

Abstract
Chronic stress induces both functional and structural adaptations within the hypothalamo-pituitary-adrenocortical (HPA) axis, suggestive of long-term alterations in neuroendocrine reactivity to subsequent stressors. We hypothesized that prior chronic stress would produce persistent enhancement of HPA axis reactivity to novel stressors. Adult male rats were exposed to chronic variable stress (CVS) for 1 wk and allowed to recover. Plasma ACTH and corticosterone levels were measured in control or CVS rats exposed to novel psychogenic (novel environment or restraint) or systemic (hypoxia) stressors at 16 h, 4 d, 7 d, or 30 d after CVS cessation. Plasma ACTH and corticosterone responses to psychogenic stressors were attenuated at 4 d (novel environment and restraint) and 7 d (novel environment only) recovery from CVS, whereas hormonal responses to the systemic stressor were largely unaffected by CVS. CRH mRNA expression was up-regulated in the paraventricular nucleus of the hypothalamus (PVN) at 16 h after cessation of CVS, but no other alterations in PVN CRH or arginine vasopressin mRNA expression were observed. Thus, in contrast to our hypothesis, reductions of HPA axis sensitivity to psychogenic stressors manifested at delayed recovery time points after CVS. The capacity of the HPA axis to respond to a systemic stressor appeared largely intact during recovery from CVS. These data suggest that chronic stress selectively targets brain circuits responsible for integration of psychogenic stimuli, resulting in decreased HPA axis responsiveness, possibly mediated in part by transitory alterations in PVN CRH expression.
THE HYPOTHALAMO-pituitary-adrenocortical (HPA) axis is a vital regulator of homeostasis in vertebrates. Real or perceived dangers are interpreted by the brain, and appropriate physiological reactions ensue to restore homeostasis. Threats to homeostasis can be categorized based on stimulus attributes and the neurocircuitry regulating the HPA axis response to these threats. Psychogenic stressors (e.g. conditioned fear, response to novelty) are stimuli that do not constitute a direct physical threat to homeostasis and thus require active cognitive processing to achieve biological significance (1, 2). Systemic stressors (e.g. respiratory distress, hemorrhage), in contrast, constitute a direct, internally perceived disruption of homeostasis (1, 2). A wealth of data suggest that HPA axis responses to these stressor subtypes are regulated by distinct neurocircuits, with psychogenic stressors primarily targeting limbic structures (including the hippocampus and prefrontal cortex) and systemic stressors triggering interoceptive cues that are conveyed directly to the hypothalamus via efferents from the brainstem (1, 2).
Marked alterations in neuroendocrine responsiveness are evident after disruption of the HPA axis by chronic stress. Repeated experience with the same, or homotypic, stressor produces habituation, or diminution of HPA axis responses (3-8). Repeated homotypic stress followed by a novel, or heterotypic stressor, induces facilitation. In this case, HPA axis responses are the same or greater than the initial response (4, 9-13), despite elevated circulating glucocorticoids from the chronic stress exposure. Given that stress-induced habituation and facilitation cannot be fully explained by differences in glucocorticoid feedback efficiency (14, 15), it is likely that plastic, perhaps permanent, changes occur in the HPA axis after stress (16). Indeed, numerous studies indicate that limited exposure to severe stressors produces lasting alterations in HPA axis responsiveness and regulation. Prior exposure to footshock potentiates the plasma ACTH response to noise stress 14 d later (17), and CRH-induced plasma ACTH levels are augmented up to 1 wk after social defeat stress (18). Inescapable tail shock facilitates the plasma corticosterone response to an inflammatory stressor (19) or later tail shock stress (20) for at least 10 d after tail shock treatment. Further evidence for dysregulation of the HPA axis after prior stress exposure comes from studies indicating dexamethasone resistance to processive and systemic stress challenge in tail shock stressed rats up to 4 d after tail shock exposure (21). With the exception of the preceding study (21), there is a dearth of information on how stressor modality modulates the expression of persistent neuroendocrine alterations after chronic stress treatment.
In the present experiments, we therefore used the chronic variable stress (CVS) model of chronic stress in rats to: 1) investigate the impact of prior stress history on determining long-term neuroendocrine responsiveness to subsequent stressors and 2) evaluate the influence of stress challenge modality (e.g. psychogenic vs. systemic). Our data indicate that CVS initially induces a transient facilitation to novel psychogenic and systemic stressors that is accompanied by an increase in CRH mRNA in the paraventricular nucleus of the hypothalamus (PVN). This facilitation is then followed by decrements in HPA axis responses to psychogenic, but not systemic, stressors at 4–7 d after CVS cessation that is paralleled by a return of CRH mRNA expression to normal levels. These results suggest that chronic stress selectively targets psychogenic stress circuitry, producing transitory adaptations that can desensitize the HPA axis.
Materials and Methods
Experiment 1
We sought to determine whether stressor modality influences HPA axis responses to novel stressors after recovery from chronic stress. Rats underwent CVS for 1 wk and were then exposed to a novel psychogenic or systemic stressor at several time points after CVS cessation.
Subjects
Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN; 175–199 g) were housed three per cage and acclimated to the animal colony for 1 wk before initiation of experimental procedures. Rats were maintained in a temperature- and humidity-controlled room (lights on 0600–1800 h) with food and water available ad libitum. All experimental procedures were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Animals and approved by the University of Cincinnati Institutional Animal Care and Use Committee.
CVS protocol
Subjects were randomly assigned to unhandled control (n = 60) or chronic stress (n = 72) groups. Our prior work has demonstrated that handling does not affect subsequent ACTH and corticosterone responses to an acute novel stressor (22); thus, we decided not to handle rats in the control groups. The CVS paradigm consisted of twice daily exposure to alternating stressors along with occasional overnight stressors for seven consecutive days. Morning stressors were administered between 0830 and 1030 h, whereas afternoon stressors were conducted between 1430 and 1630 h. Overnight stressors began immediately after cessation of afternoon stressors and ended with initiation of the next day's morning stressor. Stressors consisted of: 1) 1 h in plastic restraint tubes, 2) 1 h in plastic restraint tubes in the cold (4 C), 3) 1 h shaker stress (100 rpm), 4) 20-min warm swim (31 C), 5) 5-min cold swim (18 C), 6) overnight social isolation (one rat/cage), and 7) overnight social crowding (six rats/cage) in a randomized order with each stressor (except the overnight stressors) represented an equivalent number of times.
Acute novel stressor
Control and CVS groups were exposed to a psychogenic (novel environment) or systemic (hypoxia) stressor at 16 h, 4 d, 7 d, or 30 d (Fig. 1). Each time point represents independent control (n = 6 for each time point) and CVS groups (n = 6 for each time point); thus, rats were not repeatedly tested during recovery. For exposure to the novel environment, rats were removed from the colony room and placed on an elevated plus-maze apparatus (4 in. wide × 40 in. long, 0.125-in. lip on open arms, 14-in. height on closed arms) for 5 min under dimly lit conditions. The elevated plus-maze was used as a novel environment to evoke plasma ACTH and corticosterone secretion, not for assessment of anxiety-like behavior. The experiment was designed so that the number of rats (n) used was appropriate for statistical analysis of the hormonal measures; this n was not adequate for determination of CVS effects on behavior, so these data are not included. The hypoxia stressor consisted of placement into a clear Plexiglas chamber with 8% O2 and 92% N2 for 20 min. The large number of animals within this experiment precluded conducting the novel environment and hypoxia exposures at the same time, so the novel environment challenge was done on an initial cohort of animals, with the hypoxia challenge done on another cohort of animals a few months later. The CVS paradigm was identical for the novel environment and hypoxia groups.
Fig. 1.
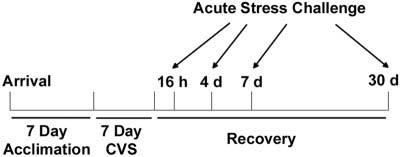
Timeline of experimental procedure. Rats arrived and acclimated to the animal colony for 1 wk before the onset of the CVS protocol. Rats were exposed to CVS for 1 wk or remained undisturbed in their home cages as unhandled controls. Independent groups of control or chronically stressed rats were given an acute stress challenge (novel environment, restraint, or hypoxia exposure) after 16 h, 4 d, 7 d, or 30 d recovery from chronic stress.
Tail blood was collected by tail vein nick at 20 and 40 min after the end of novel environment exposure or placement into the hypoxia chamber. Trunk blood was collected after decapitation at 60 min after the end of novel environment exposure or onset of hypoxia. Brains were removed, frozen in isopentane cooled on dry ice to −45 C, and stored at −80 C. Brains were cryosectioned (14 μm) on a Microm cryostat, thaw-mounted on Gold Seal Ultrastick slides (Portsmouth, NH), and stored at −20 C until in situ hybridization analysis of CRH and arginine vasopressin (AVP) mRNA expression were conducted. Separate groups of control (16 h recovery, n = 12) and CVS (16 h recovery, n = 12; 7 d recovery, n = 12) rats were not exposed to the novel stressors and thus represent basal HPA axis activation at these time points.
Physiological measures
The efficacy of the CVS paradigm was determined by examining alterations in several physiological measures. Body weight (BW) was recorded immediately before CVS initiation and at each recovery time point (16 h, 4 d, 7 d, and 30 d). Adrenal and thymus glands were removed and weighed. Physiological measures from the novel environment and hypoxia exposure groups were not different from each other, so these data were pooled and presented together in Table 1. In addition, the physiological measures from the unstressed groups at 16 h and 7 d were not different from acutely stressed groups, so data from these groups were pooled and presented together in Table 1.
TABLE 1.
Effect of CVS on physiological measures in experiment 1
| BW change (%) |
Thymus weight (mg) |
Thymus weight (mg/g BW) × 100 |
Adrenal weight (mg) |
Adrenal weight (mg/g BW) × 100 |
|
|---|---|---|---|---|---|
| 16 h | |||||
| Control (n = 24) | 14.3 ± 0.6 | 521 ± 20 | 184 ± 6 | 43.5 ± 1.0 | 15.5 ± 0.3 |
| CVS (n = 24) | 4.8 ± 0.6a | 460 ± 17 | 179 ± 7 | 44.8 ± 0.9 | 17.4 ± 0.3a |
| 4d | |||||
| Control (n = 12) | 18.5 ± 0.8 | 537 ± 34 | 189 ± 12 | 41.2 ± 0.9 | 14.1 ± 0.3 |
| CVS (n = 12) | 11.4 ± 1.3a | 493 ± 34 | 177 ± 10 | 46.6 ± 1.4a | 16.8 ± 0.3a |
| 7d | |||||
| Control (n = 12) | 22.0 ± 1.0 | 498 ± 22 | 163 ± 5 | 42.6 ± 1.6 | 14.0 ± 0.4 |
| CVS (n = 24) | 17.3 ± 0.8a | 489 ± 17 | 167 ± 4 | 46.5 ± 0.6a | 15.9 ± 0.2a |
| 30 d | |||||
| Control (n = 12) | 50.3 ± 3.7 | 430 ± 22 | 117 ± 5 | 46.8 ± 2.0 | 12.7 ± 0.4 |
| CVS (n = 12) | 49.0 ± 1.7 | 444 ± 21 | 122 ± 5 | 47.9 ± 1.5 | 13.3 ± 0.5 |
Rats were exposed to CVS for 7 d or were unhandled controls. At each recovery time point, six rats per group received novel environment exposure (5 min), whereas a different set of six rats per group were exposed to hypoxia (20 min). The total number of rats in each group is listed. Unstressed rats from the 16 h control (n = 12), 16 h CVS (n = 12), and 7 d CVS (n = 12) groups are included in this table. Time points reflect length of time after cessation of stress, and values represent the mean ± SEM of each group.
CVS is significantly different from corresponding control group (P < 0.05).
Experiment 2
This experiment was designed to ascertain whether the severity of the psychogenic stress challenge influences neuroendocrine responsiveness during recovery from CVS. Thus, rats underwent CVS and were exposed to restraint, instead of a novel environment, at the same recovery time points (see Fig. 1).
Methods for experiment 2 are identical with those of experiment 1, with the following exceptions. Weight-matched rats were assigned to control (n = 32) or CVS (n = 32) groups. Hypoxia (8% O2 for 30 min) and cold room exposure (1 h) replaced restraint and cold restraint as stressors so that restraint (30 min) could be used as a novel psychogenic stress challenge. At the time of stress challenge (16 h, 4 d, 7 d, or 30 d), rats were placed in plastic restraint tubes, and blood samples were immediately taken by tail clip (0 min). Tail blood samples were taken again 30 min after restraint initiation (30 min), and the rats were then released from the restraint tubes into their home cages. Additional tail blood samples were taken at 60 and 120 min after initial placement in the restraint tubes. This restraint stress sampling protocol was used because we have determined that it results in peak plasma ACTH and corticosterone levels comparable with those observed after the hypoxia stress protocol used in experiment 1 (23).
Blood collection
Tail blood was collected into tubes containing EDTA and trunk blood into Vacutainer tubes (BD Biosciences, Franklin Lakes, NJ) containing K3EDTA. Blood samples were immediately placed on ice. Plasma was obtained by centrifugation (1500 × g, 15 min, 4 C) and stored at −20 C for subsequent analysis of plasma ACTH and corticosterone.
RIAs
Plasma corticosterone levels were measured using 125I RIA kits (MP Biomedicals, Inc., Orangeburg, NY). Plasma ACTH concentrations were determined by an RIA that used a specific antiserum generously donated by Dr. William Engeland (University of Minnesota, Minneapolis, MN) at a dilution of 1:120,000 (novel environment and restraint challenge samples) or 1:210,000 (hypoxia challenge samples), with [125I] ACTH (Amersham Biosciences, Piscataway, NJ) as labeled tracer (24). Plasma samples from the novel environment and hypoxia groups were run in separate RIAs due to the extremely large number of samples. Plasma ACTH could not be assayed at the 16 h or 30 d recovery time points in experiment 2 due to technical difficulties.
CRH and AVP in situ hybridizations
A one-in-10 series of brain sections from the novel environment exposed rats was fixed in 4% phosphate-buffered paraformaldehyde for 10 min and rinsed twice in 5 mm potassium PBS (KPBS) for 5 min, twice in KPBS containing 0.2% glycine for 5 min, and twice in KPBS for 5 min. Sections were then acetylated in 0.25% acetic anhydride [suspended in 0.1 m triethanolamine (pH 8)] for 10 min, rinsed twice in 2× standard saline citrate (SCC) for 5 min, and dehydrated through graded alcohols.
Antisense cRNA probes complementary to CRH (765 bp) (25) and AVP (161 bp) (26) were generated by in vitro transcription using 35S-UTP. The CRH fragment was cloned into a pGem3 vector, linearized with HindIII, and transcribed with T7 RNA polymerase (Fisher Scientific Co., Pittsburgh, PA). The AVP fragment was cloned into a pCR 4-TOPO vector, linearized with NotI, and transcribed with T3 RNA polymerase. Each transcription reaction (15 μl) consisted of 1× transcription buffer, 62.5 μCi 35S-UTP, 330 μm ATP, 330 μm GTP, 330 μm CTP, 10 μm cold UTP, 66.6 mm dithiothreitol, 40 U ribonuclease inhibitor, 20 U T7 RNA polymerase, and 2.5 μg linearized DNA. The transcription reaction was incubated at 37 C for 60 min, and the labeled probe was separated from free nucleotide by ammonium acetate precipitation.
35S-probe was diluted in hybridization buffer [50% formamide, 20 mm Tris-HCl (pH 7.5), 1 mm EDTA, 335 mm NaCl, 1× Denhardt's solution, 200 μg/ml herring sperm DNA, 100 μg/ml yeast tRNA, 20 mm dithiothreitol, and 10% dextran sulfate] to yield 1,000,000 cpm/50 μl buffer. A 50-μl aliquot of diluted probe was applied to each slide. Slides were then coverslipped and incubated overnight at 55 C in humidified chambers containing 50% formamide. The next morning, coverslips were removed in 2× SCC, and slides were incubated in 100 μg/ml ribonuclease A for 30 min at 37 C. Slides were rinsed in 2× SCC, incubated in 0.2× SCC (65 C) for 1 h, dehydrated through graded alcohols, and exposed to Kodak Biomax MR-2 film (Eastman Kodak, Rochester, NY) for 7 d or 6 h for CRH and AVP probes, respectively. For each probe, all tissue sections were processed in a single assay to minimize inter-experimental error. Slides hybridized with sense probe were used as a negative control. In addition, all autoradiographs included ARC 146-14C standard slides (American Radiolabeled Chemicals, Inc., St. Louis, MO) as internal controls. Subsequently, slides hybridized with the AVP probe were coated with Kodak NTB2 liquid emulsion and exposed for 4 d at 4 C in the dark. Emulsion-dipped slides were developed with Kodak D-19 for 4 min, rinsed in distilled water for 1 min, fixed in Kodak fixer for 5 min, and then rinsed in running distilled water. Slides were counterstained with cresyl violet, dehydrated in graded alcohols, placed in xylene, and coverslipped with DPX Mountant (Fluka, Milwaukee, WI).
Image analysis
Semiquantitative analyses of in situ hybridization autoradiographs were conducted using Scion Image 1.62 software (Scion, Frederick, MD). Anatomical regions of interest were determined based on the Paxinos and Watson rat brain atlas (27). Exposure to the acute novel stressor at 16 h and 7 d did not alter CRH and AVP mRNA expression from the levels observed in unstressed rats, so these data were pooled together for analyses.
CRH mRNA expression was assessed in the PVN. Background signal was determined over a nonhybridized area and subtracted from total gray level to obtain corrected gray level units. The mean value of two to four sections through a given region (four to eight individual measurements) was calculated for each rat and used in the statistical analysis. All in situ hybridization analyses were performed by an observer unaware of group assignments.
AVP mRNA expression in parvocellular PVN was determined by areal densitometry as previously described (26). Medial parvocellular AVP mRNA-containing neurons were differentiated from magnocellular AVP mRNA neurons based upon their overall smaller size, relatively low level of AVP expression, and localization within the CRH neuron-containing region of the PVN. Emulsion-dipped sections for each animal were matched to the midlevel of the medial parvocellular PVN. Mean values for each animal were determined from one to two sections per animal and then used in subsequent analysis of group effects. Gray level measurements were taken over the medial parvocellular PVN, with care taken to avoid magnocellular neurons. Background was subtracted from the gray level measurements, and this value was multiplied by the area sampled, yielding integrated gray levels units. AVP mRNA expression in magnocellular neurons was not quantifiable because the exposure time had been optimized for analysis of AVP mRNA expression in parvocellular neurons.
Statistical analyses
Data are expressed as mean ± sem. Organ weights are reported as actual and adjusted weights (milligrams organ weight/grams total BW × 100). Body and organ weight data were analyzed by two-way factorial ANOVA with stress (control, CVS) and recovery (16 h, 4 d, 7 d, and 30 d) as main factors. The effect of CVS on unstressed morning hormone levels in experiment 1 was determined by one-way factorial ANOVA. Stress hormone levels were analyzed by separate two-way ANOVAs for each recovery day with stress as a main factor and time as the repeated measure (experiment 1: 20, 40, and 60 min; and experiment 2: 0, 30, 60, and 120 min). Alterations in basal HPA axis function in experiment 2 were determined by analyzing differences in plasma ACTH and corticosterone at the 0-min time point in the 16 h recovery group using a priori planned Student's t tests. Area under the curve was calculated to yield a measure of integrated plasma hormone secretion and then analyzed by individual Student's t tests at each recovery time point. CRH and AVP mRNA expression was analyzed by two-way ANOVA with stress and recovery day as main factors. Significant main effects or interactions from the ANOVAs were further analyzed using Fisher's least significant difference test. All data were analyzed by GB-Stat software (Dynamic Microsystems Inc., Silver Spring, MD). Statistical significance was set as P < 0.05. Outliers were detected using the Dixon-Massey method (28), and data were reanalyzed after outlier removal.
Results
Experiment 1
Chronic stress effects on physiological indices
BW gain decreased during CVS exposure [stress, F(1,123) = 37.36; P < 0.0001] but increased during the recovery period [recovery, F(3,123) = 373.09; P < 0.0001] (Table 1). A stress-recovery interaction was observed [F(3,123) = 3.51, P = 0.017]. Post hoc analyses indicated that CVS groups displayed lower BW gain at the 16 h, 4 d, and 7 d recovery time points (P < 0.01), but this difference had resolved by 30 d recovery.
CVS exposure increased both adrenal weight [stress, F(1,123) = 11.45; P = 0.001] and adjusted adrenal weight [stress, F(1,122) = 67.98; P < 0.0001] (Table 1). During recovery, adrenal weight increased [recovery, F(3,123) = 3.48, P = 0.018], and adjusted adrenal weight declined [recovery, F(3,122) = 42.94, P < 0.0001] (Table 1). A stress-recovery interaction was observed for adjusted adrenal weight [F(3,122) = 4.02, P = 0.0091] but not for actual adrenal weight. Post hoc analysis revealed that actual adrenal weight was elevated in the 4 d and 7 d stressed recovery groups, and adjusted adrenal weight was greater in the 16 h, 4 d, and 7 d stressed recovery groups.
CVS did not stimulate thymic involution in these experiments in this strain of rats (Table 1), consistent with other studies from our laboratory (29, 30). Actual and adjusted thymus weight declined during the stress recovery period, as would be expected during the normal aging process [recovery, actual, F(3,124) = 4.11, P = 0.0081; adjusted, F(3,123) = 33.82, P < 0.0001] (Table 1).
Unstressed AM plasma ACTH and corticosterone levels
Chronic stress did not alter unstressed AM plasma ACTH levels (Tables 2 and 3) in either the novel environment or hypoxia cohorts, but elevations in unstressed AM plasma corticosterone levels were observed in the novel environment cohort [F(2,17) = 6.86, P = 0.0077] (Table 2). Post hoc analysis indicated that plasma corticosterone levels were increased 16 h after cessation of chronic stress (P < 0.01), but this hypersecretion returned to control levels by 7 d recovery. Although plasma corticosterone for the hypoxia cohort appeared to be elevated in the 16 h CVS group, this hypersecretion did not reach statistical significance.
TABLE 2.
Effect of CVS on unstressed plasma hormone levels (novel environment challenge)
| Plasma ACTH (pg/ml) |
Plasma corticosterone (ng/ml) |
|
|---|---|---|
| 16 h Control (n = 6) | 55 ± 12 | 28 ± 10 |
| 16 h CVS (n = 6) | 52 ± 5 | 109 ± 31a |
| 7 d CVS (n = 6) | 39 ± 5 | 17 ± 7 |
Plasma hormone levels in the unstressed control or CVS treated rats not exposed to the novel environment were measured at 16 h or 7 d after cessation of CVS. Values represent the mean ± SEM of each group.
Significant difference from 16 h control group (P < 0.05).
TABLE 3.
Effect of CVS on unstressed plasma hormone levels (hypoxia challenge)
| Plasma ACTH (pg/ml) |
Plasma corticosterone (ng/ml) |
|
|---|---|---|
| 16 h Control (n = 6) | 21 ± 8 | 38 ± 12 |
| 16 h CVS (n = 6) | 13 ± 2 | 75 ± 20 |
| 7 d CVS (n = 6) | 28 ± 10 | 42 ± 17 |
Plasma hormone levels in the unstressed control or CVS-treated rats not exposed to hypoxia were measured at 16 h or 7 d after cessation of CVS. Values represent the mean ± SEM of each group.
Hormonal responses to novel environment
CVS rats tested at 7 d recovery exhibited a blunted plasma ACTH response after novel environment exposure [stress, F(1,35) = 10.67, P = 0.0085] (Fig. 2). Plasma ACTH responses changed within the sampling session at 16 h and 30 d recovery [16 h, time, F(2,35) = 14.09, P = 0.0002; 30 d, time, F(2,35) = 7.73, P = 0.0033], and there was a stress-time interaction at 16 h recovery [stress-time, F(2,35) = 3.96, P = 0.036]. Post hoc analysis indicated greater plasma ACTH levels in the 16 h recovery chronic stress group at 20 min after the end of novel environment. Conversely, lower plasma ACTH levels in the 7 d recovery CVS group were observed 40 min after psychogenic stress challenge. Integrated plasma ACTH secretion did not reach statistical significance at 7 d recovery time point (P = 0.072), and this measure also did not differ at any other recovery time point (data not shown).
Fig. 2.
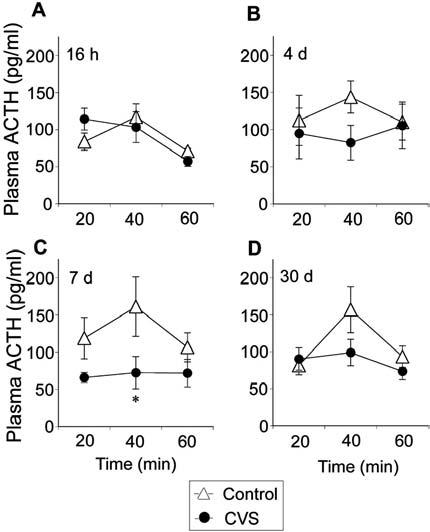
Chronically stressed rats tested at 16 h recovery exhibit transient augmentation of plasma ACTH, whereas chronically stress rats challenged at 7 d recovery exhibit an attenuated plasma ACTH response to novel environment exposure. Plasma ACTH levels at 20, 40, and 60 min after the end of a 5-min exposure to a novel environment at 16 h (A), 4 d (B), 7 d (C), or 30 d (D) recovery from CVS. Values represent mean ± sem; n = 6 per group. *, P < 0.05 vs. control group.
Prior CVS experience attenuated the plasma corticosterone response to novel environment at 7 d recovery [stress, F(1,35) = 8.81, P = 0.014] (Fig. 3). Plasma corticosterone responses changed during the stress sampling session at all recovery time points [16 h, time, F(2,35) = 19.22, P < 0.0001; 4 d, time, F(2,35) = 23.88, P < 0.0001; 7 d, time, F(2,35) = 5.51, P = 0.012; 30 d, time, F(2,35) = 23.77, P < 0.0001], and there was a stress-time interaction at the 4 d recovery time point [stress-time, F(2,35) = 4.79, P = 0.020]. Post hoc analysis revealed that plasma corticosterone levels in CVS rats were lower at 60 min after novel environment at 4 d recovery and at 40 and 60 min after stress challenge at 7 d recovery. Integrated plasma corticosterone secretion was markedly decreased in CVS rats after 7 d recovery from chronic stress (P = 0.014), but no differences were observed at other recovery time points (data not shown).
Fig. 3.
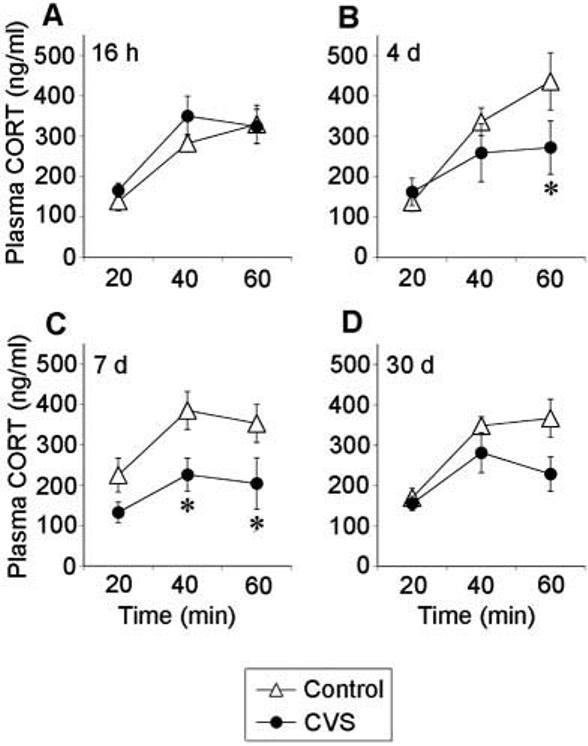
Chronically stressed rats tested at 4 d and 7 d recovery exhibit an attenuated plasma corticosterone response to novel environment exposure. Plasma corticosterone levels at 20, 40, and 60 min after the end of a 5 min exposure to a novel environment at 16 h (A), 4 d (B), 7 d (C), or 30 d (D) recovery from CVS. Values represent mean ± sem; n = 6 per group. *, P < 0.05 vs. control group.
Hormonal responses to hypoxia
Plasma ACTH levels were lower after hypoxia exposure in CVS rats at 16 h recovery [stress, F(1,35) = 5.59, P = 0.040], but not at other recovery time points, and were greater in CVS rats at 7 d recovery [stress, F(1,35) = 6.44, P = 0.030] (Fig. 4). Plasma ACTH levels decreased during the stress sampling session at all recovery time points except 30 d recovery [time, 16 h, F(2,35) = 7.69, P = 0.0033; 4 d, F(2,35) = 8.77, P = 0.0018; 7 d, F(2,35) = 23.33, P < 0.0001]. A stress-time interaction was observed at the 7 d recovery time point [stress-time, F(2,35) = 7.98, P = 0.0028]. Post hoc analysis indicated that plasma ACTH levels in CVS groups are lower than control groups 20 min after hypoxia stress challenge at 16 h recovery (P < 0.05) and are greater 20 min after hypoxia at 7 d recovery (P < 0.05). No differences in integrated plasma ACTH secretion were observed at any recovery time point (data not shown).
Fig. 4.
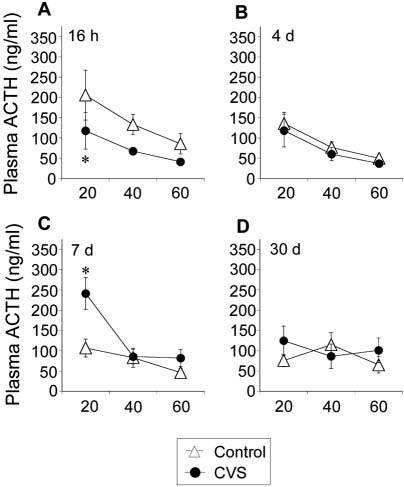
Chronically stressed rats tested at 16 h recovery exhibit slightly lower plasma ACTH levels than controls 20 min after placement into a hypoxia chamber, whereas chronically stressed rats tested at 7 d recovery exhibit higher plasma ACTH levels. Plasma ACTH levels at 20, 40, and 60 min after placement into the hypoxia chamber at 16 h (A), 4 d (B), 7 d (C), or 30 d (D) recovery from CVS. Values represent mean ± sem; n = 6 per group. *, P < 0.05 vs. control group.
Previous chronic stress did not alter the plasma corticosterone response to hypoxia at any recovery time point (stress, P > 0.05 on all days) (Fig. 5). Plasma corticosterone values increased across the sampling session on all recovery days [time, 16 h, F(2,35) = 8.80, P = 0.0018; 4 d, F(2,35) = 11.59, P = 0.0005; 7 d, F(2,35) = 5.64, P = 0.011; 30 d, F(2,35) = 18.60, P < 0.0001], and there were no stress-time interactions at any recovery time point. No differences in integrated plasma corticosterone secretion were observed at any recovery time point (data not shown).
Fig. 5.
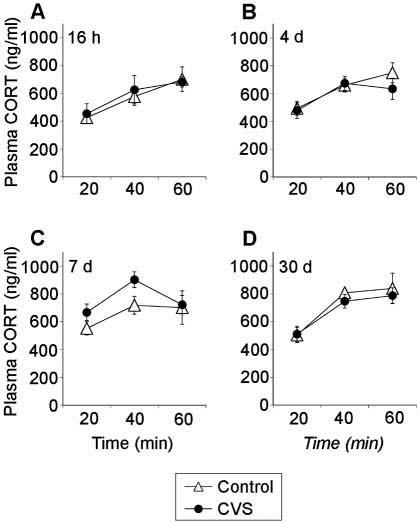
Plasma corticosterone levels in chronically stressed rats do not differ from controls after placement into a hypoxia chamber. Plasma corticosterone levels at 20, 40, and 60 min after placement into the hypoxia chamber at 16 h (A), 4 d (B), 7 d (C), or 30 d (D) recovery from CVS. Values represent mean ± sem; n = 6 per group.
CRH and AVP mRNA expression
A stress-recovery interaction was observed for CRH mRNA expression in the PVN [F(4,65) = 3.66, P = 0.0097], and post hoc analysis revealed that PVN CRH mRNA expression was up-regulated 16 h after the end of CVS exposure (Fig. 6A). PVN CRH mRNA expression was not altered at any other recovery time point. AVP mRNA expression in the parvocellular portion of the PVN was not affected by previous stress exposure at any recovery time point (Fig. 6B).
Fig. 6.
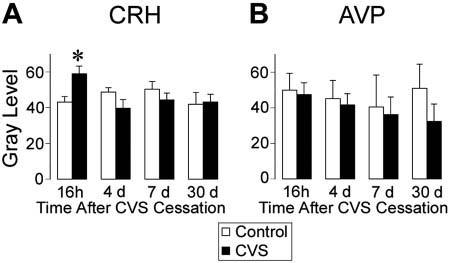
CRH mRNA expression in the PVN is up-regulated 16 h after CVS cessation, but no alterations are observed at any other recovery time point. AVP mRNA expression in the parvocellular PVN is not altered by prior CVS exposure. Semiquantitative analysis of CRH (A) and AVP (B) mRNA expression. Values represent mean ± sem; n = 6–12 per group. *, P < 0.05 vs. control group.
Experiment 2
Chronic stress effects on physiological indices
CVS exposure decreased percent BW gain [stress, F (1,63) = 46.76; P < 0.0001] (Table 4). Percent BW gain increased during recovery [recovery, F(3,63) = 343.84; P < 0.0001] (Table 4), and a stress-recovery interaction was noted [stress-recovery, F(3,63) = 6.62; P = 0.0007]. Post hoc analyses indicated that percent BW gain was lower in the CVS groups at the 16 h, 4 d, and 7 d recovery time points (P < 0.01), but this difference had resolved by the 30 d recovery time point.
TABLE 4.
Effect of CVS on physiological measures in experiment 2
| Body weight change (%) |
Thymus weight (mg) |
Thymus weight (mg/g BW) × 100 |
Adrenal weight (mg) |
Adrenal weight (mg/g BW) × 100 |
|
|---|---|---|---|---|---|
| 16 h | |||||
| Control (n = 8) | 9.5 ± 1.6 | 521 ± 28 | 167 ± 8 | 48.8 ± 1.1 | 15.7 ± 0.5 |
| CVS (n = 8) | 1.6 ± 0.3a | 534 ± 31 | 184 ± 11 | 51.4 ± 1.5 | 17.7 ± 0.5a |
| 4d | |||||
| Control (n = 8) | 13.2 ± 0.9 | 711 ± 44 | 221 ± 14 | 50.9 ± 1.6 | 15.8 ± 0.5 |
| CVS (n = 8) | 6.2 ± 0.6a | 604 ± 43 | 201 ± 13 | 49.8 ± 1.4 | 16.7 ± 0.4 |
| 7d | |||||
| Control (n = 8) | 17.9 ± 0.9 | 544 ± 37 | 161 ± 9 | 46.7 ± 0.8 | 13.9 ± 0.4 |
| CVS (n = 8) | 13.2 ± 1.0a | 601 ± 53 | 184 ± 12 | 50.7 ± 1.5 | 15.6 ± 0.2a |
| 30 d | |||||
| Control (n = 8) | 35.2 ± 1.0 | 472 ± 38 | 127 ± 10 | 56.4 ± 1.8 | 14.6 ± 0.6 |
| CVS (n = 8) | 35.4 ± 1.1 | 461 ± 25 | 120 ± 7 | 53.2 ± 2.1 | 13.9 ± 0.5 |
Effect of CVS on physiological measures in experiment 2. Rats were exposed to CVS for 7 dor were unhandled controls. At each recovery time point, eight rats from each group were restrained for 30 min. The total number of rats in each group is listed. Time points reflect length of time after cessation of stress; values represent the mean ± SEM of each group.
CVS is significantly different from corresponding control group (P < 0.05).
CVS exposure increased adjusted adrenal weight [stress, F(1,63) = 8.30; P = 0.0056] (Table 4), but actual adrenal weight was not altered. During recovery, actual adrenal weight increased [recovery, F(3,63) = 5.98, P = 0.0013], and adjusted adrenal weight declined [recovery, F(3,63) = 12.29, P < 0.0001]. A stress-recovery interaction was observed for adjusted adrenal weight [stress-recovery, F(3,63) = 3.34, P = 0.026] but not for actual adrenal weight. Post hoc analysis revealed that adjusted adrenal weight was greater at 16 h (P < 0.01) and 7 d (P < 0.05) recovery.
Thymic involution was not observed after chronic stress exposure, and thymus size decreased during the course of the recovery period (Table 4). Actual and adjusted thymus weight declined during the stress recovery period [recovery, actual, F(3,63) = 8.77, P < 0.0001; adjusted, F(3,63) = 22.34, P < 0.0001].
Hormonal responses to restraint
Responsiveness of the HPA axis to restraint, a more severe psychogenic stress challenge, was evaluated at the same recovery time points as in experiment 1. Plasma ACTH secretion in the CVS group was attenuated at 4 d recovery [stress, F(1,63) = 4.95, P = 0.043] but was not affected at 7 d recovery (Fig. 7). Plasma ACTH secretion increased during the stress sampling sessions [time, 4d,F(3,63) = 21.42, P < 0.0001; 7 d, F(3,63) = 18.42, P < 0.0001], and a stress-time interaction was observed for plasma ACTH and corticosterone at 4 d recovery [stress-time, F(3,63) = 5.48, P = 0.0029]. Post hoc analysis indicated that plasma ACTH secretion in the CVS group was attenuated relative to controls 30 min after initiation of restraint stress at 4 d recovery (P < 0.05). In addition, integrated plasma ACTH secretion in the CVS group was blunted at 4 d recovery from CVS (data not shown).
Fig. 7.
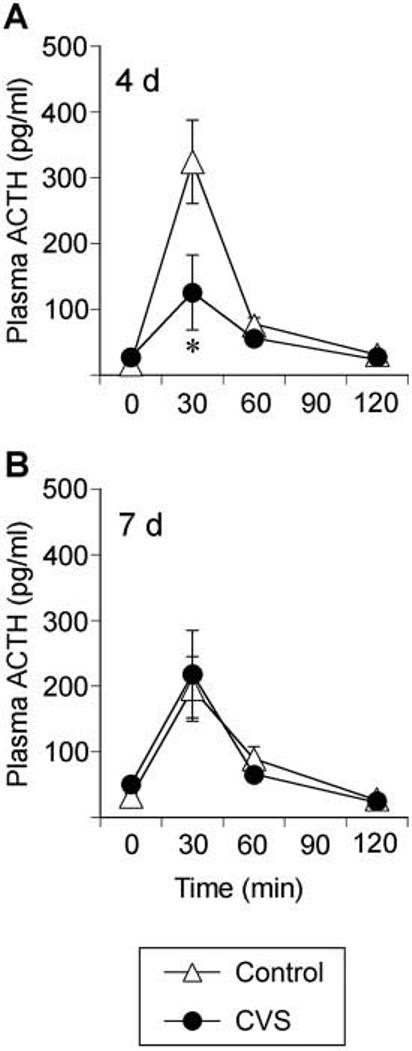
Chronically stressed rats tested at 4 d recovery exhibit an attenuated plasma ACTH response 30 min after novel restraint stress challenge. Plasma ACTH levels could not be determined at 16 h or 30 d recovery due to technical difficulties with the ACTH assay. Plasma ACTH levels at 0, 30, 60, and 120 min after placement into plastic restrainers for 30 min at 4 d (A) or 7 d (B) recovery from CVS. Values represent mean ± sem; n = 8 per group. *, P < 0.05 vs. control group.
Hypersecretion of plasma corticosterone was observed at the 0-min time point in the 16 h recovery CVS group (Fig. 8). Plasma corticosterone secretion in the CVS group was attenuated at 4 d recovery [stress, F(1,63) = 4.63, P = 0.049], but plasma corticosterone secretion was not affected by prior chronic stress at other recovery time points (Fig. 8). Plasma corticosterone levels increased during the stress sampling sessions [time, 16 h, F(3,63) = 65.62, P < 0.0001; 4 d, F(3,63) = 105.94, P < 0.0001; 7 d, F(3,63) = 130.81, P < 0.0001; 30 d, F(3,63) = 114.94, P < 0.0001], and a stress-time interaction was observed for plasma corticosterone at 4 d recovery [stress-time, F(3,63) = 5.21, P = 0.0038]. Post hoc analysis indicated that plasma corticosterone secretion in the CVS group was blunted relative to controls 30 and 60 min after initiation of restraint stress at 4 d recovery (P < 0.01). Integrated plasma corticosterone secretion in the CVS group was attenuated at 4 d recovery from CVS (data not shown).
Fig. 8.
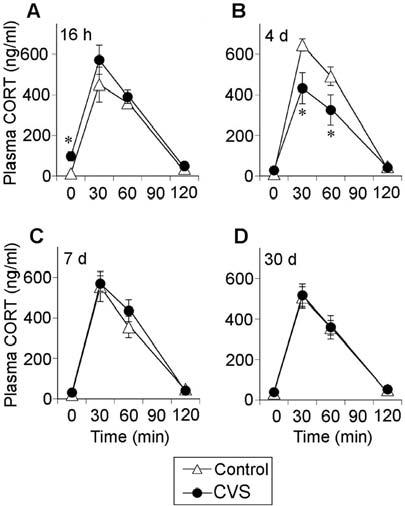
Chronically stressed rats tested at 4 d recovery exhibit an attenuated plasma corticosterone response 30 and 60 min after novel restraint stress challenge. Plasma corticosterone levels at 0, 30, 60, and 120 min after placement into plastic restrainers for 30 min at 16 h (A), 4 d (B), 7 d (C), or 30 d (D) recovery from CVS. Values represent mean ± sem; n = 8 per group. *, P < 0.05 vs. control group.
Discussion
Contrary to our hypothesis, the present study demonstrates that CVS produces a delayed manifestation of HPA axis hyporesponsiveness to novel psychogenic stressors. This deficit is observed following two different psychogenic stimuli of different intensities (novel environment and restraint), supporting a general action of chronic stress on psychogenic stress processing in brain. Importantly, hormonal responses to a novel systemic stressor (hypoxia) are largely unaffected by prior stress history, suggesting that HPA axis response capacity to homeostatic perturbation is maintained. Finally, CRH gene transcription in the PVN is up-regulated immediately after CVS cessation (16 h recovery), but no other differences in PVN CRH or AVP mRNA expression were observed.
Our finding of attenuated HPA axis responses to novel psychogenic stressors during recovery from CVS is quite surprising. Indeed, the only report we have found of HPA axis hyporesponsiveness to novel stress occurs in rats exposed to prolonged withdrawal (8 d) from intermittent morphine (31-33). In these studies, plasma ACTH, but not plasma corticosterone, responses to novel restraint stress are lower in rats undergoing withdrawal. Numerous reports, however, indicate that potentiation of HPA axis responses to subsequent novel stressors occurs in subjects with prior stress experience. For example, subjects with a single prior exposure to a severe stressor (foot shock or IL-1) exhibit enhanced plasma ACTH and corticosterone responses to a subsequent novel challenge stressor for several days to weeks (17, 19, 34). Potentiation of HPA axis responses to novel stressors also occurs after brief (12–24 h) recovery from repeated homotypic stress (10, 15). Our studies differ fundamentally in several ways from the above reports. We administered several different stressors in an unpredictable sequence over the course of a week and then examined HPA axis responsivity at several recovery time points. It is possible that the CVS-induced hyporesponsiveness we observed is due to: 1) the interval between chronic stress cessation and stress challenge, 2) the nature of the chronic stress paradigm, or 3) an interaction of these two factors. Future studies will therefore determine whether long-lasting attenuation of stress responsiveness is a general feature of CVS-related adaptation or pathology.
Facilitation of HPA axis responses is typically observed in chronically stressed rats confronted with a novel stressor 16 h after chronic stress cessation (4, 10, 15). Although overt potentiation of the HPA axis responses at 16 h recovery was not observed in the current study, responses to acute novel stressors are equivalent to those of unstressed controls. Maintenance of the HPA response in the face of increased negative feedback effects of repeated and cumulative glucocorticoid exposure is consistent with stress-induced facilitation, as defined by Dallman (13, 35, 36). The attenuation of psychogenic stress responses in CVS groups at later recovery time points indicates that stress-induced facilitation, if present, is: 1) short-lived after CVS, or 2) the magnitude is so small as to be overridden by the HPA axis hyporesponsiveness.
In the current studies, diminished HPA axis responses to restraint are observed only at 4 d recovery from CVS, whereas attenuation of HPA axis responses to novel environment is seen at both 4 and 7 d recovery. The different time course of HPA hyporesponsiveness in the two experiments may be related to the intensity and/or controllability of the challenge stressors. Our data clearly indicate that restraint produces a larger HPA axis response than novel environment exposure, which may permit an earlier escape from the poststress hyporesponsiveness. Stressor controllability may also be a factor in the differential time course; in the novel environment, the animal has the opportunity to limit exposure to aversive components by entering the closed arms, whereas restraint is an imposed and inescapable stress. Finally, subtle differences in the CVS paradigms used in two experiments may have influenced the expression of HPA axis hyporesponsiveness.
Increased CRH gene transcription was observed in the PVN at 16 h recovery from chronic stress, consistent with our prior work (30, 37) and other reports of chronic stress-induced up-regulation of CRH mRNA expression (38, 39). Our data further demonstrate that this chronic stress-induced enhancement of PVN CRH mRNA expression returns to control levels within 4 d of recovery from CVS. Thus, HPA axis hyporesponsiveness at 4 and 7 d recovery may be related to falling levels of CRH biosynthesis, which may reduce the capacity for pituitary ACTH release in response to stress. Notably, reduced CRH mRNA levels are related to HPA axis nonresponsiveness to psychogenic stress in the visible burrow model of chronic social stress, further supporting a relationship between CRH synthesis and HPA axis responsivity (40). Indeed, Hellhammer and Wade (41) have suggested that chronic stress is associated with increased hypothalamic CRH secretion. After a period of recovery during which hypothalamic CRH secretion normalizes, there is diminished ACTH release ultimately resulting in diminished corticosterone production (42). The fact that the responsivity of the HPA axis to hypoxia is retained suggests an alternative mechanism of activation in this pathway, perhaps mediated by differential release of other secretagogues, such as AVP. Given the different circuitries involved in communicating psychogenic and systemic stressors (43), it is plausible that the different types of stimuli may invoke different subpopulations of PVN neurons.
Alterations in glucocorticoid negative feedback inhibition might also underlie the neuroendocrine hyporesponsiveness that we observed. Indeed, chronically stressed rats exhibit enhanced corticosterone feedback of acute stress-induced HPA activity (44), and blunting of the ACTH stress response after chronic morphine treatment/withdrawal is associated with enhanced sensitivity to glucocorticoids (32). Decreased pituitary CRH responsiveness and increased negative feedback inhibition are, however, unlikely to be the sole mechanism(s) underlying the delayed HPA axis hyporesponsivity because the systemic stress challenge elicited similar, or perhaps facilitated, plasma ACTH and corticosterone responses at these recovery time points. Therefore, it is likely that the observed hyporesponsivity may also be related to altered activity or function of neurocircuits regulating psychogenic stress responses.
It is well known that psychogenic and systemic stressors use primarily different neural circuitry. Psychogenic stressors (e.g. novel environment, restraint) are believed to recruit forebrain limbic structures, including the hippocampus, medial prefrontal cortex, and amygdala (1, 2, 45). These regions appear to relay information to the PVN via intermediary neurons in the bed nucleus of the stria terminalis, hypothalamus, and perhaps brainstem (1, 2). Systemic stressors (e.g. hypoxia) activate brainstem structures such as the nucleus of the solitary tract, which directly activate PVN neurons (1, 2, 46). Thus, the differential responsiveness of the HPA axis to novel environment and restraint vs. hypoxia in the current study suggests that relevant neuroadaptations likely occur in forebrain regions mediating psychogenic stress responses. Numerous alterations in central stress circuitry are observed 16 h after cessation of CVS, including decreased hippocampal adrenocorticosteroid receptor and 5-hydroxytryptamine 1A receptor expression (37, 47, 48), enhanced hypothalamic glutamic acid decarboxylase expression (49), and altered expression of γ-aminobutyric acid-A, NMDA, and non-NMDA receptors in the PVN (50, 51). Although the persistence of these changes remains to be assessed, the possibility for long-term changes in neurotransmission clearly exists.
Overall, our results are consistent with long-lasting, chronic stress-induced neuroplastic changes in HPA axis responsiveness. However, there are alternative interpretations that may explain these data. First, the decreased neuroendocrine responsiveness to novel psychogenic stressors in our experiments may be associated with an “increase in the threshold of activation of the stress axis” (32), such that a more intense stressor is required for activation of the HPA axis. This is not likely because we observed diminution of peak and integrated plasma ACTH and corticosterone levels after both mild (novel environment) and severe (restraint) psychogenic stressors. Indeed, the restraint stress challenge provokes hormonal responses that are nearly equivalent to those seen after hypoxia stress challenge. Second, the temporal dynamics of the plasma ACTH response to the psychogenic (novel environment and restraint) and systemic (hypoxia) stress challenges in experiment 1 are markedly different, suggesting that the selection of sampling times may have missed differences in response to hypoxia. However, the temporal profile of plasma corticosterone secretion is very similar between the novel environment and hypoxia stress challenges, and we did not observe any indication of altered plasma corticosterone levels in the CVS groups.
Modest hypersecretion of basal plasma corticosterone was observed in groups exposed to novel environment, but this hypersecretion did not reach statistical significance for the hypoxia challenge groups. Basal levels of plasma ACTH were not altered by CVS in either the novel environment or hypoxia challenge groups. These data are congruent with our previous findings and those from other laboratories, indicating that chronic stress produces modest, and inconsistent, elevations in basal plasma corticosterone (22, 29, 30, 37, 47, 49, 50, 52-56), without altering basal plasma ACTH levels (29, 37, 50, 52, 53). Dissociations between ACTH and corticosterone data are not uncommon (57, 58) and can be explained by several factors. First, although it has been classically thought that glucocorticoid production is driven solely by ACTH release, recent studies suggest that glucocorticoid production is also modulated by non-ACTH mechanisms, such as adrenal splanchnic innervation (59). Second, the ACTH RIA we used measures immunoreactive ACTH, not bioactive ACTH. Because dissociations between bioactive and immunoreactive ACTH levels have been previously reported (60), it is possible that bioactive ACTH levels may have corresponded more closely with the corticosterone data. Finally, there is the possibility that CVS may have enhanced responsiveness of the adrenal glands to ACTH, such that a given amount of ACTH provokes a greater corticosterone response. Indeed, there is some evidence to suggest that chronic stress does increase adrenocortical responsiveness to a single dose of ACTH (61, 62).
Until recently, research on HPA axis dysfunction in psychopathologies has focused primarily on hyperactivity of the HPA axis. It is becoming increasingly clear, however, that hypoactivity of the HPA axis may also be important in patho-physiological conditions (42, 63). Hypocortisolism has been reported in posttraumatic stress disorder (64), chronic fatigue syndrome (65), and fibromyalgia (66), and stress appears to be a contributing factor in the development of these disease states (42). These findings have led Heim et al. (42) to suggest that these stress-related disorders represent a class of diseases with the common endocrine dysfunction of hypocortisolism. The present finding of HPA hyporesponsiveness after CVS is consistent with Heim's contention and suggests that this may be an appropriate model for some aspects of HPA dysfunction that often accompany these disorders.
In summary, CVS provoked robust physiological adaptations in HPA axis responsiveness to subsequent novel stress challenge; psychogenic stress challenge after CVS, but not systemic stress challenge, elicits delayed neuroendocrine hyporesponsivity. This HPA axis hyporesponsiveness after CVS may represent normal recovery from chronic stress or perhaps represents a period of increased vulnerability to further insult. These data suggest that the CVS model may provide valuable insights into mechanisms of neuroendocrine dysfunction in chronic stress-related disorders, such as depression and posttramautic stress disorder.
Acknowledgments
We thank Mark Dolgas, Ben Packard, Ingrid Thomas, and Laurie Burck for technical expertise and Dr. Helmer Figueiredo for helpful discussions.
Abbreviations
- AVP
Arginine vasopressin
- BW
body weight
- CVS
chronic variable stress
- HPA
hypothalamo-pituitary-adrenocortical
- KPBS
potassium PBS
- PVN
paraventricular nucleus of the hypothalamus
- SCC
standard saline citrate
Footnotes
This work was supported by National Institutes of Health Grants MH049698, MH069860, and AG012962 (to J.P.H.) and DA016778 (to N.M.R.), National Institutes of Health Postdoctoral Fellowships DA016466 (to M.M.O.) and DK067820 (to Y.M.U.), and by the Medical Research Service Department of Veterans Affairs (to N.M.R.).
All authors have nothing to declare.
References
- 1.Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 3.Armario A, Lopez-Calderon A, Jolin T, Balasch J. Response of anterior pituitary hormones to chronic stress. The specificity of adaptation. Neurosci Biobehav Rev. 1986;10:245–250. doi: 10.1016/0149-7634(86)90011-4. [DOI] [PubMed] [Google Scholar]
- 4.Hauger RL, Lorang M, Irwin M, Aguilera G. CRF receptor regulation and sensitization of ACTH responses to acute ether stress during chronic intermittent immobilization stress. Brain Res. 1990;532:34–40. doi: 10.1016/0006-8993(90)91738-3. [DOI] [PubMed] [Google Scholar]
- 5.Li HY, Sawchenko PE. Hypothalamic effector neurons and extended circuitries activated in “neurogenic” stress: a comparison of footshock effects exerted acutely, chronically, and in animals with controlled glucocorticoid levels. J Comp Neurol. 1998;393:244–266. [PubMed] [Google Scholar]
- 6.Odio M, Brodish A. Age-related adaptation of pituitary-adrenocortical responses to stress. Neuroendocrinology. 1989;49:382–388. doi: 10.1159/000125142. [DOI] [PubMed] [Google Scholar]
- 7.Pitman DL, Ottenweller JE, Natelson BH. Plasma corticosterone levels during repeated presentation of two intensities of restraint stress: chronic stress and habituation. Physiol Behav. 1988;43:47–55. doi: 10.1016/0031-9384(88)90097-2. [DOI] [PubMed] [Google Scholar]
- 8.Viau V, Sawchenko PE. Hypophysiotropic neurons of the paraventricular nucleus respond in spatially, temporally, and phenotypically differentiated manners to acute vs. repeated restraint stress: rapid publication. J Comp Neurol. 2002;445:293–307. doi: 10.1002/cne.10178. [DOI] [PubMed] [Google Scholar]
- 9.Bhatnagar S, Meaney MJ. Hypothalamic-pituitary-adrenal function in chronic intermittently cold-stressed neonatally handled and non handled rats. J Neuroendocrinol. 1995;7:97–108. doi: 10.1111/j.1365-2826.1995.tb00672.x. [DOI] [PubMed] [Google Scholar]
- 10.Bhatnagar S, Dallman M. Neuroanatomical basis for facilitation of hypothalamic-pituitary-adrenal responses to a novel stressor after chronic stress. Neuroscience. 1998;84:1025–1039. doi: 10.1016/s0306-4522(97)00577-0. [DOI] [PubMed] [Google Scholar]
- 11.Akana SF, Dallman MF. Chronic cold in adrenalectomized, corticosterone (B)-treated rats: facilitated corticotropin responses to acute restraint emerge as B increases. Endocrinology. 1997;138:3249–3258. doi: 10.1210/endo.138.8.5291. [DOI] [PubMed] [Google Scholar]
- 12.Ottenweller JE, Natelson BH, Pitman DL, Drastal SD. Adrenocortical and behavioral responses to repeated stressors: toward an animal model of chronic stress and stress-related mental illness. Biol Psychiatry. 1989;26:829–841. doi: 10.1016/0006-3223(89)90123-6. [DOI] [PubMed] [Google Scholar]
- 13.Young EA, Akana S, Dallman MF. Decreased sensitivity to glucocorticoid fast feedback in chronically stressed rats. Neuroendocrinology. 1990;51:536–542. doi: 10.1159/000125388. [DOI] [PubMed] [Google Scholar]
- 14.Jaferi A, Nowak N, Bhatnagar S. Negative feedback functions in chronically stressed rats: role of the posterior paraventricular thalamus. Physiol Behav. 2003;78:365–373. doi: 10.1016/s0031-9384(03)00014-3. [DOI] [PubMed] [Google Scholar]
- 15.Bhatnagar S, Vining C. Facilitation of hypothalamic-pituitary-adrenal responses to novel stress following repeated social stress using the resident/intruder paradigm. Horm Behav. 2003;43:158–165. doi: 10.1016/s0018-506x(02)00011-9. [DOI] [PubMed] [Google Scholar]
- 16.Dallman MF, Bhatnagar S. Handbook of physiology. Oxford UP; New York: 2000. Chronic stress: role of the hypothalamo-pituitary-adrenal axis; pp. 179–210. [Google Scholar]
- 17.van Dijken HH, de Goeij DC, Sutanto W, Mos J, de Kloet ER, Tilders FJ. Short inescapable stress produces long-lasting changes in the brain-pituitary-adrenal axis of adult male rats. Neuroendocrinology. 1993;58:57–64. doi: 10.1159/000126512. [DOI] [PubMed] [Google Scholar]
- 18.Buwalda B, de Boer SF, Schmidt ED, Felszeghy K, Nyakas C, Sgoifo A, Van der Vegt BJ, Tilders FJ, Bohus B, Koolhaas JM. Long-lasting deficient dexamethasone suppression of hypothalamic-pituitary-adrenocortical activation following peripheral CRF challenge in socially defeated rats. J Neuroendocrinol. 1999;11:513–520. doi: 10.1046/j.1365-2826.1999.00350.x. [DOI] [PubMed] [Google Scholar]
- 19.Johnson JD, O'Connor KA, Deak T, Spencer RL, Watkins LR, Maier SF. Prior stressor exposure primes the HPA axis. Psychoneuroendocrinology. 2002;27:353–365. doi: 10.1016/s0306-4530(01)00057-9. [DOI] [PubMed] [Google Scholar]
- 20.Servatius RJ, Ottenweller JE, Bergen MT, Soldan S, Natelson BH. Persistent stress-induced sensitization of adrenocortical and startle responses. Physiol Behav. 1994;56:945–954. doi: 10.1016/0031-9384(94)90328-x. [DOI] [PubMed] [Google Scholar]
- 21.O'Connor KA, Johnson JD, Hammack SE, Brooks LM, Spencer RL, Watkins LR, Maier SF. Inescapable shock induces resistance to the effects of dexamethasone. Psychoneuroendocrinology. 2003;28:481–500. doi: 10.1016/s0306-4530(02)00035-5. [DOI] [PubMed] [Google Scholar]
- 22.Herman JP, Watson SJ, Spencer RL. Defense of adrenocorticosteroid receptor expression in rat hippocampus: effects of stress and strain. Endocrinology. 1999;140:3981–3991. doi: 10.1210/endo.140.9.6962. [DOI] [PubMed] [Google Scholar]
- 23.Vahl TP, Ulrich-Lai YM, Ostrander MM, Dolgas CM, Elfers EE, Seeley RJ, D'Alessio DA, Herman JP. Comparative analysis of ACTH and corticosterone sampling methods in rats. Am J Physiol Endocrinol Metab. 2005;289:E823–E828. doi: 10.1152/ajpendo.00122.2005. [DOI] [PubMed] [Google Scholar]
- 24.Jasper MS, Engeland WC. Synchronous ultradian rhythms in adreno-cortical secretion detected by microdialysis in awake rats. Am J Physiol. 1991;261:R1257–R1268. doi: 10.1152/ajpregu.1991.261.5.R1257. [DOI] [PubMed] [Google Scholar]
- 25.Figueiredo HF, Bruestle A, Bodie B, Dolgas CM, Herman JP. The medial prefrontal cortex differentially regulates stress-induced c-fos expression in the forebrain depending on type of stressor. Eur J Neurosci. 2003;18:2357–2364. doi: 10.1046/j.1460-9568.2003.02932.x. [DOI] [PubMed] [Google Scholar]
- 26.Herman JP. In situ hybridization analysis of vasopressin gene transcription in the paraventricular and supraoptic nuclei of the rat: regulation by stress and glucocorticoids. J Comp Neurol. 1995;363:15–27. doi: 10.1002/cne.903630103. [DOI] [PubMed] [Google Scholar]
- 27.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1998. [DOI] [PubMed] [Google Scholar]
- 28.Dixon W, Massey F. Introduction to statistical analysis. McGraw-Hill; New York: 1969. [Google Scholar]
- 29.Herman JP, Renda A, Bodie B. Norepinephrine-γ-aminobutyric acid (GABA) interaction in limbic stress circuits: effects of reboxetine on GABAergic neurons. Biol Psychiatry. 2003;53:166–174. doi: 10.1016/s0006-3223(02)01449-x. [DOI] [PubMed] [Google Scholar]
- 30.Ziegler DR, Cass WA, Herman JP. Excitatory influence of the locus coeruleus in hypothalamic-pituitary-adrenocortical axis responses to stress. J Neuroendocrinol. 1999;11:361–369. doi: 10.1046/j.1365-2826.1999.00337.x. [DOI] [PubMed] [Google Scholar]
- 31.Houshyar H, Cooper ZD, Woods JH. Paradoxical effects of chronic morphine treatment on the temperature and pituitary-adrenal responses to acute restraint stress: a chronic stress paradigm. J Neuroendocrinol. 2001;13:862–874. doi: 10.1046/j.1365-2826.2001.00713.x. [DOI] [PubMed] [Google Scholar]
- 32.Houshyar H, Galigniana MD, Pratt WB, Woods JH. Differential responsivity of the hypothalamic-pituitary-adrenal axis to glucocorticoid negative-feedback and corticotropin releasing hormone in rats undergoing morphine withdrawal: possible mechanisms involved in facilitated and attenuated stress responses. J Neuroendocrinol. 2001;13:875–886. doi: 10.1046/j.1365-2826.2001.00714.x. [DOI] [PubMed] [Google Scholar]
- 33.Houshyar H, Manalo S, Dallman MF. Time-dependent alterations in mRNA expression of brain neuropeptides regulating energy balance and hypothalamo-pituitary-adrenal activity after withdrawal from intermittent morphine treatment. J Neurosci. 2004;24:9414–9424. doi: 10.1523/JNEUROSCI.1641-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt ED, Aguilera G, Binnekade R, Tilders FJ. Single administration of interleukin-1 increased corticotropin releasing hormone and corticotropin releasing hormone-receptor mRNA in the hypothalamic paraventricular nucleus which paralleled long-lasting (weeks) sensitization to emotional stressors. Neuroscience. 2003;116:275–283. doi: 10.1016/s0306-4522(02)00555-9. [DOI] [PubMed] [Google Scholar]
- 35.Dallman MF, Akana SF, Scribner KA, Bradbury MJ, Walker CD, Strack AM, Cascio CS. Stress, feedback, and facilitation in the hypothalamo-pituitary-adrenal axis. J Neuroendocrinol. 1992;4:517–526. doi: 10.1111/j.1365-2826.1992.tb00200.x. [DOI] [PubMed] [Google Scholar]
- 36.Akana SF, Hanson ES, Horsley CJ, Strack AM, Bhatnagar S, Bradbury MJ, Milligan ED, Dallman MF. Clamped corticosterone (B) reveals the effect of endogenous B on both facilitated responsivity to acute restraint and metabolic responses to chronic stress. Stress. 1996;1:33–49. doi: 10.3109/10253899609001094. [DOI] [PubMed] [Google Scholar]
- 37.Herman JP, Adams D, Prewitt C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology. 1995;61:180–190. doi: 10.1159/000126839. [DOI] [PubMed] [Google Scholar]
- 38.Makino S, Smith MA, Gold PW. Increased expression of corticotropin-releasing hormone and vasopressin messenger ribonucleic acid (mRNA) in the hypothalamic paraventricular nucleus during repeated stress: association with reduction in glucocorticoid receptor mRNA levels. Endocrinology. 1995;136:3299–3309. doi: 10.1210/endo.136.8.7628364. [DOI] [PubMed] [Google Scholar]
- 39.Imaki T, Nahan JL, Rivier C, Sawchenko PE, Vale W. Differential regulation of corticotropin-releasing factor mRNA in rat brain regions by glucocorticoids and stress. J Neurosci. 1991;11:585–599. doi: 10.1523/JNEUROSCI.11-03-00585.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albeck DS, McKittrick CR, Blanchard DC, Blanchard RJ, Nikulina J, McEwen BS, Sakai RR. Chronic social stress alters levels of corticotropin-releasing factor and arginine vasopressin mRNA in rat brain. J Neurosci. 1997;17:4895–4903. doi: 10.1523/JNEUROSCI.17-12-04895.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hellhammer DH, Wade S. Endocrine correlates of stress vulnerability. Psychother Psychosom. 1993;60:8–17. doi: 10.1159/000288675. [DOI] [PubMed] [Google Scholar]
- 42.Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25:1–35. doi: 10.1016/s0306-4530(99)00035-9. [DOI] [PubMed] [Google Scholar]
- 43.Figueiredo HF, Bodie BL, Tauchi M, Dolgas CM, Herman JP. Stress integration after acute and chronic predator stress: differential activation of central stress circuitry and sensitization of the hypothalamo-pituitary-adrenocortical axis. Endocrinology. 2003;144:5249–5258. doi: 10.1210/en.2003-0713. [DOI] [PubMed] [Google Scholar]
- 44.Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui DH, Tabira T. Chronic stress differentially regulates glucocorticoid negative feedback response in rats. Psychoneuroendocrinology. 2001;26:443–459. doi: 10.1016/s0306-4530(01)00004-x. [DOI] [PubMed] [Google Scholar]
- 45.Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 2001;14:1143–1152. doi: 10.1046/j.0953-816x.2001.01733.x. [DOI] [PubMed] [Google Scholar]
- 46.Sawchenko PE, Li HY, Ericsson A. Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog Brain Res. 2000;122:61–78. doi: 10.1016/s0079-6123(08)62131-7. [DOI] [PubMed] [Google Scholar]
- 47.Paskitti ME, McCreary BJ, Herman JP. Stress regulation of adrenocorticosteroid receptor gene transcription and mRNA expression in rat hippocampus: time-course analysis. Brain Res Mol Brain Res. 2000;80:142–152. doi: 10.1016/s0169-328x(00)00121-2. [DOI] [PubMed] [Google Scholar]
- 48.Lopez JF, Vazquez DM, Chalmers DT, Watson SJ. Regulation of 5-HT receptors and the hypothalamic-pituitary-adrenal axis. Implications for the neurobiology of suicide. Ann NY Acad Sci. 1997;836:106–134. doi: 10.1111/j.1749-6632.1997.tb52357.x. [DOI] [PubMed] [Google Scholar]
- 49.Bowers G, Cullinan WE, Herman JP. Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J Neurosci. 1998;18:5938–5947. doi: 10.1523/JNEUROSCI.18-15-05938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziegler DR, Cullinan WE, Herman JP. Organization and regulation of paraventricular nucleus glutamate signaling systems: N-methyl-d-aspartate receptors. J Comp Neurol. 2005;484:43–56. doi: 10.1002/cne.20445. [DOI] [PubMed] [Google Scholar]
- 51.Cullinan WE, Wolfe TJ. Chronic stress regulates levels of mRNA transcripts encoding β subunits of the GABA(A) receptor in the rat stress axis. Brain Res. 2000;887:118–124. doi: 10.1016/s0006-8993(00)03000-6. [DOI] [PubMed] [Google Scholar]
- 52.Kiss A, Aguilera G. Regulation of the hypothalamic pituitary adrenal axis during chronic stress: responses to repeated intraperitoneal hypertonic saline injection. Brain Res. 1993;630:262–270. doi: 10.1016/0006-8993(93)90665-a. [DOI] [PubMed] [Google Scholar]
- 53.Herman JP, Larson BR, Speert DB, Seasholtz AF. Hypothalamo-pituitary-adrenocortical dysregulation in aging F344/Brown-Norway F1 hybrid rats. Neurobiol Aging. 2001;22:323–332. doi: 10.1016/s0197-4580(00)00242-6. [DOI] [PubMed] [Google Scholar]
- 54.Armario A, Restrepo C, Castellanos JM, Balasch J. Dissociation between adrenocorticotropin and corticosterone responses to restraint after previous chronic exposure to stress. Life Sci. 1985;36:2085–2092. doi: 10.1016/0024-3205(85)90304-2. [DOI] [PubMed] [Google Scholar]
- 55.Simpkiss JL, Devine DP. Responses of the HPA axis after chronic variable stress: effects of novel and familiar stressors. Neuro Endocrinol Lett. 2003;24:97–103. [PubMed] [Google Scholar]
- 56.Haile CN, GrandPre T, Kosten TA. Chronic unpredictable stress, but not chronic predictable stress, enhances the sensitivity to the behavioral effects of cocaine in rats. Psychopharmacology (Berl) 2001;154:213–220. doi: 10.1007/s002130000650. [DOI] [PubMed] [Google Scholar]
- 57.Kiss A, Jezova D, Aguilera G. Activity of the hypothalamic pituitary adrenal axis and sympathoadrenal system during food and water deprivation in the rat. Brain Res. 1994;663:84–92. doi: 10.1016/0006-8993(94)90465-0. [DOI] [PubMed] [Google Scholar]
- 58.Dempsher DP, Gann DS. Increased cortisol secretion after small hemorrhage is not attributable to changes in adrenocorticotropin. Endocrinology. 1983;113:86–93. doi: 10.1210/endo-113-1-86. [DOI] [PubMed] [Google Scholar]
- 59.Ulrich-Lai YM, Engeland WC. Hyperinnervation during adrenal regeneration influences the rate of functional recovery. Neuroendocrinology. 2000;71:107–123. doi: 10.1159/000054527. [DOI] [PubMed] [Google Scholar]
- 60.Engeland WC, Miller P, Gann DS. Dissociation between changes in plasma bioactive and immunoreactive adrenocorticotropin after hemorrhage in awake dogs. Endocrinology. 1989;124:2978–2985. doi: 10.1210/endo-124-6-2978. [DOI] [PubMed] [Google Scholar]
- 61.Armario A, Hidalgo J, Giralt M. Evidence that the pituitary-adrenal axis does not cross-adapt to stressors: comparison to other physiological variables. Neuroendocrinology. 1988;47:263–267. doi: 10.1159/000124921. [DOI] [PubMed] [Google Scholar]
- 62.Riegle GD. Chronic stress effects on adrenocortical responsiveness in young and aged rats. Neuroendocrinology. 1973;11:1–10. doi: 10.1159/000122114. [DOI] [PubMed] [Google Scholar]
- 63.McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338:171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- 64.Yehuda R. Biology of posttraumatic stress disorder. J Clin Psychiatry. 2001;62(Suppl 17):41–46. [PubMed] [Google Scholar]
- 65.Poteliakhoff A. Adrenocortical activity and some clinical findings in acute and chronic fatigue. J Psychosom Res. 1981;25:91–95. doi: 10.1016/0022-3999(81)90095-7. [DOI] [PubMed] [Google Scholar]
- 66.Crofford LJ, Pillemer SR, Kalogeras KT, Cash JM, Michelson D, Kling MA, Sternberg EM, Gold PW, Chrousos GP, Wilder RL. Hypothalamic-pituitary-adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994;37:1583–1592. doi: 10.1002/art.1780371105. [DOI] [PubMed] [Google Scholar]