apoptotic blebs, and that the more diversified clone members inherited and maintained this specificity (Figures 5 and 6).
The specificity for apoptotic blebs of the branch point and of even earlier precursors of the 3H9 clone is supported by the observation that the germline revertant of 3H9 binds to annexin V-positive, apoptotic Jurkat cells rather than to viable cells (Cocca et al., 2001). Specificity for apoptotic cells is thus a characteristic that is maintained throughout the evolution of the 3H9 clone, indicating that positive selection of all 3H9 clone members is mediated by binding, processing, and presentation of epitopes derived from dying cells. We propose that the relevant antigens associated with the apoptotic blebs are components of the nucleosome core particle.
That the mutations in L chain CDR1 of 1A11 had little or no effect on binding is consistent with earlier data supporting the asymmetry of the 3H9 combining site. The 3H9 H chain plays the dominant role in DNA and chromatin binding, whereas a wide variety of L chains are compatible with binding, as evidenced by in vitro and in vivo H and L chain recombination experiments (Radic et al., 1991; Ibrahim et al., 1995). The H chain dominance accounts for L chain editing in mice expressing the 3H9 H chain (Radic et al., 1993a). In H chain-only mice, most endogenous L chains fail to suppress the autoreactivity of the H chain and multiple rounds of V to J rearrangements may be required to install one of the few L chains that prevent binding to nuclear autoantigens (Li et al., 2004). Interestingly, the few effective L chain editors most consistently exhibit negatively charged residues in CDR3 (Li et al., 2001). The structural basis for this result lies in the fact that the L chain CDR3 is nearest to the H chain binding determinants, whereas L chain CDR1 is the most remote. In that light, it is not surprising that the L chain CDR1 glycosylation had minimal or no effect on chromatin binding.
A closer look at the H chain thus holds the key for identifying determinants of chromatin binding. We propose that arginine 96 (R96) in the H chain CDR3, a residue that 3H9 shares with its clonal relatives, represents the essential H chain CDR3 determinant for nucleosome binding. Because the nucleosome is tightly constrained by multiple ionic interactions between the backbone phosphates of DNA and positively charged residues within the globular and tail domains of histones (Luger et al., 1997), R96 may occupy a unique position in the combining site from which it can assist in nucleosome binding. This possibility is supported by the fact that replacement of the 3H9 VH CDR3 by other CDR3 containing arginines at alternative positions fails to restore nucleosome binding (Seal et al., 2000). A suitable site for 3H9 binding to the nucleosome may be the acidic patch along the H2A/H2B interface that is crucial for chromatin packing interactions and becomes exposed once nucleosomes are released from chromatin (Davey et al., 2002). Therefore, binding to nucleosomes, chromatin, and apoptotic blebs, properties that define the 3H9 clone of B cells as a whole, may depend profoundly on R96 in CDR3 of the H chain.
If the binding to chromatin is unchanged by the R53 mutation that gave 3H9 its unique specificity for dsDNA, then what is its benefit to the B cell? We propose that R53 was a mutation that enabled the 3H9 B cell to receive help from a wider range of T cells. By its capacity for binding to free DNA, 3H9 may have acquired the potential for binding to non-histone protein-DNA complexes. These may include complexes between DNA and p53 (Herkel et al., 2001), or DNA repair complexes including DNA-PK (Suwa et al., 1996). Provided that peptides from one or more of these DNA binding proteins could evade T cell tolerance and elicit T cell help, the 3H9 B cell could have paved the way for the clonal expansion of B cells specific for such additional SLE autoantigens. In that manner, 3H9 may have provided crucial assistance for epitope spreading. In conclusion, we present evidence that B cells that recognize and bind to apoptotic cells are stimulated to proliferate and incur somatic mutations. Some of the mutations are neutral for binding, whereas others, such as the glycine 53 to arginine replacement in VH3H9, create additional specificities, thus providing a mechanism for clonal divergence and epitope spread.
Acknowledgments
We thank Dr. Amy Cline and other members of the Radic lab for critical comments and Mr. Tim Higgins for expert assistance with preparing figures. This work was supported by a Research Grant from the Lupus Research Institute, the UT Center of Excellence for Diseases of Connective Tissue, and the UT Rheumatic Disease Research Core Center of the National Institutes of Health.
Glossary
- AP
alkaline phosphatase
- ANA
anti-nuclear antibody
- PNGase F
Protein: N-glycosidase F
- SLE
systemic lupus erythematosus
- ScFv
single chain variable fragment
- SPR
surface plasmon resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 2006;311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, Mitchell DA, Cook HT, Butler PJ, Walport MJ, Pepys MB. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med. 1999;5:694–697. doi: 10.1038/9544. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Burlingame RW, Rubin RL, Balderas RS, Theofilopoulos AN. Genesis and evolution of antichromatin autoantibodies in murine lupus implicates T-dependent immunization with self antigen. J Clin Invest. 1993;91:1687–1696. doi: 10.1172/JCI116378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline AM, Radic MZ. Apoptosis, subcellular particles, and autoimmunity. Clin Immunol. 2004a;112:175–182. doi: 10.1016/j.clim.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Cline AM, Radic MZ. Murine lupus autoantibodies identify distinct subsets of apoptotic bodies. Autoimmunity. 2004b;37:85–93. doi: 10.1080/0891693042000196219. [DOI] [PubMed] [Google Scholar]
- Cocca BA, Seal SN, D’Agnillo P, Mueller YM, Katsikis PD, Rauch J, Weigert M, Radic MZ. Structural basis for autoantibody recognition of phosphatidylserine-β2 glycoprotein I and apoptotic cells. Proc Natl Acad Sci U S A. 2001;98:13826–13831. doi: 10.1073/pnas.241510698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocca BA, Cline AM, Radic MZ. Blebs and apoptotic bodies are B cell autoantigens. J Immunol. 2002;169:159–166. doi: 10.4049/jimmunol.169.1.159. [DOI] [PubMed] [Google Scholar]
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 A resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- Guth AM, Zhang X, Smith D, Detanico T, Wysocki LJ. Chromatin specificity of anti-double-stranded DNA antibodies and a role for Arg residues in the third complementarity-determining region of the heavy chain. J Immunol. 2003;171:6260–6266. doi: 10.4049/jimmunol.171.11.6260. [DOI] [PubMed] [Google Scholar]
- Haines BB, Angeles CV, Parmelee AP, McLean PA, Brodeur PH. Germline diversity of the expressed BALB/c VhJ558 gene family. Mol Immunol. 2001;38:9–18. doi: 10.1016/s0161-5890(01)00049-9. [DOI] [PubMed] [Google Scholar]
- Hall JC, Casciola-Rosen L, Rosen A. Altered structure of autoantigens during apoptosis. Rheum Dis Clin North Am. 2004;30:455–471. doi: 10.1016/j.rdc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Herkel J, Mimran A, Erez N, Kam N, Lohse AW, Marker-Hermann E, Rotter V, Cohen IR. Autoimmunity to the p53 protein is a feature of systemic lupus erythematosus (SLE) related to anti-DNA antibodies. J Autoimmun. 2001;17:63–69. doi: 10.1006/jaut.2001.0518. [DOI] [PubMed] [Google Scholar]
- Ibrahim SM, Weigert M, Basu C, Erikson J, Radic MZ. Light chain contribution to specificity in anti-DNA antibodies. J Immunol. 1995;155:3223–3233. [PubMed] [Google Scholar]
- Jovelin F, Mostoslavsky G, Amoura Z, Chabre H, Gilbert D, Eilat D, Bach JF, Koutouzov S. Early anti-nucleosome autoantibodies from a single MRL+/+ mouse: fine specificity, V gene structure and pathogenicity. Eur J Immunol. 1998;28:3411–3422. doi: 10.1002/(sici)1521-4141(199811)28:11<3411::aid-immu3411>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Li H, Jiang Y, Prak EL, Radic M, Weigert M. Editors and Editing of Anti-DNA Receptors. Immunity. 2001;15:947–957. doi: 10.1016/s1074-7613(01)00251-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Louzoun Y, Weigert M. Editing anti-DNA B cells by Vlambdax. J Exp Med. 2004;199:337–346. doi: 10.1084/jem.20031712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Fasy TM, Novick KE, Massa M, Monestier M. Nucleosome-specific antibody from an autoimmune MRL/Mp-lpr/lpr mouse. Arthritis Rheum. 1993;36:552–560. doi: 10.1002/art.1780360417. [DOI] [PubMed] [Google Scholar]
- Losman MJ, Fasy TM, Novick KE, Monestier M. Monoclonal autoantibodies to subnucleosomes from a MRL/Mp(−)+/+ mouse. Oligoclonality of the antibody response and recognition of a determinant composed of histones H2A, H2B, and DNA. J Immunol. 1992;148:1561–1569. [PubMed] [Google Scholar]
- Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Monestier M, Kotzin BL. Antibodies to histones in systemic lupus erythematosus and drug-induced lupus syndromes. Rheum Dis Clin North Am. 1992;18:415–436. [PubMed] [Google Scholar]
- Monestier M, Novick KE. Specificities and genetic characteristics of nucleosome-reactive antibodies from autoimmune mice. Mol Immunol. 1996;33:89–99. doi: 10.1016/0161-5890(95)00115-8. [DOI] [PubMed] [Google Scholar]
- Navratil JS, Sabatine JM, Ahearn JM. Apoptosis and immune responses to self. Rheum Dis Clin North Am. 2004;30:193–212. doi: 10.1016/S0889-857X(03)00110-8. [DOI] [PubMed] [Google Scholar]
- Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172:6692–6700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol. 1991;146:176–182. [PubMed] [Google Scholar]
- Radic MZ, Erikson J, Litwin S, Weigert M. B lymphocytes may escape tolerance by revising their antigen receptors. J Exp Med. 1993a;177:1165–1173. doi: 10.1084/jem.177.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic MZ, Mackle J, Erikson J, Mol C, Anderson WF, Weigert M. Residues that mediate DNA binding of autoimmune antibodies. J Immunol. 1993b;150:4966–4977. [PubMed] [Google Scholar]
- Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol. 1994;12:487–520. doi: 10.1146/annurev.iy.12.040194.002415. [DOI] [PubMed] [Google Scholar]
- Radic MZ, Seal SN. Selection of recurrent V genes and somatic mutations in autoantibodies to DNA. Methods. 1997;11:20–26. doi: 10.1006/meth.1996.0383. [DOI] [PubMed] [Google Scholar]
- Radic MZ, Shah K, Zhang W, Lu Q, Lemke G, Hilliard GM. Heterogeneous nuclear ribonucleoprotein P2 is an autoantibody target in mice deficient for Mer, Axl, and Tyro3 receptor tyrosine kinases. J Immunol. 2006;176:68–74. doi: 10.4049/jimmunol.176.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A. Autoantibodies, lupus and the science of sabotage. Rheumatology (Oxford) 2004;43:1326–1336. doi: 10.1093/rheumatology/keh354. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- Seal SN, Monestier M, Radic MZ. Diverse roles for the third complementarity determining region of the heavy chain (H3) in the binding of immunoglobulin Fv fragments to DNA, nucleosomes and cardiolipin. Eur J Immunol. 2000;30:3432–3440. doi: 10.1002/1521-4141(2000012)30:12<3432::AID-IMMU3432>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik MJ, Aucoin AH, Pisetsky DS, Weigert MG. Structure and function of anti-DNA autoantibodies derived from a single autoimmune mouse. Proc Natl Acad Sci USA. 1987;84:9150–9154. doi: 10.1073/pnas.84.24.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stollar BD. Molecular analysis of anti-DNA antibodies. FASEB J. 1994;8:337–342. doi: 10.1096/fasebj.8.3.7511550. [DOI] [PubMed] [Google Scholar]
- Suwa A, Hirakata M, Takeda Y, Okano Y, Mimori T, Inada S, Watanabe F, Teraoka H, Dynan WS, Hardin JA. Autoantibodies to DNA-dependent protein kinase. Probes for the catalytic subunit. J Clin Invest. 1996;97:1417–1421. doi: 10.1172/JCI118562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana H, Shirahata S, Murakami H. Generation of specificity-variant antibodies by alteration of carbohydrate in light chain of human monoclonal antibodies. Biochem Biophys Res Commun. 1992;189:625–632. doi: 10.1016/0006-291x(92)92246-t. [DOI] [PubMed] [Google Scholar]
- Thiebe R, Schable KF, Bensch A, Brensing-Kuppers J, Heim V, Kirschbaum T, Mitlohner H, Ohnrich M, Pourrajabi S, Roschenthaler F, Schwendinger J, Wichelhaus D, Zocher I, Zachau HG. The variable genes and gene families of the mouse immunoglobulin kappa locus. Eur J Immunol. 1999;29:2072–2081. doi: 10.1002/(SICI)1521-4141(199907)29:07<2072::AID-IMMU2072>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Tillman DM, Jou NT, Hill RJ, Marion TN. Both IgM and IgG anti-DNA antibodies are the products of clonally selective B cell stimulation in (NZB x NZW)F1 mice. J Exp Med. 1992;176:761–779. doi: 10.1084/jem.176.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlak P, Garrard WT. Discovery, regulation, and action of the major apoptotic nucleases DFF40/CAD and endonuclease G. J Cell Biochem. 2005;94:1078–1087. doi: 10.1002/jcb.20409. [DOI] [PubMed] [Google Scholar]