

Abstract
Virus-induced membrane fusion can be subdivided into three phases defined by studies of class I and class II fusion proteins. During Phase I, two membranes are brought into close apposition. Phase II marks the mixing of the outer membrane leaflets leading to formation of a hemifusion intermediate. A fusion pore stably forms and expands in Phase III, thereby completing the fusion process. Herpes simplex virus type 1 (HSV-1) requires four glycoproteins to complete membrane fusion, but none has been defined as class I or II. Therefore, we investigated whether HSV-1-induced membrane fusion occurred following the same general phases as those described for class I and II proteins. In this study we demonstrate that glycoprotein D (gD) and the glycoprotein H and glycoprotein L complex (gHL) mediated lipid mixing indicative of hemifusion. However, content mixing and full fusion required glycoprotein B (gB) to be present along with gD and gHL. Our results indicate that, like class I and II fusion proteins, fusion mediated by HSV-1 glycoproteins occurred through a hemifusion intermediate. In addition, both gB and gHL are probably directly involved in the fusion process. From this, we propose a sequential model for fusion via HSV-1 glycoproteins whereby gD is required for Phase I, gHL is required for Phase II, and gB is required for Phase III. We further propose that glycoprotein H and gB are likely to function sequentially to promote membrane fusion in other herpesviruses such as Epstein–Barr virus and human herpesvirus 8.
Keywords: lipid transfer, membrane fusion, fluorescence microscopy
Studies using class I and class II fusion proteins have demonstrated that virus-induced membrane fusion can be subdivided into three phases (1, 2). Phase I involves bringing opposing membranes into close proximity through a viral glycoprotein binding a cellular receptor. Phase II involves the initiation of lipid mixing between the two apposed membranes and is completed when the outer membrane leaflets are mixed to form an intermediate called hemifusion. Phase III begins when the inner membrane leaflets are mixed and continues the pore formation and expansion. The completion of Phase III signifies the completion of the fusion process. The three phases of membrane fusion (close apposition, hemifusion, and complete fusion) are useful to characterize functions of viral glycoproteins in the fusion process (1, 3, 4). Fusion proteins that have Phase I function bring membranes in close apposition and ultimately result in the initiation of the fusion process. Fusion proteins that have Phase II function are capable of mixing outer membrane leaflets leading to hemifusion. Phase III fusion proteins are capable of forming and expanding a fusion pore. For many viruses, one or two fusion proteins can carry out all phases of membrane fusion. The fusion process has yet to be characterized for viruses that require more than two fusion glycoproteins, and a major issue is whether multiple glycoproteins mediate fusion through a hemifusion intermediate.
Herpes simplex virus type 1 (HSV-1) is a member of the α-herpesvirus subfamily and requires four membrane glycoproteins to mediate fusion. Glycoprotein D (gD), glycoprotein B (gB), and the glycoprotein H and L complex (gHL) are essential for fusion. HSV-1 mutants lacking one of those four glycoproteins are not infectious, and their replication is blocked at membrane fusion (5). Three of these glycoproteins are membrane-spanning [gB, gD, and glycoprotein H (gH)], whereas glycoprotein L (gL) is a soluble glycoprotein expressed at the virion or cell surface only in a heterodimer with gH (6). HSV-1 gH is not trafficked to the cell surface in the absence of gL (6). The current paradigm for HSV-1 fusion is that the required glycoproteins form a fusion complex (7, 8), where gD binds a cell surface receptor (herpesvirus entry mediator, nectin-1, or modified heparin sulfate), inducing a conformational change that leads to fusion mediated by gB and/or gHL (8–11). Because it is hypothesized that the main function of gD resides in Phase I (8, 12) and gL functions solely as a gH chaperone (13), gB and gH are the most likely candidates to be involved in Phases II and III of the fusion process. However, their exact role in the fusion mechanism has yet to be elucidated. There are two models for fusion: the regulatory/structural model, where one glycoprotein serves as a regulator or a support for the fusogenic activity of the other (14, 15); and the sequential model, where each glycoprotein functions at a different stage in the fusion process (7). If the sequential model is correct, it may be possible to separate the function of the glycoproteins in fusion through assays that detect a fusion intermediate, such as hemifusion.
We wanted to investigate the mechanism of HSV-1 fusion by examining which glycoprotein(s) function for each phase of fusion. Current fusion and entry assays for HSV-1 are insufficient to detect fusion intermediates; therefore, we developed a cell–cell fusion assay and a virus–cell fusion assay capable of detecting hemifusion. Using these two assays, we demonstrate that HSV-1-induced membrane fusion occurred through a hemifusion intermediate and that gD and gHL were sufficient to mediate hemifusion but not complete fusion. These results suggest that gD and gHL were required for Phases I and II of the fusion process. gB is likely necessary for Phase III functions of fusion because it was not required for hemifusion, and complete fusion was ablated in the absence of gB.
Results
gB Is Not Required for Hemifusion.
Little is understood about the mechanism by which the four HSV-1 envelope glycoproteins mediate fusion. We assumed that fusion induced by HSV-1 followed the same basic mechanism as that characterized for class I and class II fusion proteins, i.e., mediated through Phases I, II, and III (1, 3, 4). We hypothesized that HSV-1 fusion occurs via a sequential model for the fusion process whereby each glycoprotein functions at a different stage in the process, specifically, that gD functioned to bring cells into close proximity and trigger the initiation of fusion (Phase I), after which either gB or gHL functioned to mediate hemifusion (Phase II), and the other glycoprotein completed the process (Phase III).
Based on our hypothesis, fusion would be arrested at hemifusion in the absence of one of the glycoproteins. To test this, we developed a cell–cell fusion assay to detect lipid mixing (hemifusion) as well as content mixing (full fusion) based on a soluble NSF attachment receptor (SNARE) hemifusion assay reported previously (16). CHO cells lack the lipid ganglioside GM1 (16), so lipid mixing could be detected by using a TRITC-conjugated form of cholera toxin β-subunit (CTX) when CHO cells were fused to cells that express GM1. CTX binds GM1 with high affinity (17), and GM1 transfer to CHO cells was detected by CTX binding. A common method to study hemifusion involves the use of lipid dyes to detect lipid mixing. However, those dyes can rapidly internalize from the plasma membrane of living cells thereby decreasing the ability to detect lipid mixing at the plasma membrane. The abundance of GM1 in the plasma membrane of Vero cells (18) enables it to be an efficient natural marker of membrane transfer.
We transfected CHO cells with plasmids expressing GFP and gBgDgHgL (ENV), gDgHgL (gB−), gBgHgL (gD−), gBgDgL (gH−), gBgDgH (gL−), or GFP alone. We incubated the transfected CHO cells with Vero-BG20 cells. Vero-BG20 cells express GM1 (18) and have been engineered to overexpress human herpesvirus entry mediator. If hemifusion occurs, GM1 transfer would be detected in the absence of cytoplasmic GFP transfer resulting in single GFP+ cells that stain GM1+. We found a significant number of GM1+ and GFP+ cells in gB− transfections, and those cells contained only one nucleus, indicating that lipid transfer/hemifusion occurred in the absence of full fusion (Fig. 1). This differed from ENV transfections where GM1- and GFP-positive multinucleated cells were also detected. We quantified those results by randomly imaging five fields of a coverslip and counting the number of GFP-single-positive and GFP/GM1-double-positive cells. Quantification was performed for three independent experiments, and the results in Table 1 are a representative experiment. A sample low-magnification field of each transfection is shown in supporting information (SI) Fig. 6. From this, we determined the percentage of the GFP+ cells that were single-nuclei hemifused cells and the percentage that were multinucleated fully fused cells. There were significantly (P < 0.05) more GFP/GM1-double-positive cells for gB− transfections than for gD−, gH−, gL−, or GFP transfections (Table 1). In the ENV samples we observed hemifused cells at approximately the same efficiency as gB− transfections. Those instances of hemifusion may have been the result of an intrinsic inefficiency in the HSV-1 fusion process or possibly the result of cells not transiently expressing sufficient levels of gB to promote full fusion. These results provide direct visual evidence that gD and gHL are sufficient to mediate hemifusion and that gB is not required to observe hemifusion. Furthermore, because gH− and gL− transfections are incapable of mediating hemifusion, gHL are the mediators of Phase II of the fusion process. An alternate possibility is that gD mediates Phases I and II and gB represses its Phase II activity in the absence of gHL. This is unlikely because gD alone with GFP (gD-GFP) displayed no significant hemifusion (Table 1). We observed results similar to those described in Table 1 using Vero cells instead of Vero-BG20, where lipid mixing was observed in cells expressing gD and gHL, but not for others (data not shown). However, the efficiency of lipid transfer as well as syncytium formation was reduced by 50% for Vero cells compared with Vero-BG20 cells, demonstrating an increase in hemifusion and fusion as gD receptor levels increased.
Fig. 1.

Glycoproteins necessary for hemifusion and complete fusion in the cell hemifusion assay. CHO cells were transfected with plasmids expressing GFP and all four fusion glycoproteins (ENV: gB, gD, gH, and gL) or three of the four (gB−, gD−, gH−, and gL−) glycoproteins. Transfected cells were overlaid with Vero-BG20 cells, fixed, and stained with TRITC-conjugated CTX (CTX-555) to detect GM1. Images were taken at ×100 magnification on a Zeiss Axiovert microscope. The experiment was performed three independent times, and images are from one representative experiment.
Table 1.
Quantitation of cell–cell hemifusion assay
| Transfection | % hemifusion ± SD | % fusion ± SD |
|---|---|---|
| GFP | 1 ± 1.3 | 0 |
| gB− | 9.3 ± 3.4 | 0.4 ± 0.8 |
| gD− | 1.3 ± 2.0 | 0 |
| gH− | 1.1 ± 1.7 | 0 |
| gL− | 1.3 ± 1.2 | 0 |
| ENV | 9.7 ± 2.4 | 12.1 ± 6.3 |
| gD-GFP | 2.4 ± 3.4 | 0 |
Lipid Transfer Occurs for gB− Virus.
Cell–cell fusion assays are used to model virus–cell fusion; however, discrepancies have been observed between virus entry and cell fusion for HSV-1 (5, 7, 19, 20). We wanted to examine whether gD and gHL were required for lipid transfer from virus to cells, so we analyzed GM1 transfer from viral to cellular membranes. Our approach was to measure GM1 transfer to CHO cells using virus isolates lacking gB, gD, gH, or gL. We generated virus lacking a particular envelope glycoprotein (gB−, gD−, gH−, or gL−) by inoculating Vero cells with mutant virus grown on complementing cells. This complemented virus will infect the Vero cells and produce noninfectious virus particles because those particles will lack one of the four required fusion proteins. Because Vero cells contain GM1 (18), we could observe GM1 transfer from the viral lipid envelope to CHO membranes using a FITC-conjugated CTX (CTX-FITC).
We examined the ability of mutant viruses lacking one of the four required envelope glycoproteins to mediate lipid transfer to susceptible cells. We inoculated CHO cells expressing nectin-1 (HveC-1 cells) (21) with wild-type or mutant virus preparations to determine the glycoprotein requirements for virus–cell hemifusion. Equivalent levels of wild-type and mutant viruses bound to HveC-1 cells after inoculation at 4°C for 2 h and incubation with CTX-FITC (data not shown). This result is in agreement with data previously published showing that viruses deleted for gB, gD, gH, or gL bind cells equivalently to wild-type HSV-1 (22–25). The HveC-1 cells were labeled with Celltracker blue to aid in the identification of cells and to stain the cytoplasm. The results seen in Fig. 2 (and lower magnification images in SI Fig. 7) show that, along with the wild-type lab isolate HSV-1(KOS), virus lacking gB (gB−) transferred GM1 to HveC-1 cells. Furthermore, viruses lacking gD (gD−), gH (gH−), or gL (gL−) were incapable of transferring significant levels of GM1. These results were obtained by using virus concentrated from culture supernatants. Identical results were obtained by using viruses semipurified by pelleting through a sucrose cushion (SI Fig. 8C). To determine whether GM1 associated with virus particles, wild-type virus was subjected to sucrose gradient centrifugation. The fractions that contained peak levels of viral protein also contained peak GM1 levels by dot blot analysis, suggesting that GM1 was associated with viral particles (SI Fig. 8A). No GM1 was detected with virus produced from CHO HveC-1 cells as expected (SI Fig. 8B).
Fig. 2.
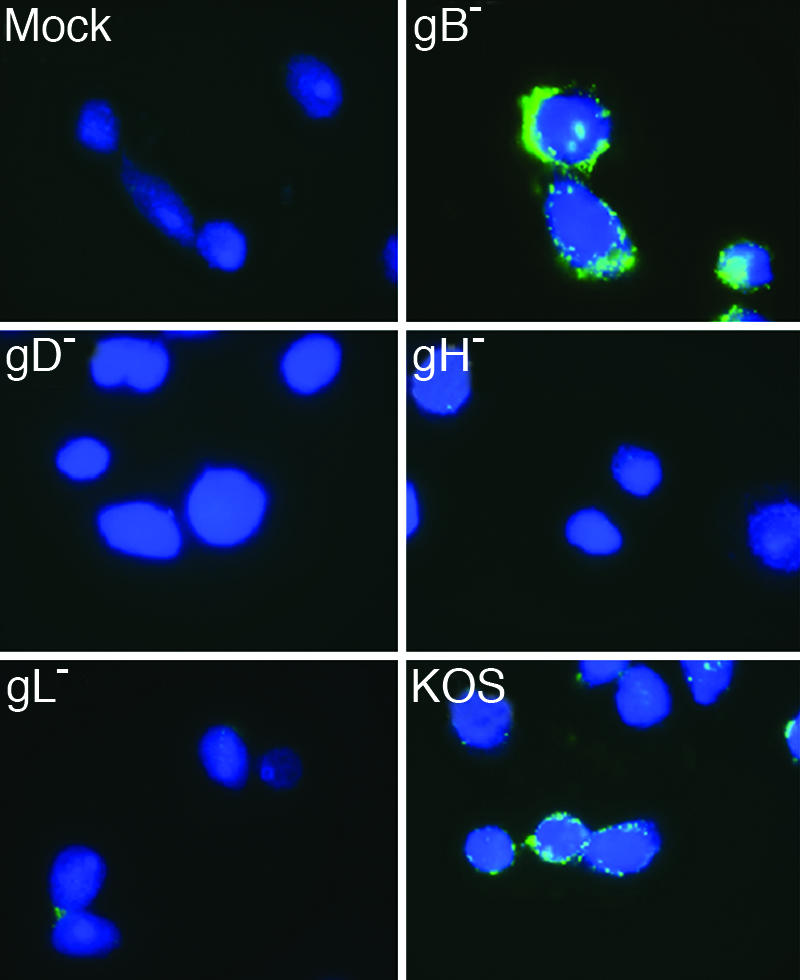
Glycoproteins required for GM1 transfer from virus to cell in virus fusion assay. Celltracker blue-labeled HveC-1 cells were inoculated with supernatants as depicted. The cells were treated with citrate buffer, fixed, and stained with FITC-CTX. Images were taken at ×100 magnification on a Zeiss Axiovert microscope. The experiment was performed three independent times, and images are from one representative experiment.
The staining pattern that we observed on the surface of HveC-1 cells was nonuniform. This pattern could represent GM1 that has not fully diffused into the cell membrane or that has coalesced into lipid microdomains because GM1 is commonly found in lipid microdomains (26). We observed lipid transfer using wild-type HSV-1(KOS) at multiplicity of infection from ≈0.01 to 1 pfu per cell with the greatest lipid transfer at the highest multiplicity of infection (data not shown). We were surprised to efficiently detect GM1 transfer at such a relatively low multiplicity of infection. However, the number of viral particles capable of transferring GM1 to the HveC-1 cells was probably much greater than the actual amount of infectious virus present in the supernatants. Particle:infectivity ratios for HSV-1 have been determined to range from 10 to 50 or greater (27, 28). Virus–cell GM1 transfer for HSV-1(KOS) and gB− viruses was most efficient at 37°C but also occurred at 23°C but not at 4°C (data not shown).
We quantified the GM1 transfer results by using flow cytometry (Fig. 3) and found that gB− viruses consistently transferred GM1 comparably to wild-type KOS, if not slightly more efficiently than KOS. We also found that gB− and KOS viruses were incapable of transferring GM1 in the absence of a gD receptor [KOS (CHO)] (Fig. 3). Increased signal was observed with gH− and gL− viruses that was not detected by using microscopy. Because the gD− signal was considerably lower than gH− and gL− viruses, this higher fluorescence may have been residual bound particles or spontaneous transfer of GM1 to the cells by virus bound to gD. We repeated the previous experiment using DiI-labeled wild-type and mutant viruses (SI Fig. 9). DiI is a lipophilic dye that intercalates into membranes and provides an alternative to GM1 for lipid transfer detection. We observed similar results to those obtained monitoring GM1 transfer such that DiI transfer to target HveC-1 cells was increased for the wild-type KOS and gB− viruses compared with the gH−, gL−, and gD− viruses. However, the relative DiI fluorescence for the gH− and gL− viruses was increased over that observed for GM1 transfer likely because of endocytosis of bound virus (SI Fig. 9B). Endocytosis and degradation of HSV-1 bound to CHO cells occur regardless of whether the cells express a gD receptor and are susceptible to virus entry (29, 30). Unlike the DiI experiments, the GM1 transfer assay should detect GM1 only on the cell surface, so GM1-containing virus internalized by cells should not be detected.
Fig. 3.

Relative GM1 transfer of HSV-1 and mutants viruses. HveC-1 cells were inoculated with culture medium (Blank) or supernatants from virus preparations. Cells were treated with citrate buffer, removed with Accutase, fixed, and stained with FITC-CTX to detect GM1 transfer. CHO-K1 cells were inoculated with KOS as a negative control [KOS (CHO)]. Results shown are the percentage of GM1+ relative to KOS GM1+ values. KOS GM1+ values ranged from 65% to 85% GM1+ cells of 30,000 total cells. Error bars represent the standard deviation of an average of two duplicate independent experiments.
We next examined the virus-containing supernatants to determine levels of virus proteins for the various mutant and wild-type isolates. Because we could not quantify mutant virus production using standard virus titer techniques, we performed a Western blot to compare levels of viral capsid protein, VP5. The results in Fig. 4 demonstrate that, although KOS was typically produced more efficiently, levels of the viral capsid protein in the mutant virus supernatants were similar (within 2-fold) for any given mutant and virus preparation. Importantly, the small variation in VP5 levels for the mutant viruses did not correlate with an increase in GM1 transfer. We examined levels of each transmembrane glycoprotein (gB, gD, and gH) to confirm that our mutant viruses lacked the appropriate glycoprotein, and deletion of one of the glycoproteins did not affect the surface expression of the other glycoproteins. All viruses expressed similar levels of each glycoprotein as has been shown for similar deletion mutants (31). Finally, we conducted quantitative PCR amplification of the glycoprotein G ORF from the virus-containing supernatants as a measure of viral particles. The results of the quantitative PCR closely paralleled the determination of VP5 levels in Fig. 4 (data not shown). Therefore, the transfer of GM1 to cells from gB− viral supernatants could not be due to an elevated amount of virus compared with gD−, gH−, and gL− viral supernatants.
Fig. 4.

Virus quantification. Equal volumes of concentrated virus supernatants were subjected to SDS/PAGE on an 8–16% gradient gel, transferred to nitrocellulose, and incubated with anti-VP5, anti-gD, anti-gH, or anti-gB. Blots were visualized as described in Materials and Methods. Quantitation was performed by using VP5 for each virus preparation made. Results are representative of those conducted on eight different sets of virus preparations. Anti-gB, anti-gD, and anti-gH blots are representative blots taken from one of three separate virus preparations.
Anti-gH Antibody Blocks Hemifusion.
If the lipid transfer we observed was mediated by gHL, then antibodies to gH that block virus entry may block lipid transfer. Importantly, antibodies to gH block virus entry but do not interfere with virus binding (32). Incubation of gB− and KOS virus-containing supernatants with a mAb to gH (52S) that blocks virus entry (32) abrogated GM1 transfer to HveC-1 cells (Fig. 5). Treatment of KOS virus-containing supernatants with an anti-gB mAb (H1817) that blocks virus entry (33) did not block GM1 transfer to HveC-1 cells. In parallel experiments, incubation of supernatants containing HSV-1(KOS) with 52S or H1817 eliminated virus entry, confirming that the concentration of mAbs used in these experiments blocked virus entry (SI Fig. 10). GM1 transfer was blocked from 25 to 100 μg/ml, with virtually complete entry blocking for both mAbs at 100 μg/ml. The level of 52S used was within the range previously reported to block virus entry (32). Taken together, these results indicate that gH function was critical to mediate hemifusion.
Fig. 5.

Ab blocking of lipid transfer from virus to cells. Antibody (100 μg) or no antibody was added to ≈1 × 107 pfu of concentrated gB− or KOS virus supernatant for 1 h, followed by addition of supernatants to Celltracker red-labeled HveC-1 cells as described previously. (A) KOS, no Ab. (B) KOS, 100 μg of 52S. (C) KOS, 100 μg of H1817. (D) gB−, no Ab. (E) gB−, 100 μg of 52S. (F) Mock, no Ab. Images were taken at ×63 magnification on a Leica SP1 AOBS confocal microscope. The experiment was performed three independent times, and images are from one representative experiment.
Discussion
Currently, there are two theories regarding the mechanism of HSV-1 fusion. One mechanism focuses on either gB or gH being the major fusogenic glycoprotein, leaving the other with a peripheral role as either a regulatory protein or a structural protein in a gDgBgHL fusion complex (the regulatory/structural model) (10, 15, 34–37). Here we report evidence for a sequential model for fusion, whereby each glycoprotein is required for a phase of the fusion process. It would appear that gD has Phase I function to bring membranes into proximity and activate gHL and gB for fusion (8, 10, 12). Receptor binding may cause a conformational change in gD that induces gHL to initiate lipid mixing to the hemifusion intermediate (Phase II). Phase III would then begin as gB stabilizes and expands the fusion pore to allow content mixing and complete the fusion process. Our model does not limit gD to having only Phase I function, gHL to Phase II, or gB to Phase III. The fusogenic activity of gHL and gB may overlap during the hemifusion intermediate whereby gHL may have some pore expansion activity, while gB may have some hemifusion activity. Also, gH and gB may have cellular receptors that aid in the close apposition steps. Finally, gD may trigger gB to complete Phase III because it triggers gHL to complete Phase II. Regardless, we have provided evidence that all three transmembrane envelope glycoproteins function in the fusion process and that each glycoprotein is sequentially required for a separate phase of the fusion process.
A sequential model for HSV-1 fusion requires gB to function during Phase III of fusion. There is evidence that gB plays an active role in Phase III. Mutations in the cytoplasmic tail of gB enhance cell–cell fusion of HSV-1 isolates (38–40) and also enhance cell–cell fusion of HSV glycoproteins in plasmid-based fusion assays (20, 41, 42) in a manner similar to that seen for mutations in the cytoplasmic tail of the fusion protein of HIV, gp120/gp41 (43). Furthermore, the recently solved crystal structure of HSV-1 gB lacking the transmembrane domain and cytoplasmic tail revealed a striking homology to the structure of vesicular stomatitis virus G fusion protein (15). Both gp120/gp41 and G proteins are active during Phase III of fusion. Finally, an EBV gB mutant is solely capable of mediating cell–cell fusion upon overexpression at the cell surface (44).
gH and gB are required for membrane fusion for herpesviruses such as EBV and human herpesvirus 8 (HHV-8) (45, 46), and fusogenic domains have been found in both glycoproteins (15, 34–37, 41, 44). We hypothesize that gH and gB will also function sequentially in EBV and HHV-8 membrane fusion. Therefore, strategies developed to inhibit hemifusion by HSV glycoproteins may be generally applicable to inhibit EBV and HHV-8 fusion.
A neutralizing mAb to gH, 52S, blocked lipid transfer between HSV-1 particles and the CHO-HveC-1 plasma membrane, indicating a direct role for gH in the formation of the hemifusion intermediate. HSV-1 gH is present on the surface of infected cells and virus particles as a heterodimer with gL (6), and gL is absolutely required for cell–cell and virus–cell fusion (24, 47). It is not clear at present whether gL has a role in hemifusion other than to ensure the appropriate folding and localization of gH. mAbs to HSV-1 gL can block cell–cell fusion but not virus entry, suggesting that gL may be directly involved in the fusion process (48). However, all gL mutants that mediate gH trafficking out of the endoplasmic reticulum also mediate membrane fusion, and no mutant has yet been isolated that mediates gH trafficking to the cell surface and fails to function in fusion (13, 49, 50). The latter results suggest that the sole role for gL in fusion is to chaperone gH to the cell or virion surface and argue against a direct role for gL in hemifusion. Class II fusion proteins bind a companion protein to ensure appropriate folding and trafficking, and the companion protein must be dissociated for the fusion protein to function (51–53). Whether gL functions similarly to the class II companion proteins or plays another role in membrane fusion remains to be determined.
HSV-1 is thought to enter CHO cells, as well as HeLa and keratinocytes, through a pH-dependent pathway after endocytosis of virus bound to the cell surface (29, 30). This mode of virus entry is in contrast to that observed in Vero and neuronal cell lines, where the virus is thought to fuse directly with the plasma membrane at the cell surface (29, 30, 54). The results presented here suggest that transfer of the lipid GM1 from the virus particle to CHO cells occurs at the cell surface. Intracellular GM1 would not be detected because the cells were fixed with paraformaldehyde (a nonpermeabilizing agent) before CTX-FITC addition. The simplest explanation for our results is that we detected HSV-1 that fused directly at the cell surface, suggesting that virus entry occurs by both direct fusion at the cell surface and receptor-mediated endocytosis in CHO cells. We are currently analyzing virus–cell lipid transfer using agents that block receptor-mediated endocytosis and virus entry in CHO cells to more carefully explore this possibility. Our experiments included incubation in citrate buffer (pH 3) to remove virus bound to cell surface that had not undergone fusion or hemifusion. Therefore, we cannot rule out the possibility that bound virus was briefly exposed to an intermediate pH that induced lipid mixing and fusion at the cell surface. A recent report, however, found that low pH treatment did not facilitate entry of HSV-1 bound to cells (55). Alternatively, virus–cell lipid mixing could occur at the surface of CHO cells, whereas complete fusion would occur after endocytosis and a possible reduction in pH. A similar mechanism has been proposed for avian sarcoma and leukosis virus, where hemifusion was detected at neutral pH and full fusion after a reduction in pH (56). However, those results were not independently verified when avian sarcoma and leukosis virus hemifusion and full fusion were found to depend on endocytosis and a reduction in pH (57).
Materials and Methods
Cells, Plasmids, and Antibodies.
CHO-K1 cells (American Type Culture Collection, Manassas, VA) and CHO HveC-1 cells (provided by P. Spear, Northwestern University) were grown in F12 media containing 7% serum and penicillin/streptomycin. Vero cells (provided by P. Spear) and Vero-BG20 cells were grown in DMEM containing 7% serum and penicillin/streptomycin. Vero-BG20 cells are Vero cells that stably overexpress human herpesvirus entry mediator via the expression plasmid pBG20. Cells were selected by using G418, and a pool of cells was sorted via flow cytometry until they homogeneously expressed herpesvirus entry mediator with a mean fluorescence intensity ≈5-fold greater than wild-type Vero cells (data not shown). HveC-1 and Vero-BG20 cells were maintained in 500 μg/ml G418. Antibodies against HSV-1 proteins were mouse mAbs anti-gH, 52S (58), anti-gB H1817 (Virusys, Sykesville, MD) (33), and anti-VP5 (Virusys), and rabbit antiserum against gH, R137 (49), and gD, R7 (59). The HSV glycoprotein plasmids used in this study were as published (47), and pAcGFP, a GFP expression plasmid, was purchased from BD Clontech (Mountain View, CA). Virus isolates used were HSV-1(KOS)tk-12 (60), HSV-1(KOS)gBKO82 (61), HSV-1(KOS)gD6 (60), HSV-1(KOS)gH87, and HSV-1(KOS)gL86 (62) (provided by P. Spear).
Transfections.
In each well of a six-well plate, 60–80% confluent CHO-K1 cells were incubated with 1.5 μg of plasmid DNA and 5 μl of LipofectAMINE (Gibco/BRL, Grand Island, NY) in OPTIMEM (Gibco/BRL), according to the manufacturer's instructions. The cells were incubated for 6–8 h, and medium was replaced with F12 medium containing 20% FBS and penicillin/streptomycin.
Cell Hemifusion Assay.
CHO cells were transfected with plasmids expressing the required envelope glycoproteins and GFP. Two days after transfection 2 × 106 Vero cells were added to a well of a six-well plate. These cells were coincubated at 37°C for 18 h and then fixed with 4% paraformaldehyde. Cells were washed with PBS, and 2 μg/ml TRITC-conjugated CTX (CTX-555; Molecular Probes, Eugene, OR) was added for 30 min at 37°C. Cells were washed with PBS containing 2% heat-inactivated serum (FACS buffer) and mounted with Mowiol (Calbiochem, San Diego, CA). Pictures were taken on a Zeiss Axiovert microscope. Quantitation of cell hemifusion assay was performed by blinding transfections, randomly capturing five fields of a coverslip, visualizing the red and green channels together, and counting GFP- and double-positive cells. Typically, 50 GFP-positive events were found per field. Cells were also stained with DAPI to determine the number of nuclei in GM1+/GFP+ events.
Mutant Virus Generation.
A confluent 10-cm2 dish of Vero cells was inoculated with 2 × 107 pfu/ml complemented gBK082 (gB−), gD6 (gD−), gH87 (gH−), gL86 (gL−), or KOStk-12 isolates (provided by P. Spear) or were mock-infected with PBS Ca2+ Mg2+ for 2 h at 37°C. Cells were then treated with 0.1 M sodium citrate buffer (pH 3.0) for 5 min and washed with DMEM containing 2% heat-inactivated serum (DMEV). Upon incubation for 48 h in DMEV, supernatant was removed and centrifuged to pellet cell debris. Supernatant was concentrated to remove small-molecular-mass debris by using a Centri-plus spin concentrator with a 100-kDa cutoff (Millipore, Billerica, MA).
Western Blotting.
Virus supernatant was lysed by boiling in loading buffer (2% SDS/175 mM 2-mercaptoethanol) and loaded onto a 8–16% gradient gel (Bio-Rad, Hercules, CA). The gel was transferred onto nitrocellulose and blotted by using anti-VP5 antibody and an IRDye 800 secondary (Rockland Immunochemicals, Gilbertsville, PA). gD and gH blots were first probed with R7 or R137, respectively, followed by a secondary anti-rabbit Alexa Fluor 647-nm antibody (Molecular Probes). Blots were imaged by using Odyssey Infrared Imaging System (LI-COR, Lincoln, NE) and quantified by using Scion Image densitometry analysis. gB blot was probed with H1817, followed by secondary anti-mouse peroxidase-conjugated antibody. Blot was visualized by using enhanced chemiluminescence.
Virus Fusion Assay.
Concentrated virus-containing supernatant generated from Vero cell infections was added to a well of a six-well plate of HveC-1 cells labeled with either 5 μM Celltracker red CMTPX or 35 μM Celltracker blue CMAC (Molecular Probes). Cells were incubated with virus for 2 h at 37°C at an approximate multiplicity of infection of 1. Cells were then treated with 0.1 M sodium citrate buffer (pH 3.0) or 10–1,000 μg/ml PK and fixed with 4% paraformaldehyde. Paraformaldehyde was washed out with FACS buffer, and 2 μg/ml FITC-CTX (Sigma, St. Louis, MO) was added. Cells were mounted onto slides by using Mowiol and imaged by using a Leica SP1 AOBS confocal microscope. For flow cytometry, cells were removed with Accutase (Sigma) before fixation, washed with PBS, fixed as before, and washed with FACS buffer.
SI.
Colocalization of virus/GM1, DiI virus fusion, quantitative PCR, mAb blocking, and X-Gal assays are described in SI Methods.
Supplementary Material
Acknowledgments
We thank Sara Bair, Kankana Chava, and Allison Henley for maintenance of cell lines and Rebecca Dutch, Anthony Sinai, and Yuri Klyachkin for helpful advice and discussion of the manuscript. This investigation was funded by National Institutes of Health Grant AI51476.
Abbreviations
- HSV-1
herpes simplex virus type 1
- gB
glycoprotein B
- gD
glycoprotein D
- gH
glycoprotein H
- gL
glycoprotein L
- gHL
gH and gL complex
- CTX
cholera toxin β-subunit.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0608374104/DC1.
References
- 1.Chernomordik LV, Kozlov MM. Cell. 2005;123:375–382. doi: 10.1016/j.cell.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 2.Jardetzky TS, Lamb RA. Nature. 2004;427:307–308. doi: 10.1038/427307a. [DOI] [PubMed] [Google Scholar]
- 3.Blumenthal R, Clague MJ, Durell SR, Epand RM. Chem Rev. 2003;103:53–69. doi: 10.1021/cr000036+. [DOI] [PubMed] [Google Scholar]
- 4.Cohen FS, Melikyan GB. Methods. 1998;16:215–226. doi: 10.1006/meth.1998.0670. [DOI] [PubMed] [Google Scholar]
- 5.Spear PG. Cell Microbiol. 2004;6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 6.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. J Virol. 1992;66:2240–2250. doi: 10.1128/jvi.66.4.2240-2250.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subramanian RP, Dunn JE, Geraghty RJ. Virology. 2005;339:176–191. doi: 10.1016/j.virol.2005.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G. Proc Natl Acad Sci USA. 2004;101:7445–7450. doi: 10.1073/pnas.0401883101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fusco D, Forghieri C, Campadelli-Fiume G. Proc Natl Acad Sci USA. 2005;102:9323–9328. doi: 10.1073/pnas.0503907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zago A, Jogger CR, Spear PG. Proc Natl Acad Sci USA. 2004;101:17498–17503. doi: 10.1073/pnas.0408186101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones NA, Geraghty RJ. Virology. 2004;324:213–228. doi: 10.1016/j.virol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 13.Klyachkin YM, Stoops KD, Geraghty RJ. J Gen Virol. 2006;87:759–767. doi: 10.1099/vir.0.81563-0. [DOI] [PubMed] [Google Scholar]
- 14.Gianni T, Fato R, Bergamini C, Lenaz G, Campadelli-Fiume G. J Virol. 2006;80:8190–8198. doi: 10.1128/JVI.00504-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 16.Giraudo CG, Hu C, You D, Slovic AM, Mosharov EV, Sulzer D, Melia TJ, Rothman JE. J Cell Biol. 2005;170:249–260. doi: 10.1083/jcb.200501093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heyningen SV. Science. 1974;183:656–657. [Google Scholar]
- 18.Majoul I, Schmidt T, Pomasanova M, Boutkevich E, Kozlov Y, Soling HD. J Cell Sci. 2002;115:817–826. doi: 10.1242/jcs.115.4.817. [DOI] [PubMed] [Google Scholar]
- 19.Cairns TM, Milne RS, Ponce-de-Leon M, Tobin DK, Cohen GH, Eisenberg RJ. J Virol. 2003;77:6731–6742. doi: 10.1128/JVI.77.12.6731-6742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muggeridge MI. J Gen Virol. 2000;81:2017–2027. doi: 10.1099/0022-1317-81-8-2017. [DOI] [PubMed] [Google Scholar]
- 21.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 22.Cai W, Person S, Warner SC, Zhou J, DeLuca NA. J Virol. 1987;61:714–721. doi: 10.1128/jvi.61.3.714-721.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forrester A, Farrell H, Wilkinson G, Kaye J, Davis-Poynter N, Minson T. J Virol. 1992;66:341–348. doi: 10.1128/jvi.66.1.341-348.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roop C, Hutchinson L, Johnson DC. J Virol. 1993;67:2285–2297. doi: 10.1128/jvi.67.4.2285-2297.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ligas MW, Johnson DC. J Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Badizadegan K, Wolf AA, Rodighiero C, Jobling M, Hirst TR, Holmes RK, Lencer WI. Int J Med Microbiol. 2000;290:403–408. doi: 10.1016/S1438-4221(00)80052-1. [DOI] [PubMed] [Google Scholar]
- 27.Cai W, Schaffer PA. J Virol. 1992;66:2904–2915. doi: 10.1128/jvi.66.5.2904-2915.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everett RD, Boutell C, Orr A. J Virol. 2004;78:1763–1774. doi: 10.1128/JVI.78.4.1763-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicola AV, McEvoy AM, Straus SE. J Virol. 2003;77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicola AV, Straus SE. J Virol. 2004;78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodger G, Boname J, Bell S, Minson T. J Virol. 2001;75:710–716. doi: 10.1128/JVI.75.2.710-716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuller AO, Santos RE, Spear PG. J Virol. 1989;63:3435–3443. doi: 10.1128/jvi.63.8.3435-3443.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapsal JM, Pereira L. Virology. 1988;164:427–434. doi: 10.1016/0042-6822(88)90556-9. [DOI] [PubMed] [Google Scholar]
- 34.Galdiero S, Falanga A, Vitiello M, Browne H, Pedone C, Galdiero M. J Biol Chem. 2005;280:28632–28643. doi: 10.1074/jbc.M505196200. [DOI] [PubMed] [Google Scholar]
- 35.Galdiero S, Vitiello M, D'Isanto M, Falanga A, Collins C, Raieta K, Pedone C, Browne H, Galdiero M. J Gen Virol. 2006;87:1085–1097. doi: 10.1099/vir.0.81794-0. [DOI] [PubMed] [Google Scholar]
- 36.Gianni T, Martelli PL, Casadio R, Campadelli-Fiume G. J Virol. 2005;79:2931–2940. doi: 10.1128/JVI.79.5.2931-2940.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gianni T, Menotti L, Campadelli-Fiume G. J Virol. 2005;79:7042–7049. doi: 10.1128/JVI.79.11.7042-7049.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bzik DJ, Fox BA, DeLuca NA, Person S. Virology. 1984;137:185–190. doi: 10.1016/0042-6822(84)90022-9. [DOI] [PubMed] [Google Scholar]
- 39.Cai WH, Gu B, Person S. J Virol. 1988;62:2596–2604. doi: 10.1128/jvi.62.8.2596-2604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gage PJ, Levine M, Glorioso JC. J Virol. 1993;67:2191–2201. doi: 10.1128/jvi.67.4.2191-2201.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan Z, Grantham ML, Smith MS, Anderson ES, Cardelli JA, Muggeridge MI. J Virol. 2002;76:9271–9283. doi: 10.1128/JVI.76.18.9271-9283.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foster TP, Melancon JM, Kousoulas KG. Virology. 2001;287:18–29. doi: 10.1006/viro.2001.1004. [DOI] [PubMed] [Google Scholar]
- 43.Wyss S, Dimitrov AS, Baribaud F, Edwards TG, Blumenthal R, Hoxie JA. J Virol. 2005;79:12231–12241. doi: 10.1128/JVI.79.19.12231-12241.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McShane MP, Longnecker R. Proc Natl Acad Sci USA. 2004;101:17474–17479. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haan KM, Lee SK, Longnecker R. Virology. 2001;290:106–114. doi: 10.1006/viro.2001.1141. [DOI] [PubMed] [Google Scholar]
- 46.Pertel PE. J Virol. 2002;76:4390–4400. doi: 10.1128/JVI.76.9.4390-4400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pertel PE, Fridberg A, Parish ML, Spear PG. Virology. 2001;279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 48.Novotny MJ, Parish ML, Spear PG. Virology. 1996;221:1–13. doi: 10.1006/viro.1996.0347. [DOI] [PubMed] [Google Scholar]
- 49.Peng T, Ponce de Leon M, Novotny MJ, Jiang H, Lambris JD, Dubin G, Spear PG, Cohen GH, Eisenberg RJ. J Virol. 1998;72:6092–6103. doi: 10.1128/jvi.72.7.6092-6103.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cairns TM, Landsburg DJ, Whitbeck JC, Eisenberg RJ, Cohen GH. Virology. 2005;332:550–562. doi: 10.1016/j.virol.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 51.Stadler K, Allison SL, Schalich J, Heinz FX. J Virol. 1997;71:8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wengler G, Wengler G. J Virol. 1989;63:2521–2526. doi: 10.1128/jvi.63.6.2521-2526.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Fugere M, Day R, Kielian M. J Virol. 2003;77:2981–2989. doi: 10.1128/JVI.77.5.2981-2989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wittels M, Spear PG. Virus Res. 1991;18:271–290. doi: 10.1016/0168-1702(91)90024-p. [DOI] [PubMed] [Google Scholar]
- 55.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. J Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Earp LJ, Delos SE, Netter RC, Bates P, White JM. J Virol. 2003;77:3058–3066. doi: 10.1128/JVI.77.5.3058-3066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melikyan GB, Barnard RJ, Markosyan RM, Young JA, Cohen FS. J Virol. 2004;78:3753–3762. doi: 10.1128/JVI.78.7.3753-3762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Showalter SD, Zweig M, Hampar B. Infect Immun. 1981;34:684–692. doi: 10.1128/iai.34.3.684-692.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Isola VJ, Eisenberg RJ, Siebert GR, Heilman CJ, Wilcox WC, Cohen GH. J Virol. 1989;63:2325–2334. doi: 10.1128/jvi.63.5.2325-2334.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. Virology. 1998;246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 61.Cai WZ, Person S, Warner SC, Zhou JH, DeLuca NA. J Virol. 1987;61:714–721. doi: 10.1128/jvi.61.3.714-721.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montgomery RI, Warner MS, Lum BJ, Spear PG. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}