

Abstract
Apelin, a ligand for apelin-angiotension receptor-like 1 (APJ), has recently been shown to be a potent positive inotropic agent in normal hearts. In humans, levels of apelin have been shown to rise in early-stage heart failure and to fall in late-stage heart failure. In this study, we tested the hypothesis that apelin augments contraction directly in failing rat cardiac muscle. Right ventricular heart failure secondary to pulmonary hypertension was induced by exposing the rats to hypoxia (10% O2 inhaled air) for 14–16 weeks. Trabeculae were dissected and mounted between a force transducer and a motor arm, superfused with Krebs-Henseleit (K-H) solution (pH 7.4, 22 °C), and loaded with fura-2. Both force development and [Ca2+]i transient amplitude increased in a dose-dependent manner in the presence of Apelin-12 (10~70 nM, [Ca2+]o=0.5 mM) in failing muscles as compared to control (36±7% vs 7.4±5% at 70 nM, P<0.05). Also, [Ca2+]i transients increased up to 18.4±9.5% as compared to control (4.5±1.9%, P<0.05). The increases in contraction in the presence of apelin were also maintained over a range of external Ca2+ (0.5–2.0 mM). Steady-state force-[Ca2+]i relation of the failing muscles reveals decreased maximal Ca2+-activated force (Fmax) (51.45±5.3 vs 98.5±11.5 mN/mm2, P<0.001), with no changes in Ca2+ required for 50% of maximal activation (Ca50) (0.45±0.07 vs 0.30±0.04 μM, P>0.05) and Hill coefficient (4.60±0.73 vs 3.17±0.92, P>0.05). Apelin (70 nM) had no effect on the steady-state force-[Ca2+]i relation in failing muscles (Fmax: 63.03±3.5 mN/mm2; Ca50: 0.50±0.08 μM; Hill coefficient: 4.73±0.89;). These results indicate that apelin exerts a selective positive inotropic action in failing myocardium. The increased force development is the result of increased [Ca2+]i transients rather than changes in myofilament calcium responsiveness.
Keywords: Apelin, contractility, heart failure, e-c coupling, Ca2+ responsiveness
1. Introduction
Apelin (Tatemoto et al., 1998), a newly discovered endogenous peptide ligand for the apelin-angiotension receptor-like 1 (APJ), has been the focus of several recent studies, including studies in human heart failure. APJ is a Gi coupled transmembrane receptor (O'Dowd et al., 1993) and shares significant homology with angiotension II receptor type-1 (AT-1). Unlike the angiotension II-AT-1 system, the role of the apelin-APJ system has not been well characterized. Recently, the apelin-APJ system has been postulated to play an important role in cardiovascular homeostasis (Kleinz and Davenport, 2005). In patients, left ventricular apelin mRNA was increased several-fold in end-stage heart failure secondary to either coronary artery disease or idiopathic dilated cardiomyopathy (Foldes et al., 2003). Plasma apelin levels were found to be increased in patients with early stages of heart failure and decreased in late stages of heart failure (Chen et al., 2003). The fact that apelin exerts the most potent positive inotropic action (among all identified inotropic agent) in normal hearts (Szokodi et al., 2002) suggests a role for reduced apelin levels in the pathogenesis of heart failure. Indeed, in rat failing hearts, administration of apelin augmented pressure development and cardiac output (Berry et al., 2004).
The mechanism of the positive inotropic effect of apelin is not known. In normal hearts, the apelin-induced inotropic effect was attenuated by inhibition of phospholipase C, protein kinase C, Na+-H+ exchanger, and Na+-Ca2+ exchanger (Szokodi et al., 2002). These data suggest that apelin causes activation of phospholipase C-dependent signal transduction pathway, which ultimately affects Ca2+ availability and/or Ca2+ responsiveness of the myofilaments. In this study, we investigated the effect of apelin on contraction in both normal and failing cardiac muscles with a view to defining the mechanism of apelin-induced augmentation of contraction by measuring intracellular Ca2+ and assessing myofilament Ca2+ responsiveness in these intact muscles. We found that apelin increased force development disproportionately in failing muscles. The increases in force development were due to increased Ca2+ availability, rather than changes in myofilament Ca2+ responsiveness. A preliminary report of this study has been presented (Dai et al., 2005).
2. Methods and Materials
2.1. Drugs
Apelin-12 peptide was purchased from Phoenix Pharmaceuticals, INC. (USA) and was rehydrated to a stock solution in distilled water before use. Ryanodine (Tocris Bioscience, UK) was dissolved in dimethysulfoxide (DMSO) to a stock of 1.0 mM and stored in 0.5 ml volumes at −20 °C. Fura-2 potassium salt (1.0 mM, Molecular Probe, USA) was dissolved in distill water and stored in −20 °C before use. All other chemicals were obtained from Sigma Co. (USA).
2.2. Animals
Rats (LBN-F1 rats, 200–250g, n=30) were used in these experiments. The care of the animals and the experiment protocol were approved by the Animal Care and Use Committee of The Johns Hopkins University.
2.3. In vivo chronic hypoxia
A rat hypoxia protocol (Ping et al., 1999; Xue and Johns, 1996) was use to induce right heart failure. Briefly, rats were exposed to either a normobaric Plexiglas chamber maintained at 10% inspired O2 (hypoxia group) or a similar chamber open to air (21% inspired O2, normoxic group) for about 14–16 weeks. Both groups of animals were kept in the same room and maintained at 20–24 °C with a 12h/12h light:dark cycle. Hypoxia was maintained using a Pro-ox Model 350 unit (Reming Bioinstruments, Refield, NY) which controlled fraction of O2 in inspired gas by solenoid controlled infusion of N2 balanced against an inward leak of air through holes in the chamber. The hypoxia rats were exposed to room air for 10–15min daily while their cages were changed. CO2, water vapor, and ammonia were removed by pumping the atmosphere of the hypoxia chamber through Bara Lyme, Drierite (anhydrous calcium sulfate), and activated carbon. In previous studies (Jung et al., 2001; Ping et al., 1999), significant pulmonary hypertension and right ventricular hypertrophy were found after ~3 weeks of hypoxia. However, in our preliminary studies, depression of contraction was not seen until ~14 weeks after hypoxia.
2.4. Trabecular muscles
The heart was exposed via mid-sternotomy after the animal was anesthetized. 0.3–0.5 ml of heparin (300–500 units) was injected into the left atrium and the heart was then rapidly excised and placed into a dissection dish. The aorta was cannulated and the heart was perfused retrogradely (~15 ml/min) with dissecting Krebs-Henseleit (H-K) solution equilibrated with 95% O2 and 5% CO2. The dissecting K-H solution is composed of (in mM): NaCl 120, NaHCO3 20, KCl 5, MgCl 1.2, glucose 10, CaCl2 0.5, and 2,3-butanedione monoximine (BDM) 20, pH 7.35–7.45 at room temperature (21–22 °C). Trabecula from the right ventricle of the heart was dissected and mounted between a force transducer and a motor arm. Generally, one muscle from one failing right ventricle was dissected. The trabecula was then superfused with normal K-H solution (KCl, 5.0 mM) at a rate of ~10 ml/min and stimulated at 0.5 Hz. Dimensions of the muscles were measured with a calibration reticule in the ocular of the dissection microscope (×40, resolution ~10 μm).
2.5. Force and sarcomere length measurements
Force was measured using a force transducer system from SenseNor (Germany) and was expressed in millinewtons per square millimeter of cross-sectional area. Sarcomere length was measured by laser diffraction (Gao et al., 1996). Briefly, light diffracted by the central region of the muscle was detected by a reticon diode linear array system (RC0100-RG512, EG&G Reticon). The light intensity of the first order of diffraction was integrated, and sarcomere length was determined from the median of the light intensity distribution using a custom-made sarcomere length detection system (University of Calgary, Canada). Resting sarcomere length was set at 2.20–2.30 μm throughout the experiments.
2.6. Measurement of [Ca2+]i
[Ca2+]i was measured using the free acid form of fura 2 as described in our previous studies (Backx et al., 1995; Gao et al., 1994; Gao, 1998). Fura 2 potassium salt was microinjected iontophoretically into one cell and allowed to spread throughout the whole muscle (via gap junctions). The tip of the electrode (~0.2 μm in diameter) was filled with fura 2 salt (1.0 mM) and the remainder of the electrode was filled with 150 mM KCl. After a successful impalement into a superficial cell in the unstimulated muscle, a hyperpolarizing current of 5–10 nA was passed continuously for ~15 min. In some muscles, multiple injections (up to 3–4) were applied at different sites, with duration of the injection limited to <10 min at each site to achieve a good signal-to-noise ratio. As previously established, the loading did not affect force development. The epifluorescence of fura 2 was measured by exciting at 380 and 340 nm. The fluorescent light was collected at 510 nm by a photomultiplier tube (R1527, Hamamatsu). The output of photomultiplier is collected and digitized. [Ca2+]i was given by the following equation (after subtraction of the autofluorescence):
| (1) |
where R is the observed ratio of fluorescence (340/380), K’d is the apparent dissociation constant, Rmax is the ratio of 340 nm/380 nm at saturating [Ca2+], and Rmin is the ratio of 340 nm/380 nm at zero [Ca2+]. The values of K’d, Rmax, and Rmin were determined by in vivo calibrations as described previously (Gao et al., 1994; Gao, 1998).
2.7. Steady-state activation of trabeculae
Ryanodine (1.0 μM) was used to enable steady-state activation. After 15 min of exposure to ryanodine, different levels of tetanizations were induced briefly (~4–8 seconds) by stimulating the muscles at 10 Hz at varied [Ca2+]o (0.5–20 mM). All experiments were performed at room temperature (20–22 °C).
2.8. Statistics
Student’s t-test and one-way analysis of variance (ANOVA) was used for statistical analysis of the data. A value of P<0.05 was considered to indicate significant differences between groups. Unless otherwise indicated, pooled data were expressed as mean ± standard error of the mean (S.E.M.).
3. Results
3.1 Force and [Ca2+]i transients in failing muscles
Right heart failure of the rat was induced by exposure to chronic hypoxia (inspired O2 10%). After 3–4 months, most rats developed signs of right heart failure including failure to gain body weight, decreased activity, labored breathing, and clubbed claws. Table 1 shows some of the indices of right heart failure as a result of chronic hypoxia obtained after thoracotomy and animal sacrifices. Muscles from these failing right ventricles were then dissected. Figure 1 shows both the representative raw records of force development and [Ca2+]i transient from non-failing and failing muscles as well as the pooled data. Failing muscles had lower amplitudes of [Ca2+]i transients and generated less force at all external Ca2+ tested. Another cardinal feature of the failing muscle is slowed dynamics of twitch force and [Ca2+]i transients. Both the time to peak of force development and [Ca2+]i transients were significantly delayed. The relaxation times for contraction and [Ca2+]i transients were also prolonged (Figure 2). Thus, our failing muscles share the same characteristics as muscles from the failing hearts of spontaneous hypertension and heart failure (SHHF) rat and from the hearts of experimental ischemic heart failure (De Tombe et al., 1996; Perez, 1999).
Table 1.
Effect of chronic hypoxia on mean pulmonary pressure (Pm), right ventricle to left ventricle ratio (RV/LV), and liver to body weight ration (liver/body) in rats
| Pm | RV/LV | Liver/body | |
|---|---|---|---|
| Control | 11 ± 0.5 | 0.232 ± 0.005 | 0.026 ± 0.001 |
| Hypoxia | 23 ± 1* | 0.433 ± 0.01* | 0.035 ± 0.002* |
Data are mean ±S.E.M. n = 15/group, temp 22 °C.
, P<0.05 vs. control.
Figure 1.
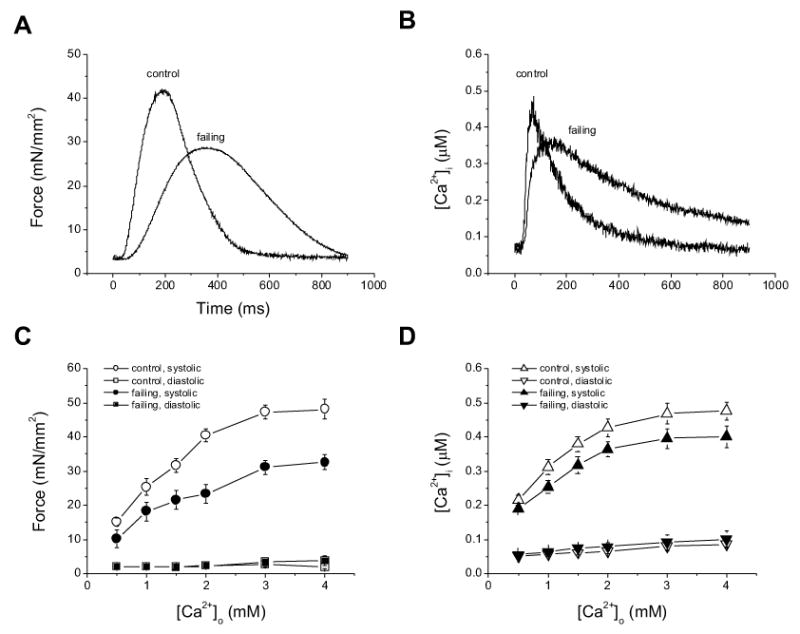
Representative tracings of twitch force (A) and [Ca2+]i transients (B) from control and failing trabeculae. The muscles were superfused with K-H solution and stimulated at 0.5 Hz. [Ca2+]=2.0 mM, temp 22 °C. Note that the failing muscle generated lower amplitudes of twitch force and Ca2+ transient. Pooled data of twitch force (C) and amplitudes of [Ca2+]i transients (D) of control and failing muscles at varied external [Ca2+]s. Force development and Ca2+ transients remain depressed at all [Ca2+]os tested (P<0.014). n = 9 in each group.
Figure 2.
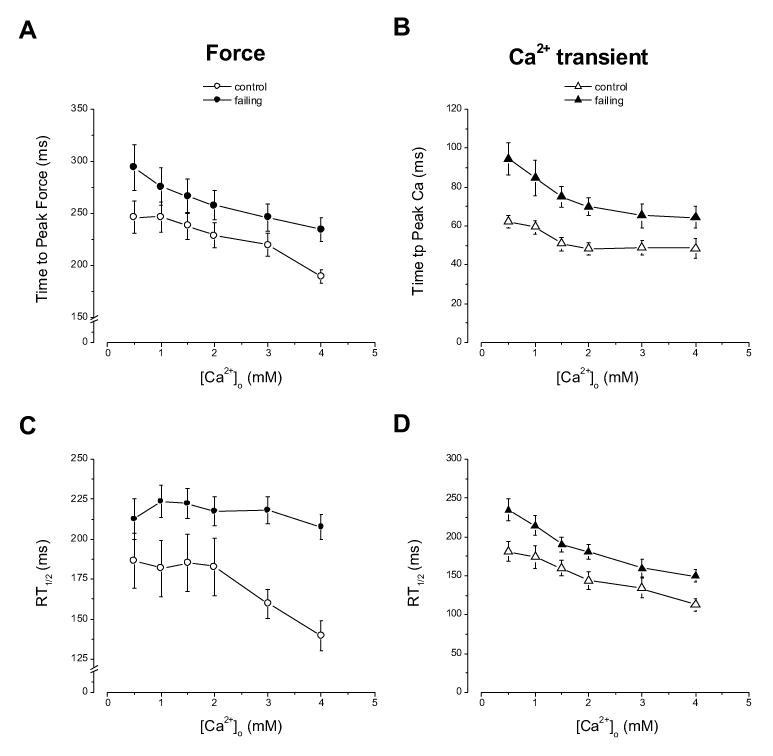
Time to peak force (A) and time to peak Ca2+ transient (B) of control and failing muscles at varied [Ca2+]os. The times to peak for both twitch force and [Ca2+]i transient are significantly prolonged (p<0.014). The relaxation times from peak to 50% of peak levels of force (C) and Ca2+ transients (D) are also significantly prolonged in each group (P<0.014). n = 9.
3.2. Effect of apelin on force and [Ca2+]i transients in failing cardiac muscle
Apelin was shown to be a very potent inotropic agent in isolated rat heart with a EC50 of ~33 pM (Szokodi et al., 2002). In the failing hearts, apelin also increased contractility (Ashley et al., 2005; Berry et al., 2004). To investigate the mechanism of apelin’s action, we measured twitch force and [Ca2+]i transients directly in both normal and failing trabecular muscles (Figure 3). First, apelin was added to the perfusate in cumulative doses and after about 15 min, measurements of force and [Ca2+]i were performed. In normal muscles, apelin increased force development marginally from 12.5±1.4 to 13.2±1.3 mN/mm2 (an average of 1.00±0.32 mN/mm2 increase, n=6). When normalized to the force before apelin treatment, the increase in force was 7.4±5% over baseline at the highest dose tested (70 nM) (P>0.05, paired t-test). The amplitude of [Ca2+]i transients was also increased minimally (4.5±1.9%). In failing muscles, on the other hand, force development started to increase at lower doses and reached a maximum at 50–70 nM of apelin (from 6.35±0.64 to 8.72±0.71 mN/mm2, with an averaged increase of 2.32±0.48 mN/mm2, n=6), corresponding to 36±7% over baseline (P<0.05 paired t-test). The increases in force were also significantly higher in failing muscles after apelin when compared to control muscles (P<0.05 vs control). [Ca2+]i transients were also increased significantly (18.4±9.5% increase vs 4.5±1.9%, P<0.05). Thus, apelin disproportionately augments contraction in failing muscles.
Figure 3.
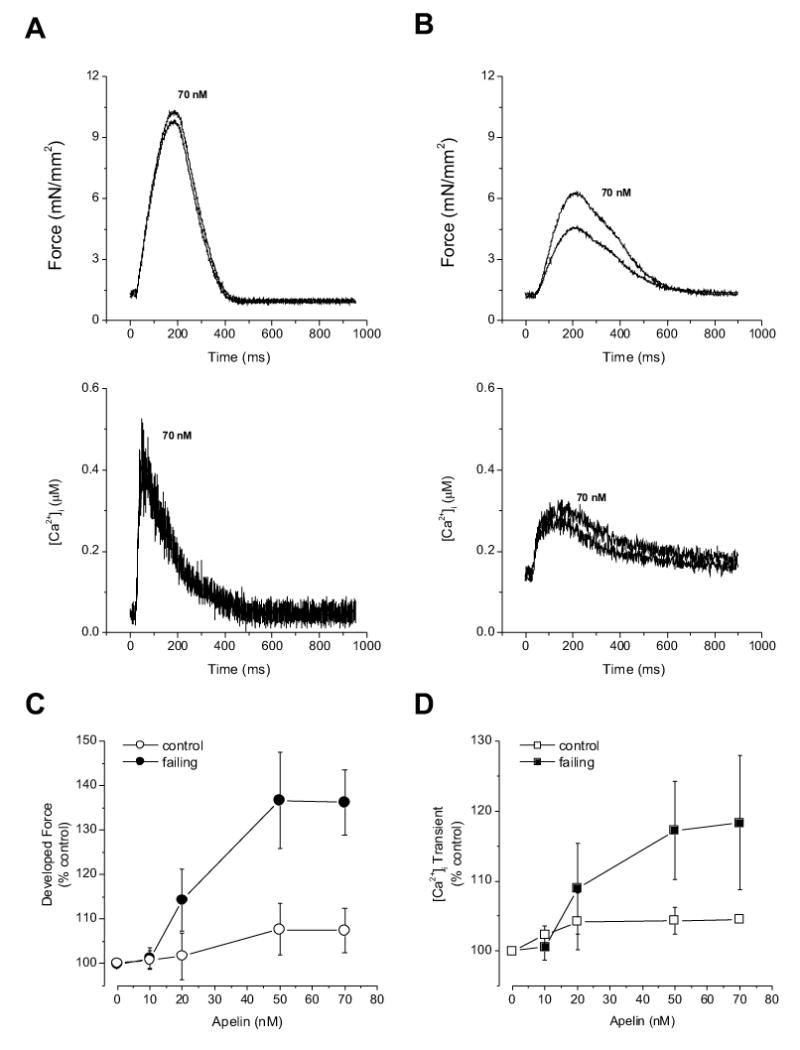
Representative tracings of twitch force and [Ca2+]i transient in a control (A) and failing (B) trabeculae before and after apelin exposure. Apelin had minimal effect on contraction and [Ca2+]i transient in the control muscle. Apelin increased amplitudes of force development and [Ca2+]i transient without affecting the time course of force and [Ca2+]i transient. Pooled data of increases of developed forces (C) and [Ca2+]i transients (D) after varied doses of apelin in control and failing muscles. The failing muscles were more responsive to apelin than control muscles. [Ca2+] = 0.5 mM, temperature: 22 °C, n=6 in each group.
Increased force development can be either a result of increased availability of activator Ca2+ or increased sensitivity of the myofilaments to Ca2+, or both. To assess the two possibilities, we plotted the peak force developments and peak [Ca2+]i transients in failing muscles before and after apelin (70 nM), measured at varied [Ca2+]os. The data were overlapped and there were no differences in force at any given [Ca2+]i. To simplify the analysis, we assumed a linear relationship between peak force developments and peak [Ca2+]i transients, the slopes of the linear relationships before and after apelin treatment are not different (Figure 4). These data support the notion that increases in force development after apelin is caused by increased availability of activator Ca2+.
Figure 4.
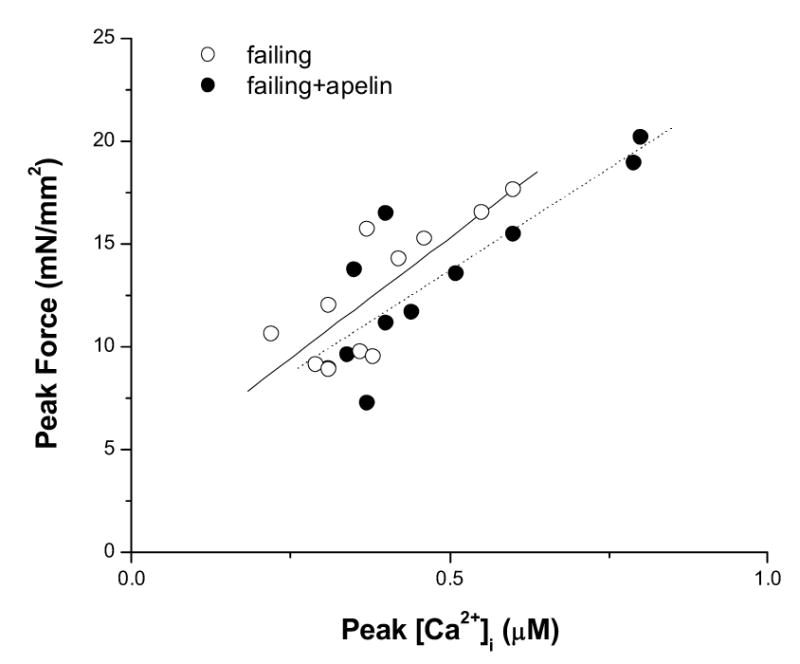
Pooled data of peak force versus peak [Ca2+]i transient of failing muscles before and after apelin treatment (70 nM). Varied levels of twitch forces were achieved by exposing the muscles to different [Ca2+]os. A linear relationship between force and [Ca2+]i was assumed and fit by least-squares minimization. n = 4 in each group.
3.3. Effects of apelin on maximal Ca2+-activated force and Ca2+ sensitivity of failing cardiac muscle
The data above suggest that the responsiveness of the myofilaments to Ca2+ is not increased by apelin in failing myocardium. To assess changes in myofilament Ca2+ responsiveness directly, we tetanized the trabeculae to achieve steady-state activation of the myofilaments and to determine force-[Ca2+]i relations over a broad range of [Ca2+]os in these intact muscles. Figure 5 shows the pooled data for all the force-[Ca2+]i relations in normal muscles and in failing muscles before and after apelin. Maximal Ca2+ -activated force is depressed in failing muscles (51.45±5.3 vs 98.5±11.5 mN/mm2, control, P<0.001), and there are no changes in Hill coefficient (4.60±0.73 vs 3.17±0.92, control, P>0.05) and Ca2+ required for 50% activation (0.45±0.07 vs 0.30±0.04 μM, control, P>0.05). Apelin had no effect on the force-[Ca2+]i relationship in failing muscles (Fmax: 63.03±3.5 mN/mm2; Ca50: 0.50±0.08 μM; Hill coefficient: 4.73±0.89;). Steady-state force-[Ca2+]i relation in control muscles was also unaffected by apelin (result not shown).
Figure 5.
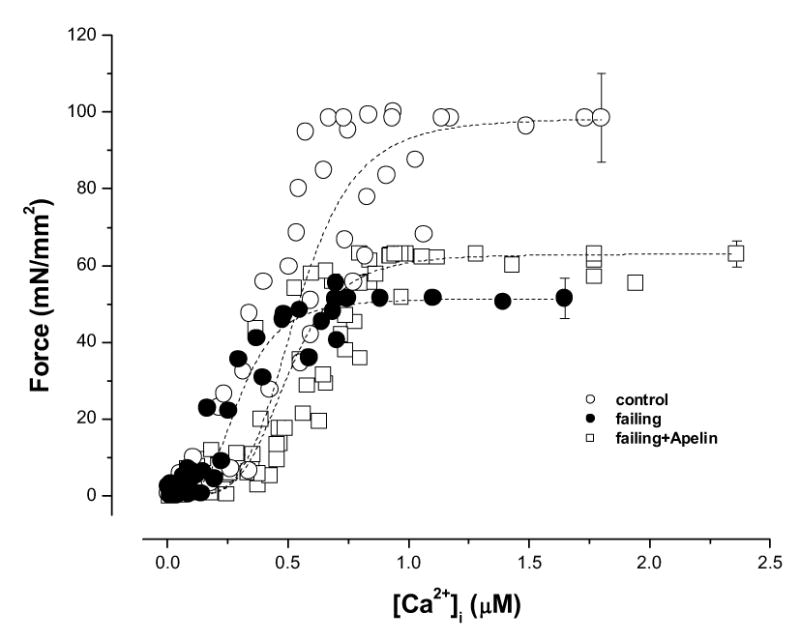
Steady-state force-[Ca2+]i relations of control, failing, and failing muscles treated with apelin (70 nM). The failing muscles exhibited depressed maximal Ca2+-activated force (P<0.001 vs. control) with no changes in Ca2+ required for 50% activation and Hill coefficient as compared with control muscles. Apelin did not change the force-[Ca2+]i relation in failing muscles. See text for details. n = 8 control, n=5 failing, and n=6 failing + apelin.
4. Discussion
This study begins to elucidate the mechanism underlying the positive inotropic action of apelin with direct measurement of intracellular Ca2+ and assessment of myofilament responsiveness to Ca2+ in intact cardiac muscles. We have found that apelin exerted positive inotropic action in cardiac muscles and disproportionately augmented force development in failing muscles. This study also shows direct evidence that increased availability of activator Ca2+, not myofilament Ca2+ sensitization, underlies the augmented contraction in intact cardiac muscle.
In this study, we obtained failing trabecula from failed right heart as a result of chronic hypoxia. The behavior of these muscles was consistent with those from the failing hearts of SHHF rats and isolated myofibrils from hearts subjected to pulmonary banding (Fan et al., 1997; Perez, 1999). Our findings are consistent with a number of studies. Apelin exerts strong inotropic action in isolated heart with an EC50 of 33 pM (Szokodi et al., 2002)]. In patients with heart failure, the plasma levels of apelin are increased in the early stages of heart failure but decreased in later stages of heart failure (Chen et al., 2003), suggesting a temporary compensatory role of apelin-AJP system, which failed as a result of progressing heart failure. Studies in animal models of heart failure have shown that administration of apelin increased cardiac output (Berry et al., 2004). In addition, the increase of contractility in failing heart after apelin infusion was more rapid and greater. Similarly, the failing muscles in our study were also more sensitive to apelin with greater increases in force development.
Increases in contraction in cardiac muscle can be a result of increased availability of activator Ca2+, and/or increased Ca2+ responsiveness of the myofilaments, or both. In this study, we have shown that the increases in force development were accompanied by increases in amplitudes of [Ca2+]i transients in failing muscles after exposure to apelin, suggesting that increases in activator Ca2+ underlie force augmentation by apelin. Consistent with this, apelin did not change the characteristics of steady-state force-[Ca2+]i relations of the muscles (Figure 5). However, the possibility of myofilament sensitization by apelin (either directly or indirectly) still exits, given the diversity of signal transduction pathways following APJ activation. For example, protein kinase C activation would suppress Ca2+ responsiveness of the myofilaments, especially in the failing myocardium thus masking apelin’s effect on myofilaments. Experiments are needed to examine the direct effect of apelin on myofilaments and to selectively block the different pathways to unveil any myofilament sensitization by apelin.
One other finding in this study is that failing myocardium responded disproportionately to apelin. Apparently, more Ca2+ is released in failing muscles after apelin exposure. Mechanism of increased Ca2+ availability by apelin in failing myocardium is not known at present. Apelin did not affect the activities of voltage-activated Ca2+ and K+ channels, thus ruling out involvement of these sarcolemmal channels. Inhibitions of phospholipase C and protein kinase C attenuated the inotropic response to apelin (Szokodi et al., 2002). Inhibition of Na+-H+ exchanger and Na+-Ca2+ exchanger also suppressed the effect of apelin. Interestingly, none of these inhibitors (either alone or in combination) resulted in complete suppression of the inotropic response to apelin. This suggests additional pathway(s) may also be involved in enhancing Ca2+ availability. In vascular tissue, apelin activates cell membrane G-protein-coupled receptor, APJ, which then activates phospholipase C-β. Activation of phospholipase C-β results in generation of inositol(1,4,5)trisphosphate and sn-1,2-diacylglycerol. Inositol(1,4,5)trisphosphate is rapidly released into the cytoplasm, activates inositol(1,4,5)trisphosphate receptors in the sarcoplamic reticulum and causes Ca2+ release from the sarcoplamic reticulum, which leads to vasoconstriction (Kleinz and Davenport, 2005). In normal cardiac cells, however, inositol(1,4,5)trisphosphate induced Ca2+ release seems rather slow (Kentish et al., 1990). Nevertheless, it is known that Ca2+ release from the sarcolpasmic reticulum is diminished as a result of hyperphosphorylation of the ryanodine receptors (Marks et al., 2002). Under this circumstance, the Ca2+ release mediated by inositol(1,4,5)trisphosphate could potentially be amplified. Indeed, it has been reported that levels of mRNA encoding inositol(1,4,5)trisphosphate receptors were elevated in patients with dilated cardiomyopathy (Go et al., 1995). Another mechanism for the increased intracellular Ca2+ is via reversed Na+/Ca2+ exchanger, which appears to be partially responsible for the inotropic action of apelin in normal rat heart (Szokodi et al., 2002). In heart failure, the role of this exchanger is still controversial. In hearts with systolic dysfunction with normal functioning Na+/Ca2+ exchanger, stimulation of Na+/Ca2+ in the reversed mode can in theory increase intracellular Ca2+ and increase contraction. In dilated failing myocardium, however, Na+/Ca2+ exchanger is already defective in the forward mode resulting in abnormal Ca2+ extrusion during diastole (Hasenfuss and Pieske, 2002), any stimulation of the reversed mode would exaggerate increases in diastolic Ca2+, which is clearly undesirable. In this study, we did not observe significant increases in diastolic Ca2+ levels after apelin exposure (results not shown). The mechanism of increases in intracellular Ca2+ availability by apelin in failing myocardium will be the focus of our future investigations.
In normal isolated trabeculae, the responses to apelin were small (only ~7.4% increases over baseline, Figure 3). This is different from other studies in which significant positive inotropic effects were observed in normal hearts (Berry et al., 2004; Szokodi et al., 2002). Nevertheless, our results do not necessarily contrast these studies. Our data did show a trend towards increases in contraction at higher apelin doses. Different experimental preparations and conditions may likely contribute to the different observations. For example, when delivered via coronary endovascular system, apelin may have been more effective in interacting with APJ receptors, present in both endothelial cell and myocytes with preference in the endothelial vasculature.
In conclusion, apelin increased contraction disproportionately in failing myocardium. The positive inotropic action of apelin is caused by increased availability of Ca2+, not by increased Ca2+ responsiveness of the myofilaments. Further studies are needed to investigate the mechanisms of increased intracellular Ca2+ by apelin in failing myocardium.
Acknowledgments
This study was supported by NIH HL-45065 (WDG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, Deng A, Eichhorn J, Mahajan R, Agrawal R, Greve J, Robbins R, Patterson AJ, Bernstein D, Quertermous T. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65:73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backx PH, Gao WD, Azan-Backx MD, Marban E. The relationship between contractile force and intracellular [Ca2+] in intact rat cardiac trabeculae. J Gen Physiol. 1995;105:1–19. doi: 10.1085/jgp.105.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MF, Pirolli TJ, Jayasankar V, Burdick J, Morine KJ, Gardner TJ, Woo YJ. Apelin has in vivo inotropic effects on normal and failing hearts. Circulation. 2004;110:II187–193. doi: 10.1161/01.CIR.0000138382.57325.5c. [DOI] [PubMed] [Google Scholar]
- Chen MM, Ashley EA, Deng DX, Tsalenko A, Deng A, Tabibiazar R, Ben-Dor R, Fenster b, Yang B, King JY, Fowler M, Robbins R, Johnson FL, Bruhn L, McDonagh T, Dargie H, Yakhini Z, Tsao PS, Quertermous T. Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction. Circulation. 2003;108:1432–1439. doi: 10.1161/01.CIR.0000091235.94914.75. [DOI] [PubMed] [Google Scholar]
- Dai T, Ramirez-Correa G, Gao WD. Apelin disproportionally increases contraction in failing cardiac muscle. Circulation. 2005;112:II–159. [Google Scholar]
- De Tombe PP, Wannenburg T, Fan D, Little WC. Right ventricular contractile protein function in rats with left ventricular myocardial infarction. Am J Physiol. 1996;271:H73–H79. doi: 10.1152/ajpheart.1996.271.1.H73. [DOI] [PubMed] [Google Scholar]
- Fan D, Wannenburg T, de Tombe PP. Decreased myocyte tension development and calcium responsiveness in rat right ventricular pressure overload. Circulation. 1997;95:2312–2317. doi: 10.1161/01.cir.95.9.2312. [DOI] [PubMed] [Google Scholar]
- Foldes G, Horkay F, Szokodi L, Vuolteenaho O, Ilves M, Lindstedt KA, Mayranpaa M, Sarman B, Seres L, Skoumal R, Lako-Futo Z, deChatel R, Ruskoaho H, Toth M. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure, Biochem. Biophys Res Commun. 2003;308:480–485. doi: 10.1016/s0006-291x(03)01424-4. [DOI] [PubMed] [Google Scholar]
- Gao WD, Backx PH, Azan-Backx M, Marban E. Myofilament Ca2+ sensitivity in intact versus skinned rat ventricular muscle. Circ Res. 1994;74:408–415. doi: 10.1161/01.res.74.3.408. [DOI] [PubMed] [Google Scholar]
- Gao WD, Liu Y, Mellgren R, Marban E. Intrinsic myofilament alterations underlying the decreased contractility of stunned myocardium. A consequence of Ca2+- dependent proteolysis? Circ Res. 1996;78:455–465. doi: 10.1161/01.res.78.3.455. [DOI] [PubMed] [Google Scholar]
- Gao WG, Perez NG, Marban E. Calcium cycling and contractile activation in intact mouse cardiac muscle. J Physiol. 1998;507:175–184. doi: 10.1111/j.1469-7793.1998.175bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. 1995;95:888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- Jung F, Weiland U, Johns RA, Ihling C, Dimmeler S. Chronic hypoxia induces apoptosis in cardiac myocytes: a possible role for Bcl-2-like proteins. Biochem Biophys Res Commun. 2001;286:419–425. doi: 10.1006/bbrc.2001.5406. [DOI] [PubMed] [Google Scholar]
- Kentish JC, Barsotti RJ, Lea TJ, Mulligan IP, Patel JR, Ferenczi MA. Calcium release from cardiac sarcoplasmic reticulum induced by photorelease of calcium or Ins(1,4,5)P3. Am J Physiol. 1990;258:H610–H615. doi: 10.1152/ajpheart.1990.258.2.H610. [DOI] [PubMed] [Google Scholar]
- Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Ther. 2005;107:198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Marks AR, Reiken S, Marx SO. Progression of heart failure: is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation. 2002;105:272–275. [PubMed] [Google Scholar]
- O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen t. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–360. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- Perez NG, Hashimoto K, McCune S, Altschuld RA, Marban E. Origin of contractile dysfunction in heart failure. Calcium cycling versus myofilaments. Circulation. 1999;99:1077–1083. doi: 10.1161/01.cir.99.8.1077. [DOI] [PubMed] [Google Scholar]
- Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafonova Z, Bolli R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, Pikkarainen S, Piuhola J, Rysa J, Toth M, Ruskoaho H. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91:434–440. doi: 10.1161/01.res.0000033522.37861.69. [DOI] [PubMed] [Google Scholar]
- Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- Xue C, Johns RA. Upregulation of nitric oxide synthase correlates temporally with onset of pulmonary vascular remodeling in the hypoxic rat. Hypertension. 1996;28:743–753. doi: 10.1161/01.hyp.28.5.743. [DOI] [PubMed] [Google Scholar]