

Abstract
Menin is the protein product of the MEN1 tumor-suppressor gene; one allele of MEN1 is inactivated in the germ line of patients with “multiple endocrine neoplasia type 1” (MEN1) cancer syndrome. Menin interacts with several proteins involved in transcriptional regulation. RNA expression analyses have identified several menin-regulated genes that could represent proximal or distal interaction sites for menin. This report presents a substantial and unbiased sampling of menin-occupied chromatin regions using Serial Analysis of Chromatin Occupancy; this method combines chromatin immuno-precipitation with Serial Analysis of Gene Expression. Hundreds of menin-occupied genomic sites were identified in promoter regions (32% of menin-occupied loci), near the 3′ end of genes (14%), or inside genes (21%), extending other data about menin recruitments to many sites of transcriptional activity. A large number of menin-occupied sites (33%) were located outside known gene regions. Additional annotation of the human genome could help in identifying genes at these loci, or these might be gene-free regions of the genome where menin occupancy could play some structural or regulatory role. Menin occupancy at many intragenic positions distant from the core promoter reveals an unexpected type of menin target region at many loci in the genome. These unbiased data also suggest that menin could play a broad role in transcriptional regulation.
Keywords: MEN1, multiple endocrine neoplasia, SACO, chromatin immunoprecipitation, ChIP
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is a cancer syndrome predisposed by heterozygous germ-line mutations in the MEN1 tumor-suppressor gene (OMIM no. 13110). Somatic inactivation of the normal MEN1 allele in a predisposed cell initiates clonal tumor of parathyroid, enteropancreatic neuroendocrine, anterior pituitary, or other tissues [1]. Biallelic somatic loss of MEN1 has also been detected commonly in sporadic tumors of similar tissues [1]. The MEN1-encoded menin protein is expressed in all normal tissues and is predominantly nuclear [2]. Menin interacts with a variety of transcription factors and chromatin-modifying proteins: AP1 transcription factor JunD; NFκB proteins p50, p52, and p65; homeobox-containing protein Pem; TGFβ-induced protein Smad3; BMP-2-induced proteins Smad1, Smad5, and Runx2; corepressor mSin3A; and the MLL1/MLL2-containing COMPASS-like protein complex (reviewed in Agarwal et al. [3]). These interactions of menin with transcriptional regulatory proteins can produce either a suppressing effect or an enhancing effect on gene expression. Therefore, transcriptional regulation (without menin necessarily binding directly to specific DNA sequence) seems to be an important physiological activity of menin.
Analysis of menin target genes is an obvious avenue to understanding menin's function. Direct participation of menin in the regulation of some genes (Hoxa7, Hoxa9, Hoxa10, Hoxc8, FoxC1, FoxC2, hTERT, IGFBP2, Meis1, p18, and p27) has been suggested by chromatin immunoprecipitation (ChIP) analyses [3–10]. Furthermore, in specific promoter-based luciferase assays, overexpression of menin modulated the promoter activity of p18, p27, rat insulin, human prolactin, human cFos, human PAI2, and mouse IGFBP2 [3,11]. In addition, cDNA or oligonucleotide microarray techniques have revealed menin-regulated genes by comparing gene expressions in cell lines (vector-transfected versus MEN1-transfected) [5,7,12–14], in Men1+/+ versus Men1-/- mouse embryos [5], or in human MEN1 tumors [15,16]. Menin target genes in one tissue have shown minimal overlap with menin target genes in other tissues.
Recent advances in applying ChIP with DNA microarray (ChIP chip) or cloning techniques have been helpful in identifying novel target genes or DNA-binding sites of several proteins in the context of the whole genome [17–21]. These methods have shown that selected proteins with binding-site specificity occupy far more DNA sites than previously suspected. Serial Analysis of Chromatin Occupancy (SACO) is one such approach that combines ChIP with Serial Analysis of Gene Expression (SAGE) technique, and it has been successfully used to identify genomewide cAMP-responsive element-binding protein (CREB) targets [18]. Unlike CREB, menin does not possess any obvious DNA-binding domain nor are there any specific recognized DNA sequences that bind menin. Thus, menin's binding to DNA may possibly be indirectly facilitated by partnership with other transcriptional regulators. In the current report, SACO is used to survey menin-binding sites in human genomic DNA.
Materials and Methods
Cell Culture and Antibodies
HeLa-S3 cells (ATCC, Manassas, VA) were grown in complete Dulbecco's modified Eagle's medium (supplemented with 10% fetal calf serum, 2 µM glutamine, and 100 mg/ml penicillin-streptomycin) at 5% CO2. Normal rabbit IgG was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA), and antimenin (BL342) was obtained from Bethyl Laboratories (Montgomery, TX).
ChIP
HeLa cells were fixed at room temperature in 1% formal-dehyde/1x phosphate-buffered saline (PBS) for 20 minutes. Cells were scraped in cold harvesting buffer (100 mM Tris-HCl pH 9.4 and 10 mM DTT) and pelleted by centrifugation at 3000g for 5 minutes at 4°C. Cell pellets were washed with cold 1x PBS, and 107 cells were lysed in 0.6 ml of lysis buffer [20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 0.5% Triton X-100, and protease inhibitors (Roche Molecular Biochemicals, Indianapolis, IN)]. Chromatin lysates were sonicated with an ultrasonic processor (Model GE 750; PGC Scientific, Frederick, MD) to an approximate DNA size of 1000 bp and below, then centrifuged for 10 minutes at 13,000 rpm at 4°C. Supernatants were transferred to fresh tubes, and each 0.6-ml aliquot of the lysate was precleared with 80 µl of washed and bovine serum albumin (BSA)-blocked 50% protein A-Sepharose (Amersham Pharmacia, Piscataway, NJ) by rocking for 1 hour at 4°C. Immunoprecipitation was performed overnight at 4°C with 4 mg of antimenin antibody or normal rabbit IgG as control. Immune complexes were captured with 80 µl of 50% protein A-Sepharose slurry for 1 hour at 4°C. Beads were collected by centrifugation at 8000 rpm for 1 minute and washed as follows: four times in lysis buffer for 10 minutes, once in LiCl buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA pH 8.0, and 10 mM Tris-HCl pH 8.0), once with 1x TE pH 8.0 for 30 minutes, and then once with 1x TE for 5 minutes. Chromatin protein/DNA complexes were eluted from the beads twice by adding 100 µl of elution buffer (1% SDS and 0.1 M NaHCO3 pH 8.0) at room temperature for 15 minutes each. The beads were collected by centrifugation at 13,000 rpm for 1 minute, and eluates were pooled and heated at 65°C overnight to reverse crosslinks. DNA fragments were purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA).
SACO Library
A SACO library was prepared using antimenin ChIP DNA obtained from 6 x 107 HeLa cells [18]. An outline of the menin-SACO library construction is shown (Figure W1). A modified version of the Long-SAGE protocol [22] was used to create ditags. Ditag concatemers were cloned into pZErO (Invitrogen, Carlsbad, CA) kanamycin vector and transformed by electroporation into E. cloni 10G electrocompetent cells (Lucigen, Middleton, WI). This antimenin plasmid SACO library was titered, and glycerol stocks were prepared from transformed bacteria. The average number of ditags in plasmids was analyzed by polymerase chain reaction (PCR) using vector primers flanking the insert.
SACO Data Analysis and Bioinformatics
Sequencing of SACO library plasmids was performed at Rexagen/Regulome (Seattle,WA). Approximately 5000 plasmids were sequenced to obtain the sequence of at least 40,000 tags. Concatemer sequences were extracted from chromatograms with the base caller “phred” using recommended settings [23]. A custom “perl” script separated ditags at all CATGs. Duplicate sequences were removed from the analysis (same set of concatemerized tags). The resulting 21-bp SACO genomic signature tags (GSTs) were matched to genomic CATG sites using a C program. GSTs with exact matches or matches with one substitution error that were uniquely assignable to a genomic location were considered as positives. GSTs without a unique genomic match or with multiple unique matches were not considered. GSTs within 2 kb of each other were taken to be associated with the same locus. A set of scripts that automate the analysis of SACO data is available online at http://genome.bnl.gov/SACO/. Menin-SACO raw data are available at http://saco.ohsu.edu/. Additional details on the analysis of SACO loci have been published [18].
ChIP-PCR Analysis of Menin-Occupied Loci
The genomic sequence (2 kb) flanking the GST median was copied from ENSEMBL. Primers were designed using MIT's (Whitehead Institute, Cambridge, MA) Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) such that primer pairs would amplify 200- to 300-bp products located close to the GST. Primer sequences are available on request. For GST confirmation, independent ChIP assays were performed using 6 x 107 HeLa cells. Quantitative PCR (qPCR; 25 µl) reactions were performed in duplicate using Mx3000P (Stratagene, La Jolla, CA) and the SYBR-green qPCR kit (Stratagene). PCR conditions were as follows: 95°C for 10 minutes; 40 cycles of 95°C for 30 seconds, 55°C for 60 seconds, and 72°C for 30 s. Antimenin and rabbit IgG ChIPs were expressed as nanograms of gel-purified (Qiagen) amplicon. Products showing a greater-than-two-fold enrichment relative to an IgG control were considered confirmed. Each amplicon was analyzed by agarose gel electrophoresis, and those yielding multiple products or no products were discarded and primers were redesigned. Other primer pairs used for ChIP-PCR were as follows: positive control primer pairs for Hoxc8 and Hoxa9 promoters, and seven negative control primer pairs (ch12: 2997, H2A, H2B, ch12:6151, LRP, MycP2, and Tubulin).
HeLa RNA Expression Analysis
Total RNA was isolated from two independent culture dishes of exponentially growing HeLa cells with Trizol (Invitrogen) and further purified using RNAeasy (Qiagen). Each sample was analyzed at the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) microarray core facility using Affymetrix microarray platform (Affymetrix, Santa Clara, CA). Labeled samples were hybridized to Affymetrix human genome U133 Plus 2.0 array. Microarray data were normalized and analyzed using Affymetrix GeneChip software Microarray Analysis Suite 5.0. Expression levels were assigned based on positive or negative signals for gene expression, as per Affymetrix's detection call of “present” or “absent” (a signal intensity of > 200 arbitrary units was considered “present”).
Results and Discussion
Menin-SACO Library
Menin expression has been detected in all tissues and cell lines studied so far, except in menin-null tumor tissues or menin-null mouse embryo fibroblasts (MEFs). HeLa cells have been used for performing transcriptional reporter assays involving menin-interacting proteins and for isolating a menin-containing COMPASS-like protein complex. Hence, menin activities and targets almost certainly exist in HeLa cells. Thus, HeLa cells seemed appropriate for the initial analysis of menin-associated targets at a genomic scale. A SACO library was prepared from a HeLa cell chromatin extract after ChIP with an antimenin antibody. The menin-SACO library contained in excess of 3 x 106 GSTs. Close to 5000 plasmids containing concatemerized ditags were sequenced, and after removing duplicate sequences (possessing the same order of tags in concatemers), the sequence from 38,743 GSTs was analyzed. This may not be sufficient for an exhaustive analysis of menin targets in the entire genome. But a representative sampling of menin occupancy across the genome was accomplished. Of the 38,743 GSTs, 32,956 were distinct, of which 22,191 (67%) could be mapped to unique loci in the current build of the human genome sequence (Hg 17). A total of 8369 (25%) GSTs mapped to multiple locations belonging to repetitive sequences, and 2396 (8%) GSTs could not be located in the human genome sequence. The specificity of SAGE-type library analysis is increased by considering loci represented by multiple tags or GSTs [24]. Single hits increase the number of menin targets by eight-fold. The number of likely false positives makes analysis of this data set of single hits difficult and unreliable. Other groups working with techniques similar to SACO also do not consider single hits in their analysis of target loci [e.g., in the analysis of p53 target loci (by ChIP PET) using two statistical analysis methods, Wei et al. [21] concluded that singletons were most likely background, and Kim et al. [19] have shown that identification of chromosomal targets by sequence tag analysis of genomic enrichment (STAGE) could be validated by independent methods when target genes were designated by multiple occurrences of STAGE tags). Therefore, all analyses of the menin-SACO library focused on loci represented by more than one GST (Figure 1). These include 2616 GSTs representing 1162 unique loci (unique locus = GSTs mapping within 2 kb of each other). The 2-kb interval was chosen based on a natural cutoff in the distribution of GSTs [18].
Figure 1.

Characterization of clones in the menin-SACO library. Partial sequencing of the menin-SACO library identified 1162 menin target loci; 778 of these loci were distributed near annotated or predicted genes within 2 kb of the 5′ end, the 3′ end, or inside genes. GSTs = genomic signature tags.
Identification of Menin-Occupied Loci
Among the 1162 menin-occupied loci represented by multiple GSTs, 778 (67%) loci mapped to at least one mRNA or gene predicted by ECgene EST and mRNA clustering annotation (UCSC genome browser). These consisted of 371 (32%) GSTs located within 2 kb of the 5′ end of annotated genes (defined as the most 5′ region of UCSC “known gene” or ENSEMBL gene annotation), 163 (14%) GSTs located within 2 kb of the 3′ end of annotated genes, and 244 (21%) GSTs located inside genes (not within 2 kb of the 5′ and 3′ ends of a gene). The rest of the 384 (33%) menin-occupied loci were located > 2 kb away from any known or predicted gene. The 778 loci represented 635 characterized genes. These 635 genes included 157 genes that encoded hypothetical/predicted proteins. Note that a single gene could be represented by multiple menin-occupied loci if the “ unique loci” mapped within or > 4 kb from each other in the same gene. This was observed for 70 genes in the current data set. The properties of these 70 genes were unremarkable.
Thus, as expected, menin was predominantly located near the 5′ ends of annotated genes; at the same time, the surprisingly high frequency of menin occupancy at the 3′ end and inside genes could be explained by the location of as yet unknown regulatory elements or unknown genes in these regions.
Confirmation of Menin Occupancy at Target Loci
ChIP followed by PCR (ChIP-PCR) was performed for 51 menin-occupied loci represented by more than one GST and located within 2 kb of the 5′ end of annotated genes. qPCR of antimenin ChIP DNA from HeLa cells confirmed menin occupancy at 94% (48 of 51) of loci, specifically enriched by more than two-fold over anti-IgG ChIP control (range of fold change, 0.12–10.0) (Figure 2). qPCR data also verified menin occupancy at the Hoxc8 and Hoxa9 promoter regions that have been previously reported as menin targets [5,9,10,25]. Negative control primer pairs (n = 7) did not show a > 1.6-fold enrichment over anti-IgG ChIP control (range of fold change, 1.1–1.6). Also tested were 26 menin-occupied loci that mapped inside genes (Figure 2), of which 24 showed a more-than-two-fold enrichment in the antimenin ChIP over IgG control ChIP (range of fold change, 1.7–16.5). Therefore, almost each of the menin-occupied genomic loci identified by sequencing the menin-SACO library was also enriched in independent ChIP-PCR analysis, confirming the robustness of menin-binding sites identified by SACO.
Figure 2.
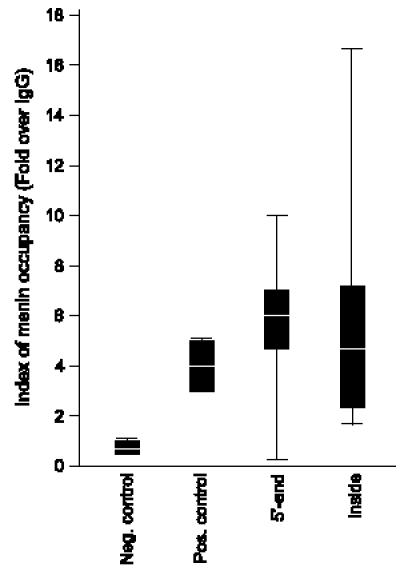
qPCR of ChIPs as an index of menin occupancy. Enrichment of menin occupancy is shown as fold change over IgG ChIP-PCR for each category. Shown are the interquartile range (black box), median (white line on the black box), and overall range (black line). Primer pairs used: Neg = negative control (n = 7); Pos = positive control (n = 2; Hoxc8 and Hoxa9); 5′ end = GSTs mapping to the 5′ end of genes (n = 51); Inside = GSTs mapping inside genes and not within 2 kb of the 5′ end or the 3′ end of a gene (n = 26). The nucleotide position of each GST analyzed by ChIP-PCR and the fold change over IgG are shown (Table W3).
Characterization of Menin-Occupied Loci
The current study has identified many menin-occupied DNA sites outside the 5′ end of genes. The SACO analysis reported here is consistent with an independent approach that we have used [26] with ChIP chip and arrays containing 20,000 human promoters, an end-to-end coverage of 381 genes, and an additional 20 Mb on chromosome 7. Both analyses showed many interaction loci of menin in promoter regions, but also many other menin interaction loci outside these regions. The sensitivity of detecting weaker direct protein-DNA interactions or weaker indirect protein-protein-DNA interactions would depend on the quality of the ChIP step that is common to both SACO and ChIP chip. Therefore, both techniques would be equally handicapped in being sensitive to weak interactions. With the current menin-SACO data set, a representative sampling of menin occupancy across the genome was accomplished. From ChIP chip menin data, there were at least 1706 promoters in HeLa cells that bound menin. In the present report, after partial sequencing of the menin-SACO library, 371 menin-occupied loci were found near promoter regions. The ChIP chip approach may indeed have missed some targets. Therefore, we estimate that approximately 22% (or less) of the genome is being interrogated with the current menin-SACO data set. A comparison has not yet been performed between SACO-identified menin-occupied sites and those sites identified by ChIP chip analysis in HeLa cells.
A location map of the 1162 menin-occupied loci using “ENSEMBL KaryoView” (http://www.ensembl.org/Homo_sapiens/karyoview) showed even distribution among subchromosomal loci (data not shown). No significant association of menin GSTs at regions of gene clustering was observed when we examined this subchromosomal distribution of menin-occupied loci.
It is important to identify the nature of menin-occupied DNA sites and to understand menin's functions at sites of menin occupancy. Menin is known to regulate AP1-activated transcription at AP1-binding sites [27,28]. A directed search for AP1-binding sites “TGAGTCA” or “TGACTAA” near the 1162 menin-occupied loci (using a 1-kb flanking sequence) revealed that these sites occurred at a frequency of 7.14 x 10-5 and 6.28 x 10-5, respectively. These frequencies were not significantly higher than the frequency in the human genome of “TGAGTCA” (8.08 x 10-5) or the frequency of a random heptanucleotide (6.52 x 10-5) with the same GC content as the AP1-binding site [29].
Sequence analysis of the DNA-binding site motifs of other menin-interacting transcription factors was not performed because either their consensus-binding sites were not known (Pem, Ches1), the sequence was small (CAGA for Smad proteins), or the consensus sequence was variable and sequence subunit specificity for the binding site was not known (GGGRNNYYCCC for NFκB-p50, NFκB-p52, and NFκB-p65). We are interested in performing these analyses when more extensive data are available. In addition, Smad proteins have been reported to regulate transcription from AP1 sites [30]. Furthermore, the AP1 transcription factor JunD is our favored candidate and is, so far, the most promising candidate as a valid menin partner. We have also tried to find a common menin-DNA interaction motif at the 1162 menin-occupied loci represented by multiple GSTs, but searches have so far not been successful (unpublished data).
It is not known if any of the 244 intragenic menin-occupied regions participates in transcriptional regulation. The functional significance of factor occupancy at intragenic loci and the role of intragenic loci in regulating transcription are being actively pursued in several laboratories [18,21,31]. Therefore, further studies might also shed light on the role of menin occupancy at intragenic loci. Given menin's presence in protein complexes that modify transcriptionally active chromatin, one possibility is that menin occupancy identified at intragenic regions may coincide with regions where menin could track the transcription process along the gene as a component of protein complexes.
Therefore, the current analysis further highlights recent evidence of the broad role of menin in transcriptional regulation.
CpG Island Analysis of Menin-Occupied Loci
Menin-occupied loci were examined for the presence of CpG islands within 2 kb of GSTs. Among the 1162 loci that could be located near mRNA, CpG islands were found near 61% of menin-occupied loci that were near the 5′ end of genes, 5% of the 3′-end loci were near CpG islands, and 6% of the loci inside genes were near CpG islands. For loci that could not be located to mRNA, only 8% were near CpG islands. Therefore, the possibility of a gene(s) being located near these orphan loci could be very low. They might end up being “inside” genes (intragenic) based on additional annotations of the human genome.
When transcriptional regulatory proteins are shown to occupy loci near the 3′ end of genes or inside genes, bidirectional or antisense transcription is generally suspected to occur near such regions [18]. Based on the less abundant occurrence of CpG islands near the menin-occupied loci that were represented by GSTs mapping near the 3′ end (5% near CpG islands) or inside genes (6% near CpG islands) compared to those near the 5′ ends (61%), it is possible that these loci may not occur near sites of bidirectional or antisense transcription because such regulatory regions are reported to be associated with CpG islands [32].
Functional Categories of Genes Occupied By Menin
To analyze the types of genes near menin-occupied loci, Gene Ontology (GO) biological process categories were assigned to the 635 menin target genes by using GOstat [33]. Hypothetical genes (n = 157) were not considered for GO assignment. Functional categories of 478 menin target genes are summarized (Table 1). In comparison to the categories of all genes, the most overrepresented categories are genes important or predicted to be important in cellular metabolism (51%), macromolecule metabolism (33%), and cell cycle (7%). In the GO function hierarchy, a few genes belong to multiple categories and were thus scored more than once. The large number of genes without annotations or functions did not allow a thorough classification of the entire sample.
Table 1.
Functional Categories of 478 Annotated Menin Target Genes Identified by SACO.
| GO Term (Biological Process) | Menin Targets* [Gene Count (%)] | Human Genome* [Gene Count (%)] | P† |
| Cellular metabolism | 242 (51) | 11,588 (41) | 1.19E-03 |
| Macromolecule metabolism | 156 (33) | 7,104 (25) | 5.13E-03 |
| Nucleobase, nucleotide, and nucleotide and nucleic acid metabolism | 115 (24) | 4,916 (18) | 4.19E-03 |
| Biopolymer metabolism | 105 (22) | 4,349 (16) | 2.73E-03 |
| Regulation of cellular physiological process | 104 (22) | 4,207 (15) | 1.31E-03 |
| Cell organization and biogenesis | 58 (12) | 1,985 (7) | 1.07E-03 |
| Cell cycle | 35 (7) | 792 (3) | 1.09E-06 |
| Transcription from RNA PolII promoter | 26 (5) | 500 (2) | 1.50E-03 |
| Morphogenesis | 24 (5) | 615 (2) | 1.19E-03 |
| Organismal physiological process | 24 (5) | 2,553 (9) | 2.88E-02 |
| G-coupled protein receptor protein signaling pathway | 10 (2) | 1,491 (5) | 2.73E-02 |
| Response to biotic stimulus | 9 (2) | 1,380 (5) | 3.19E-02 |
Functional categories are based on GO annotation (GOstat: http://gostat.wehi.edu.au).
Note that in GO function hierarchy, some genes belong to multiple categories.
GO terms 1 to 9 are overrepresented, and terms 10 to 12 are underrepresented in the list of menin target genes.
GO terms with similar representation in menin targets and in human genome are not shown.
Percentage calculation: menin targets = 478; human genome = 28,042.
Compared to human genome gene count for each category.
Correlation between Gene Occupancy and Expression
The occupancy of a locus by menin does not indicate whether the gene is expressed, nor does it specify the direction of any regulation by menin. To find out the expression status of menin-occupied target genes, total RNA preparations isolated from HeLa cells were analyzed for expression using oligonucleotide arrays. Among the 635 menin target genes identified by SACO, a comparison of HeLa RNA expression data and SACO data gave 614 genes for which both menin occupancy and gene expression status were available. Sixty-two percent of the menin-occupied genes were expressed, compared to 40% of genes expressed for the entire microarray, indicating that menin-occupied genes are more likely to be transcribed (P < .003). When restricted to expressed genes in HeLa cells, the representation of genes where menin occupied the 5′ end was similar to that observed for the genomic occupancy of menin GSTs identified near the 5′ end of genes (48% in genomic occupancy vs 52% in expressed gene list). But the representation of genes where menin occupied intragenic loci or 3′ ends was different from the genomic occupancy of menin at these sites (intragenic loci: 31% in genomic occupancy vs 40% in expressed gene list; 3′ end loci: 21% in genomic occupancy vs 9% in expressed gene list).
To further evaluate how the genes occupied by menin in HeLa cells are modulated as a consequence of menin loss in MEN1-associated human tumors or Men1-null MEFs, a comparison with published gene expression array data was made. The analysis showed that among the 15 genes that were common between the menin targets identified by SACO and the published gene expression data (Table W1), eight genes were downregulated in islet tumors (that lack menin). This correlation of menin-occupied genes and their downregulation on menin loss in islet tumors is not significant because the “islet tumor downregulated genes” category was overrepresented in the analysis compared to other categories (143 of 319 genes that were considered for comparison).
Menin shows a differential effect on transcription activated by JunD versus c-Jun—two members of the AP1 family of transcription factors [27,28]. To assess whether any of the menin-occupied loci identified in this study correlates with AP1-regulated genes, a list of 249 AP1-regulated genes [34] was compared to menin-occupied target genes. The analysis showed 10 genes that were common between the menin targets identified by SACO and the published data about AP1-regulated genes (Table W2). This correlation did not yield any important data about menin interaction and AP1-regulated genes except for Hoxa10, which has been previously shown by ChIP-PCR as a menin target [10] (Table W3), and DDR2, a gene involved in osteoarthritis that is upregulated in menin-complemented menin-null MEFs (Table W1). The 10 genes that were common targets were identified as AP1-regulated genes using various methods and various cells (as reported in Hayakawa et al. [34]). Information about the directionality of their AP1-regulated transcription is not known, and it is also not known if they are JunD-regulated or c-Jun-regulated.
Future analysis of SACO-identified menin targets and their transcriptional regulation under physiological and/or pathological conditions should help extend our findings.
Modes of Menin Action
The many interactions of menin with proteins and with “genes” suggest two alternate models of menin action. Protein interactions, mainly with JunD, indicate that menin might initiate its action into only one pathway, for example, by suppressing the activity of an oncogenic substrate JunD [35]. In contrast, the current study, together with another recent study [26], opens a new paradigm for the normal and abnormal actions of menin. The genomic interactions identified by SACO and ChIP chip point to the possibility of a more complex nature of menin action. The new paradigm involves action beginning at many genomic pathways. Further work should explore whether these two models of menin action are complementary.
Conclusions
A substantial sampling of the menin-SACO library for menin-occupied loci in genomic chromatin showed that menin was not confined, in part, to a large number of promoter regions but that menin could also occupy many other regions inside genes and at the 3′ ends of genes. These data suggest that new as yet unidentified regulatory sequences could be present at those intragenic loci occupied by menin, or they could be regions where menin tracks with transcriptional progression. Menin's functions at the 1162 genomic loci and the nature of the DNA sequence at these loci remain to be established.
Supplementary Material
Acknowledgements
We thank MEN1 collaborators in National Institutes of Health intramural laboratories and members of the Goodman laboratory for helpful discussions. We thank Margaret Cam of the NIDDK microarray core facility for HeLa expression analysis, and Tao Tao of the NCBI User Service for AP1-binding site search.
Footnotes
This research was supported, in part, by the Intramural Research Program of the National Institutes of Health (National Institute of Diabetes and Digestive and Kidney Diseases, National Human Genome Research Institute, and National Institute of Deafness and Communication Disorders) and by an extramural National Institutes of Health grant DK45423 (to R.H.G.).
This article refers to supplementary material, which is designated by “W” (i.e., Table W1, Figure W1) and is available online at www.bcdecker.com.
References
- 1.Marx SJ. Multiple endocrine neoplasia type 1. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. New York, NY: McGraw-Hill; 2002. pp. 450–475. [Google Scholar]
- 2.Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, Chandrasekharappa SC. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA. 1998;95:1630–1634. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal SK, Kennedy PA, Scacheri PC, Novotny EA, Hickman AB, Cerrato A, Rice TS, Moore JB, Rao S, Ji Y, et al. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res. 2005;37:369–374. doi: 10.1055/s-2005-870139. [DOI] [PubMed] [Google Scholar]
- 4.Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci USA. 2006;103:1018–10123. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 6.Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La P, Desmond A, Hou Z, Silva AC, Schnepp RW, Hua X. Tumor suppressor menin: the essential role of nuclear localization signal domains in coordinating gene expression. Oncogene. 2006;25:3537–3546. doi: 10.1038/sj.onc.1209400. [DOI] [PubMed] [Google Scholar]
- 8.Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–889. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 9.Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102:14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 11.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci USA. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.La P, Schnepp RW, Petersen D, Silva C, Hua A. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology. 2004;145:3443–3450. doi: 10.1210/en.2004-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnepp RW, Mao H, Sykes SM, Zong WX, Silva A, La P, Hua X. Menin induces apoptosis in murine embryonic fibroblasts. J Biol Chem. 2004;279:10685–10691. doi: 10.1074/jbc.M308073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stalberg P, Grimfjard P, Santesson M, Zhou Y, Lindberg D, Gobl A, Oberg K, Westin G, Rastad J, Wang S, et al. Transfection of the multiple endocrine neoplasia type 1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects expression of JunD, delta-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1. J Clin Endocrinol Metab. 2004;89:2326–2337. doi: 10.1210/jc.2003-031228. [DOI] [PubMed] [Google Scholar]
- 15.Dilley WG, Kalyanaraman S, Verma S, Cobb JP, Laramie JM, Lairmore TC. Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol Cancer. 2005;4:9. doi: 10.1186/1476-4598-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forsberg L, Bjorck E, Hashemi J, Zedenius J, Hoog A, Farnebo LO, Reimers M, Larsson C. Distinction in gene expression profiles demonstrated in parathyroid adenomas by high-density oligoarray technology. Eur J Endocrinol. 2005;152:459–470. doi: 10.1530/eje.1.01864. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Sadowski I. Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc Natl Acad Sci USA. 2005;102:4813–4818. doi: 10.1073/pnas.0407069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Bhinge AA, Morgan XC, Iyer VR. Mapping DNA-protein interactions in large genomes by sequence tag analysis of genomic enrichment. Nat Methods. 2005;2:47–53. doi: 10.1038/nmeth726. [DOI] [PubMed] [Google Scholar]
- 20.Sikder D, Kodadek T. Genomic studies of transcription factor-DNA interactions. Curr Opin Chem Biol. 2005;9:38–45. doi: 10.1016/j.cbpa.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 22.Saha S, Sparks AB, Rago C, Akmaev V, Wang CJ, Vogelstein B, Kinzler KW, Velculescu VE. Using the transcriptome to annotate the genome. Nat Biotechnol. 2002;20:508–512. doi: 10.1038/nbt0502-508. [DOI] [PubMed] [Google Scholar]
- 23.Ewing B, Green P. Base-calling of automated sequencer traces using phred: II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 24.Wu SM, Baxendale V, Chen Y, Pang AL, Stitely T, Munson PJ, Leung MY, Ravindranath N, Dym M, Rennert OM, et al. Analysis of mouse germ-cell transcriptome at different stages of spermatogenesis by SAGE: biological significance. Genomics. 2004;84:971–981. doi: 10.1016/j.ygeno.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS, et al. Genomewide analysis of menin binding provides insights to MEN1 tumorigenesis. PLoS Genet. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 28.Knapp JI, Heppner C, Hickman AB, Burns AL, Chandrasekharappa SC, Collins FS, Marx SJ, Spiegel AM, Agarwal SK. Identification and characterization of JunD missense mutants that lack menin binding. Oncogene. 2000;19:4706–4712. doi: 10.1038/sj.onc.1203832. [DOI] [PubMed] [Google Scholar]
- 29.Zhou H, Zarubin T, Ji Z, Min Z, Zhu W, Downey JS, Lin S, Han J. Frequency and distribution of AP-1 sites in the human genome. DNA Res. 2005;12:139–150. doi: 10.1093/dnares/12.2.139. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- 31.Lieb JD, Clarke ND. Control of transcription through intragenic patterns of nucleosome composition. Cell. 2005;123:1187–1190. doi: 10.1016/j.cell.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Engstrom PG, Suzuki H, Ninomiya N, Akalin A, Sessa L, Lavorgna G, Brozzi A, Luzi L, Tan SL, Yang L, et al. Complex Loci in human and mouse genomes. PLoS Genet. 2006;2:e47. doi: 10.1371/journal.pgen.0020047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 34.Hayakawa J, Mittal S, Wang Y, Korkmaz KS, Adamson E, English C, Ohmichi M, McClelland M, Mercola D. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol Cell. 2004;16:521–535. doi: 10.1016/j.molcel.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal SK, Novotny EA, Crabtree JS, Weitzman JB, Yaniv M, Burns AL, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc Natl Acad Sci USA. 2003;100:10770–10775. doi: 10.1073/pnas.1834524100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.