

Abstract
Senescence, a progressive degenerative process leading to age-related increase in mortality, is found in most eukaryotes. However, the molecular events underlying aging remain largely unknown. Understanding how longevity is regulated is a fundamental problem. Here we demonstrate that the respiratory function is a key factor that contributes to shortening lifespan of the filamentous fungus Podospora anserina. In this organism, senescence is systematically associated with mitochondrial DNA instabilities. We show that inactivation of the nuclear COX5 gene encoding subunit V of the cytochrome c oxidase complex leads to the exclusive use of the alternative respiratory pathway and to a decrease in production of reactive oxygen species. This inactivation results in a striking increase of longevity associated with stabilization of the mitochondrial chromosome. Moreover, accumulation of several senescence-specific mitochondrial DNA molecules is prevented in this nuclear mutant. These findings provide direct evidence of a causal link between mitochondrial metabolism and longevity in Podospora anserina.
Although senescence is a characteristic feature in most organisms, the mechanisms whereby the aging process is controlled have yet to be elucidated. Numerous studies implicate mitochondria in this process. A decrease of mitochondrial function and an increased proportion of defective mitochondrial DNA (mtDNA) molecules do indeed accompany aging in most organisms (see refs. 1–3 for reviews). That mitochondria contribute to aging appears to be a reasonable proposition. Mitochondria, whose primary function is to produce energy, play a central role in cellular metabolism and in the control of vital functions. They also represent the major source of reactive oxygen species (ROS) (4), which seem to be implicated in a variety of pathological processes and aging (see refs. 5–7 for reviews). However, few studies provide any direct evidence that mitochondria are causally involved in aging. Recently it was shown in Caenorhabditis elegans that the products of the clk-1 and mev-1 genes, which control lifespan, are located in mitochondria and that their function affects the level of respiration (8, 9).
In the filamentous fungi Podospora anserina and Neurospora crassa, senescence is always associated with the accumulation of defective mitochondrial DNA (see refs. 10 and 11 for reviews). In P. anserina, aging is systematically correlated with the accumulation of circular multimeric DNA molecules called senDNAs. Several groups of senDNAs originating from separate regions of the mitochondrial chromosome can be recovered. One of them (senDNA α) is present in all senescent cultures of wild-type strains. It exactly corresponds to the first intron (intron α) of the COX1 gene, which encodes subunit I of cytochrome c oxidase. Intron α is a mobile group II intron able to transpose in the mitochondrial chromosome (12). Analysis of mitochondrial mutants selected as escaping senescence revealed that they carry a deletion of the mitochondrial chromosome that covers part of the intron α and the first exon of the COX1 gene (13–15). As a result, these mutants are deficient for the intron α activities and unable to generate senDNA α. They also are deficient in cytochrome c oxidase and use the cyanide-resistant alternative pathway for respiration. Recently we have shown that intron α is not necessary for the occurrence of senescence, although it is an accelerator of the process, probably by means of its role in the destabilization of the mitochondrial chromosome (16). In humans and in other organisms, aging and several mitochondrial diseases (3, 17–22) are associated with respiratory defects and accumulation of mtDNA rearrangements. However, the relationships that causally link these different parameters are far from established (2, 19). Thanks to the occurrence in P. anserina of an alternative pathway of respiration, this organism provides a suitable model for studying the effects of the absence of the cytochrome respiratory pathway on senescence. Thus we decided to examine the effects of the complete absence of cytochrome c oxidase in strains containing a fully functional mitochon- drial chromosome, potentially able to give rise to the diverse senDNAs and mitochondrial modifications observed during senescence.
In the present study, we report the isolation and the disruption of the nuclear gene encoding subunit V of cytochrome c oxidase. We show that this disruption leads to the exclusive use of the alternative oxidase, to a striking increase of longevity, and to a strong stabilization of the mitochondrial chromosome. These findings provide direct evidence of a causal link between respiration and longevity and demonstrate that the cytochrome respiratory function is one of the major processes contributing to the destabilization of the mitochondrial genome and the shortening of lifespan.
Materials and Methods
Strains, Growth Conditions, and Lifespan Measurement.
Genetic and physiological properties of P. anserina have been described by Rizet and Engelmann (23) and reviewed by Esser (24). All of the strains used in this study are derived from the wild-type s strain (25). The mex16 strain was selected as a long-lived mitochondrial mutant. The mutation consists of an insertion accompanied by a short deletion covering part of the intron α and of the upstream exon COX1e1 (14). Resistance to phleomycin and hygromycin B was measured on minimal synthetic medium (M2) containing, respectively, 10 μg/ml and 100 μg/ml of the corresponding antibiotic. Sensitivity to salicylhydroxamic acid (SHAM) was tested on M2 medium supplemented with 150 μg/ml SHAM. Lifespans were measured on M2 medium in culture tubes at 27°C, on five subcultures derived from two to six independent spores of each strain, as previously described (26). The lifespan of a strain was defined as the mean length of growth of parallel subcultures between the point of incubation and the arrest edge of the culture.
Nucleic Acid Analysis.
P. anserina mitochondrial and nuclear DNA and RNA were extracted either by the minipreparation method (27) or by purification in a 4′,6-diamidino-2-phenylindole (DAPI)/CsCl gradient (28). Southern analysis followed the standard protocol (29). The specific probes used to reveal the α, β, and γ senDNAs are as described by Begel et al. (16).
Cloning and Sequencing Procedures.
The two primers used to amplify a fragment of the COX5 gene from P. anserina genomic DNA were specific N. crassa primers. They are located in highly conserved regions of the COX5 gene (30). Their sequences are, respectively, 5′-AGATGGGAGCAGATGCCCATGCAGGAGCAG-3′ and 5′-TTGCCACTCCTTGGTCATGGTGGCAGGAGGGGG-3′. Amplification was achieved by 35 cycles of touchdown PCR (92°C for denaturation, 66–62°C for hybridization, and 72°C for elongation). The 533-bp product obtained, used as a probe, revealed a 2.6-kb EcoRV genomic fragment. An EcoRV minibank was prepared and screened with the same probe, and a positive clone (more than approximately 1,800 were tested) was selected and sequenced. To complete the sequence of the region downstream of the COX5 coding sequence, an amplification product covering the end of the COX5 gene was obtained and used as a probe; it revealed a 1.6-kb MunI genomic fragment. This fragment was circularized, amplified according to the inverse PCR method, (31) and sequenced.
RNA Analysis.
Reverse transcription–PCR (RT-PCR) assays were performed to define the position of the introns and localize the polyadenylation site. Thirty micrograms of total RNA and 50 pM V(T)34 oligonucleotide (V = G, C, or A) were denatured and annealed. Reverse transcription was performed with 5 units of avian myeloblastosis virus (AMV) reverse transcriptase, 10 μmol of each dNTP in 1× AMV buffer at 42°C for 1 h. PCR amplification was carried out in 25 μl containing 1% of the cDNA obtained, 50 pM COX5-specific and V(T)34 primers, 250 μM each dNTP, and one unit of Pfu DNA polymerase (Stratagene) for 30 cycles.
Vector Constructions and Transformation.
The bacterial hygromycin B phosphotransferase gene (HPH), which confers resistance to hygromycin B, was cloned into the BamHI site of pUC18 to give pPH1. The 2.6-kb EcoRV fragment containing the COX5 gene and its flanking sequences was inserted into the SmaI site of pPH1, resulting in the plasmid pPH51. Plasmid pPH52 containing the COX5 gene deleted for 139 bp and interrupted by the insertion of the Tn5 bacterial BLE gene, which confers resistance to phleomycin (32), was obtained as described in Fig. 1A. Transformation experiments were conducted as previously described (33) except that the protoplast preparation was carried out in 2% (wt/vol) Glucanex (Novo Nordisk), a β-glucanase.
Figure 1.
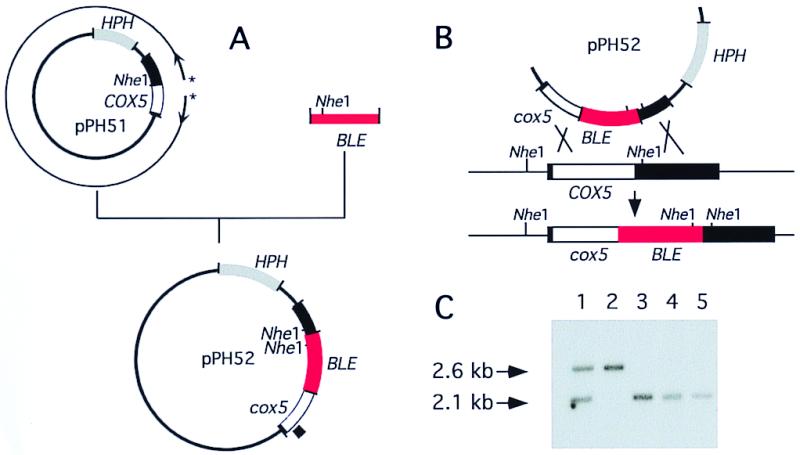
(A) Construction of the plasmid pPH52 designed for gene disruption. The plasmid pPH51 containing the HPH gene and a 2.6-kb fragment including the COX5 gene and its flanking sequences (filled bar) was amplified in a PCR experiment using two divergent primers (*). The amplification product (thick line) is deleted for 139 bp, including the start codon and the complete mitochondrial targeting sequence. It was ligated to the BLE cassette, giving plasmid pPH52. The disrupted COX5 gene is designed cox5∷BLE. (B) Diagram of integration by homologous recombination. (C) Southern analysis of transformants. Genomic DNA was digested by NheI (position shown in B) and hybridized with the probe located in the COX5 gene and indicated by a square outside pPH52 in A. Lane 1 is a primary transformant; lane 5 the recipient wild-type strain, lanes 2 to 4 are secondary monokaryotic progenies derived from the cross between the primary transformant and the wild-type strain. The 2.1- and 2.6-kb fragments respectively correspond to the wild-type and disrupted COX5 locus.
Respiration.
Spectral analysis of the cytochromes of whole cells was performed as previously described (34). The percentage of cyanide (CN)- and SHAM-sensitive respiration was measured on mycelia harvested from liquid medium in a Gilson oxygraph equipped with a Clark type O2 electrode. Assays were made in 0.6 M sorbitol/7.5 mM MgCl2/10 mM KH2PO4/10 mM imidazole, pH 7.4/0.2% BSA. KCN at 1 mM or SHAM at 2.5 mM was added, respectively, to inhibit the cytochrome c oxidase or the alternative pathway (35).
Flow Cytometric Analysis.
ROS elimination was measured by the production of dichlorofluorescein (DCF) derived from oxidization of the reduced diacetate form H2DCF-DA (36). Protoplasts were suspended at 106 per ml in a 0.6 M sorbitol/10 mM Tris⋅HCl, pH 7.5, solution and loaded with H2DCF-DA (80 μM). Measurements of the fluorescent DCF were performed every 15 min to follow ROS production in each strain. The nontoxicity of H2DCF-DA in our experimental conditions was monitored by the ability of the protoplasts to regenerate during the course of the experiment. Flow cytometric measurements were performed on a XL3C flow cytometer (Coulter, France). H2O2 was added at a 0.018% concentration, corresponding to the minimal concentration necessary to reduce the growth rate of all of the strains analyzed. KCN was added at 1 mM to inhibit the cytochrome c oxidase pathway.
Results
Construction of a Strain Inactivated for the COX5 Gene.
Cytochrome c oxidase is a large multiprotein complex (respiratory complex IV) located in the inner membrane of mitochondria. The three central catalytic subunits (Cox1p, Cox2p, and Cox3p) are mitochondrially encoded, whereas the others are encoded in the nucleus. From analysis of Saccharomyces cerevisiae and Neurospora crassa respiratory function, it appeared that Cox5p, subunit V of cytochrome c oxidase encoded by the COX5 gene, is essential for the assembly and/or the function of complex IV (30, 37). The P. anserina COX5 gene was isolated as described in Materials and Methods. It is present in only one copy in the genome (data not shown). The coding sequence of 834 bp is interrupted by two introns, 268 and 71 bp. Its analysis reveals a protein synthesized as a 168-amino acid precursor with a mitochondrial cleavage site predicted between amino acids 24 and 25 (motif RRAATT). The mature protein shows identity with subunit V of lower eukaryotes (45% with S. cerevisiae subunit A to 77% with N. crassa) and subunit IV of higher eukaryotes (20–28%).
To test the effects of the absence of functional respiratory complex IV, the COX5 gene was inactivated by replacement with a disrupted copy. Plasmid pPH52 designed to replace the wild-type allele by a defective copy is shown in Fig. 1A. It was used to transform the wild-type strain. To enrich for homologous recombination between the cox5∷BLE construction and the chromosomal COX5 gene, transformants resistant to phleomycin and sensitive to hygromycin were selected (Fig. 1B). Of 1,452 primary phleomycin-resistant transformants tested, 83 were found to be hygromycin-sensitive. Their analysis by Southern blotting revealed that one transformant contained both cox5∷BLE-disrupted and COX5 nuclei (Fig. 1C, lane 1). This primary transformant was crossed with the wild-type strain, and homokaryotic phleomycin-sensitive and -resistant spores were obtained. Southern blot analysis indicated that the sensitive spores contained a wild-type COX5 allele (Fig. 1C, lanes 3 and 4), whereas the resistant spores contained the disrupted copy (Fig. 1C, lane 2). From genetic analysis of about 100 asci, it was concluded that the COX5 gene is located at 12 centimorgans from the centromere and that the disrupted cox5∷BLE allele is recessive.
COX5 Gene Disruption Has Pleiotropic Effects.
The low-temperature spectrum of the cox5∷BLE strain is similar to that of the wild-type strain except for the absence of a peak at 607 nm characteristic of reduced cytochrome aa3 (data not shown). The absence of functional cytochrome c oxidase was confirmed by oxygraphic analysis. The respiration of the mutant was completely resistant to cyanide and sensitive to SHAM, an inhibitor of the alternative respiratory pathway. In contrast, the respiration of wild-type young mycelium is about 10% sensitive to SHAM and 90% sensitive to cyanide (Fig. 2). The cox5∷BLE mutation displays several phenotypic properties (Table 1). It leads to a severe alteration in germinating mycelium and to female sterility. It causes very thin and poorly colored growing mycelia. The mutant exhibits a thermosensitive phenotype at 35°C; its growth is also impaired at 18°C, but the impairment is less pronounced (65% of the growth at 27°C) than for the wild-type strain (43%). Its growth rate at 27°C is decreased to about half that of the wild-type strain.
Figure 2.

Effects of cyanide and SHAM on the respiration of COX5 and cox5∷BLE strains. Green arrow indicates the addition of the cells. Addition of KCN and SHAM (1 mM and 2.5 mM, respectively) is respectively indicated as red and blue arrows. The initial amount of oxygen present in the chamber corresponds to 100%; KCN and SHAM (when KCN did not completely block respiration) are respectively added when 60% and 30% of oxygen is still present. Without addition of inhibitors, 100% of the oxygen is consumed.
Table 1.
Phenotypic properties and lifespans of wild-type and mutant strains
| Genotype* | Growth rate,† cm/day
|
Female fertility | Longevity‡ | ||
|---|---|---|---|---|---|
| 27°C | 34°C§ | 18°C§ | |||
| COX5 | 0.68 ± 0.03 | 0.71 ± 0.02 | 0.29 ± 0.03 | Fertile | 11.4 ± 1.66 (20) |
| (105%) | (43%) | 10.6 ± 1.35 (20) | |||
| cox5∷BLE | 0.30 ± 0.04 | 0.11 ± 0.03 | 0.19 ± 0.03 | Sterile | >150 (12), 79 ± 14 (3) |
| (39%) | (65%) | >140 (8), 63 ± 28 (7) | |||
| cox5∷BLE[COX5] | 0.68 ± 0.08 | 0.76 ± 0.05 | 0.25 ± 0.05 | Fertile | 12.6 ± 4.65 (15) |
| (112%) | (37%) | 15.4 ± 7.41 (15) | |||
COX5, wild-type strain; cox5∷BLE, COX5-disrupted strain; cox5∷BLE[COX5], strain containing the disrupted COX5 gene and an ectopic COX5 copy.
† Growth rate are given as means ± SD. Growth rates were measured for 2 weeks on 30 subcultures of each strain.
‡ Mean lifespans are in cm ± SD. Number in parentheses indicate the number of subcultures that have stopped growing (roman type) and that are still living after 1½ year of culture (italic type). Upper value is mat+, lower one is mat−.
§ Growth rate of a strain at 34°C and 18°C is expressed in cm/day ± SD and as the percentage of the growth rate of the same strain at 27°C (in parenthesis).
Because most of the theories of aging implicate production of ROS (38), we compared cellular ROS production in the cox5∷BLE and mex16 mutants, which are both deficient in cytochrome c oxidase, with that in the wild-type strain. As shown in Fig. 3A, the intensity of fluorescence (indicating the amount of ROS elimination) was higher in COX5 than in the cox5∷BLE and mex16 protoplasts. Because H2DCF reduction to fluorescent DCF depends on the activity of a catalyst (peroxidase, cytochrome c, or Fe2+), we verified that the difference between the wild-type and the two mutant strains reflects a difference in ROS production and not in the concentration of the factors allowing H2DCF reduction. The intensity of fluorescence was therefore monitored after addition of H2O2. As shown on Fig. 3B, similar curves were obtained for the three strains, indicating that the decrease of fluorescence in the mutant strains is caused by a reduction in ROS production. Addition of 1 mM KCN in the assay medium reduces ROS amount in wild-type cells to a level similar to that of cox5∷BLE and mex16 cells (Fig. 3C), further confirming that this reduction is caused by the inactivation of cytochrome c oxidase.
Figure 3.

Measurement of ROS elimination in the different strains. COX5 (■), mex16 (♦), and cox5∷BLE (●) protoplasts from each strain were incubated with H2DCF diacetate, and the DCF fluorescence was measured every 15 min by flow cytometry. Cells with altered membranes (BET positives) were excluded from the analysis. Values of DCF fluorescence, expressed in arbitrary units, are average values calculated with at least 10,000 protoplasts. (A) ROS elimination in standard conditions. (B) ROS elimination in 0.018% H2O2 medium. (C) ROS elimination by COX5 protoplasts in the absence (blue curve) or in the presence (violet curve) of 1 mM KCN compared with the ROS elimination by cox5∷BLE protoplasts in the absence of KCN.
Inactivation of the COX5 Gene Leads to a Striking Increase of Lifespan and to a Stabilization of the Mitochondrial Chromosome.
Longevity of the cox5∷BLE strain was measured on five subcultures derived from six independent spores and compared with reference wild-type cultures. These experiments revealed a striking effect of COX5 gene inactivation. As shown in Table 1, the longevity of wild-type cultures is 11.4 ± 1.66 and 10.6 ± 1.35 cm according to the mat locus (21 to 28 days of culture), whereas 20 of 30 of the cox5∷BLE subcultures are more than 140 cm long (more than 1½ years of culture) and are still living. All of the five subcultures derived from one of the six spores analyzed demonstrated a growth arrest after 41 to 48.7 cm of culture (154 to 175 days). Five other subcultures issued from three different spores stopped growing between 62 and 112.8 cm (231 to 413 days).
Senescence in the wild-type strain is systematically correlated with the accumulation of circular, tandemly arranged mtDNA molecules (senDNAs). Three distinct classes of senDNAs (α, β, and γ) originating from three distinct regions of the mitochondrial chromosome can usually be observed (see refs. 10 and 11 for reviews). The mtDNA content of cox5∷BLE cultures was analyzed at various stages during growth. In no instance was mtDNA alteration observed, even after 160 cm of growth. The senescent state of the wild-type cultures is always paralleled by the accumulation of senDNA α and by a strong reduction of intact mitochondrial chromosome (Fig. 4, lane 3). The mtDNA content of 8 of the 10 cox5∷BLE subcultures (derived from 4 independent spores) that had stopped growing between 40 and 80 cm was analyzed 2 cm before the arrest of growth (Fig. 4, lanes 4, 6, and 7). Either no trace (Fig. 4, lane 4) or a very low amount of senDNA α and no trace of β senDNAs (Fig. 4, lane 6) was observable in the different cox5∷BLE senescent cultures, whereas variable amounts of γ senDNAs were recovered in all of the cultures (lane 7). It can be noticed that the accumulation of γ senDNAs (whose junction is show by an arrow in Fig. 4, lane 7) was not accompanied by the disappearance of intact mitochondrial chromosome, since the stoichiometry of the chromosomic fragments corresponding to the amplified region appears unaltered (see Fig. 4, ● and ♦, lanes 4, 6, and 7). Although senDNA α is barely detectable by hybridization in growing and nongrowing cox5∷BLE cultures, the 3′-5′ α junction (tandem or circular molecules) is always observed in PCR amplification experiments. This result means that accumulation of senDNAα is impaired in the mutant, whereas the generation of intron α DNA circles can still occur.
Figure 4.

Southern blot analysis of mtDNA extracted from young and senescent wild-type, cox5∷BLE, and cox5∷BLE[COX5] subcultures. DNA is digested by HaeIII. Lanes 1 and 2 respectively correspond to young cultures of wild-type and cox5∷BLE; lane 3, to a senescent wild-type culture; lanes 4, 6, and 7, to a cox5∷BLE culture that had stopped growing; and lane 5, to a senescent cox5∷BLE[COX5] culture carrying both the cox5∷BLE deletion and an ectopic integration of the COX5 gene. Hybridization was performed with probes specific for the regions of the mitochondrial chromosome from which senDNA α (lanes 1 to 5), senDNAs β (lane 6), and senDNAs γ (lane 7), respectively, originate. The black arrow indicates the expected size for senDNA α; ● corresponds to the encompassed HaeIII mtDNA fragments expected to hybridize with the α probe; ♦ corresponds to the encompassed HaeIII fragments expected to hybridize with the β and γ probes; and white arrow indicates an additional fragment generated by circularization of γ senDNA.
Reintroduction of the Wild-Type COX5 Allele Complements All of the Phenotypic Characteristics of the Mutant Strain.
Plasmid pPH1 containing the wild-type COX5 gene and the HPH reporter gene was used to transform the cox5∷BLE strain, and hygromycin-resistant transformants resulting from a nonhomologous integration of the vector were selected. The analysis of the cox5∷BLE[COX5] transformants revealed that the reintroduction of the COX5 gene reestablished a wild-type growth rate, respiratory function, mycelial aspect, and female fertility (Table 1). Interestingly, it also reestablished a wild-type lifespan and the correlation between senescence and accumulation of senDNAα (often accompanied by β and γ senDNAs) to a level comparable to wild-type senescent cultures. As in wild-type strains, this accumulation is paralleled by a strong decrease of intact mtDNA (Fig. 4, lane 5). The large variation in the lifespan values of the cox5∷BLE[COX5] transformant studied (Table 1) corresponds to few subcultures (3/30) presenting extended lifespan: 27, 34.9 and 36.9 cm. The COX5[COX5] progeny of a cross between wild-type and this mutant strain demonstrate similar variation (4/30 arrested after 31.9, 31.5, 25.8, and 22.5 cm). This result indicates that this variation likely results from an ectopic integration effect and not from an incomplete restoration of wild-type longevity.
Discussion
Causal Link Between Absence of Cytochrome c Oxidase and Longevity.
Although the physiological and molecular consequences of aging demonstrated a correlation between senescence, accumulation of mtDNA rearrangements, and mitochondrial cytochrome defects (see refs. 1–3 for reviews), particularly in filamentous fungi (see ref. 11 for a review), it was not possible until now to establish a causal relation to lifespan for any specific function. We believe that the results presented here constitute one of the first demonstrations that the respiratory function is causally related to aging. They agree with recent results that established that intron α is not essential for senescence (16) and unequivocally demonstrate that the “immortality” of the mex mutants lacking both intron α and the COX1 protein results from the lack of cytochrome c oxidase function. Because loss-of-function mutations in genes encoding essential cytochrome c oxidase subunits (COX1p in mex1 and mex16 mutants and COX5p in the cox5∷BLE mutant) considerably lengthen lifespan, it can be concluded that the normal respiratory function is one of the principal processes contributing to shortening lifespan. This result can explain why impairment of mitochondrial protein synthesis, caused by chloramphenicol treatment or the mitochondrial mutation capr1, greatly delays the onset of senescence (26, 34). The finding that the absence of a normal respiratory pathway results in a lengthened lifespan may seem paradoxical when compared with the data obtained in numerous organisms that show defects in cytochrome c oxidase associated with aging. It can also seem contradictory with the recent finding that in C. elegans, mutations in the mev-1 gene cause a severely decreased activity of succinate dehydrogenase cytochrome b and a shortened lifespan, indicating a causal link between restriction of the respiration pathway, increased superoxide production, and premature aging (8). These apparent contradictions are discussed below.
Mitochondrial Chromosome Integrity and Longevity.
In mammals, the assertion that age-related accumulation of mtDNA mutations plays a major role in aging is controversial (2, 19). However, it is obvious that the presence of nonfunctional mtDNA molecules exceeding a threshold concentration in obligate aerobes may result in death and that there is therefore an inherent relationship between the maintenance of mtDNA integrity and longevity (11, 39). This relationship is verified in the cox5∷BLE mutant, where the increased lifespan is accompanied by an increased stability of the mitochondrial genome.
In contrast to the wild-type strain, which displays a rapid and reproducible cessation of growth systematically associated with amplification of senDNA α and often of other senDNAs, the cessation of growth in the cox5∷BLE strain is not systematic; when it occurs it is significantly delayed and never associated with the accumulation of α or β senDNAs. Only variable γ senDNAs, in more or less abundant amounts, can be observed when mutant subcultures stop growing. The absence of senDNA α is not because of a notable reduction of α-spliced RNA molecules assumed to be an intermediate in the formation of senDNA α (12) and whose amount is similar in wild-type and mutant mitochondria (C. Sellem, personal communication). It appears that some steps necessary for the amplification of the intron α DNA circles, but also for amplification of β senDNAs, is compromised in the absence of normal respiration.
Recently, several nuclear mutants that do not amplify senDNA α when senescent have been described. One of them, grisea, is deficient for a transcription factor involved in the control of copper homeostasis (40); two others are affected in the cytoplasmic translational apparatus and contain high levels of β senDNAs (41). In contrast to these two last mutations hypothesized to modulate the expression of specific proteins involved in the specific accumulation of the different types of senDNAs, it seems that a more general mechanism affecting the integrity of the mitochondrial chromosome is modified in the cox5∷BLE strain, where the accumulation of all of the senDNAs seems to be compromised. This mechanism is either directly or indirectly related to respiratory metabolism.
The observation that cox5∷BLE subcultures stop growing without extensive mitochondrial DNA rearrangements and with a large concentration of intact chromosomal mtDNA raises a fundamental question because, except for the grisea mutant, in which reorganization of the mtDNA also appears to be reduced during aging (40), all cases of senescence reported until now in P. anserina are associated with gross mtDNA instabilities.
Respiratory-Dependent Parameters Controlling mtDNA Maintenance and Longevity.
The absence of cytochrome c oxidase in the cox5∷BLE strain results in a severe respiratory defect and leads to the use of a cyanide-insensitive respiratory pathway. In contrast to published results suggesting that in P. anserina the role of the alternative oxidase is assumed by the laccase protein (42), recent data obtained in our laboratory indicate that P. anserina, like N. crassa (43, 44) and Aspergillus nidulans (45), possesses an alternative oxidase related to that present in plants, protists, and some yeast species (see refs. 46 and 47 for reviews). This alternative oxidase is known to branch off the cytochrome pathway at ubiquinone and donates electrons directly to oxygen.
An important feature of this pathway is that it does not trap chemical or electrochemical energy and that electrons that flow down this pathway lose two of the three potential coupling sites. Some characteristics of the cox5∷BLE mutant (female sterility, reduced growth rate, alteration of the germination capacities, thin mycelium) are probably the consequences of this reduced level of ATP. However quantification of the total mass of a wild-type culture that stops growing around 10–15 cm and that of a mutant culture displaying a thin mycelium but growing more than 140 cm shows that the mutant cox5∷BLE strain is able to produce more mycelium than the wild-type strain. The increased mtDNA stability and longevity could be related to the deficiency in ATP production. Indeed, the idea that a slow rate of living caused by a reduced rate of energy metabolism results in an increased lifespan is currently proposed (48, 49). According to this hypothesis, the balance between the rate of molecular damage and that of its repair sets lifespan. A slower rate of living could lead to slower accumulation of unrepaired damage (50). In the same connection, a reduced metabolic rate could result in a slower production of reactive by-products of metabolism (such as ROS). Consistent with this hypothesis is the finding that the production of ROS is decreased in the cox5∷BLE strain. Because of this decrease or favored repair, the rate at which a number of genetic alterations accumulate (specially the senDNA species) could be considerably reduced in the mutant strain.
Besides a dramatic fall in ATP supply, the exclusive use of the alternate pathway may have important physiological consequences. In wild-type P. anserina, as in all organisms with a branched mitochondrial respiratory chain, numerous factors influence the positioning of electrons (44, 51). In general, it appears that the alternative oxidase is induced when the cytochrome pathway becomes restricted or inhibited either with specific inhibitors or by disruption of mitochondrial protein synthesis as shown in particular in N. crassa (44). Hypotheses about the function of the alternative oxidase are numerous. However, except for its role in supplying heat in thermogenic plant tissues, no proven function was demonstrated in nonthermogenic plants, protists, and fungi. It has been recently suggested that in addition to an “energy overflow” role, the alternative oxidase could also act as a defense system against oxidative damage (46, 52, 53) and may play a protective role in mitochondria by preventing production of harmful reactive oxygen species. Its constitutive expression in the cox5∷BLE strain could then result in better maintenance of the integrity of the mitochondrial chromosome. This hypothesis presents common points with the above “rate of living” hypothesis and is also in accordance with our results showing a decrease of generation of ROS with cox5∷BLE and mex16 strains. It could also explain why a reduction of the cytochrome pathway in organisms without alternative oxidase such as C. elegans (8, 54) causes increased ROS production and premature aging, whereas in other organisms such as P. anserina, it would cause a decrease in superoxide levels and an extended lifespan. It must be noted, however, that in P. anserina (55) as in Neurospora (56) wild-type strains, senescence is paralleled by switching from cytochrome c oxidase-mediated to cyanide-resistant respiration. This observation indicates that whereas the constitutive expression of the alternative pathway seems to prevent or to considerably delay the senescence syndrome, its expression appears to be insufficient to reverse this syndrome once it is set up.
A number of studies in different cell types have shown that alteration of the mitochondrial functions results in the induction of nuclear genes via an intracellular signaling pathway termed retrograde regulation. It seems that this kind of regulation most likely operates when mitochondrial respiratory functions are compromised (see ref. 57 for a review). Recently, Kirchman et al. (58) demonstrated that in S. cerevisiae activation of the retrograde response is correlated with an extension of lifespan. In the same way, Felkai et al. (9) proposed that in C. elegans, the clk-1 gene, which is a homologue of the yeast COQ7 (a regulator of mitochondrial function), could control lifespan by means of its contribution to the cross-talk between mitochondria and nucleus. As mentioned above, the alternative oxidase is induced when the cytochrome pathway is compromised. This induction constitutes an interesting parallel to the retrograde signaling in yeast, and it is likely that in addition to the alternate oxidase, this type of regulation influences other genes. These genes are expected to be involved in the adjustment of the metabolism to the absence of the cytochrome pathway. Another hypothesis to account for the increased longevity of the cox5∷BLE strain would therefore be that another metabolic function affected by the retrograde response plays a major role in the control of mitochondrial chromosome maintenance and longevity.
We think that the results of our investigations may have important implications for understanding the aging process of various biological systems, including more complex multicellular organisms, because relationships between mitochondrial function and aging have been recently demonstrated in other model systems (8, 58). Although there are probably “multiple avenues that may lead to senescence and these avenues may be different in different species” (6, 59), it is generally admitted that some of them are common to the different systems. The causal link established here between respiration and aging could be such a conserved factor operational over a large diversity of organisms.
Acknowledgments
We thank G. Dujardin, L. Sperling, M. Picard, B. Albert, C. Sellem, and Y. d'Aubenton-Carafat for their helpful discussions and comments on the manuscript. This work was supported by grants from the Centre National de la Recherche Scientifique, the Association Française contre les Myopathies, and the Ministère de l'Education Nationale de l'Enseignement et de la Recherche (ACC-SV4 no. 9504063).
Abbreviations
- ROS
reactive oxygen species
- senDNA
circular multimeric mtDNA associated with senescence
- SHAM
salicylhydroxamic acid
- DCF
dichlorofluorescein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The nucleotide sequence of the Podospora anserina COX5 gene has been deposited in the GenBank database (accession no. AJ245552).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.070501997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.070501997
References
- 1.Wallace D C. Sci Am. 1997;277(2):40–47. doi: 10.1038/scientificamerican0897-40. [DOI] [PubMed] [Google Scholar]
- 2.Nagley P, Wei Y H. Trends Genet. 1998;14:513–517. doi: 10.1016/s0168-9525(98)01580-7. [DOI] [PubMed] [Google Scholar]
- 3.Wallace D C. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 4.Davies K J. Biochem Soc Symp. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 5.Sohal R S, Weindruch R. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin G M, Austad S N, Johnson T E. Nat Genet. 1996;13:25–34. doi: 10.1038/ng0596-25. [DOI] [PubMed] [Google Scholar]
- 7.Wallace D C, Melov S. Nat Genet. 1998;19:105–106. doi: 10.1038/448. [DOI] [PubMed] [Google Scholar]
- 8.Ishii N, Fujii M, Hartman P S, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. Nature (London) 1998;394:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- 9.Felkai S, Ewbank J J, Lemieux J J, Labbe C, Brown G G, Hekimi S. EMBO J. 1999;18:1783–1792. doi: 10.1093/emboj/18.7.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dujon B, Belcour L. In: Mitochondrial DNA Instabilities and Rearrangements in Yeasts and Fungi. Berg D E, Howe M M, editors. Washington, DC: Am. Soc. Microbiol.; 1989. pp. 861–878. [Google Scholar]
- 11.Griffiths A. Annu Rev Genet. 1992;26:351–372. doi: 10.1146/annurev.ge.26.120192.002031. [DOI] [PubMed] [Google Scholar]
- 12.Sellem C H, Lecellier G, Belcour L. Nature (London) 1993;366:176–178. doi: 10.1038/366176a0. [DOI] [PubMed] [Google Scholar]
- 13.Belcour L, Vierny C. EMBO J. 1986;5:609–614. doi: 10.1002/j.1460-2075.1986.tb04254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sainsard-Chanet A, Begel O. Nucleic Acids Res. 1990;18:779–783. doi: 10.1093/nar/18.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vierny C, Keller A M, Begel O, Belcour L. Nature (London) 1982;297:157–159. doi: 10.1038/297157a0. [DOI] [PubMed] [Google Scholar]
- 16.Begel O, Boulay J, Albert B, Dufour E, Sainsard-Chanet A. Mol Cell Biol. 1999;19:4093–4100. doi: 10.1128/mcb.19.6.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson F B, Sinclair D A, Guarente L. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 18.Papa S. Biochim Biophys Acta. 1996;1276:87–105. doi: 10.1016/0005-2728(96)00077-1. [DOI] [PubMed] [Google Scholar]
- 19.Lightowlers R N, Jacobs H T, Kajander O A. Trends Genet. 1999;15:91–93. doi: 10.1016/s0168-9525(98)01684-9. [DOI] [PubMed] [Google Scholar]
- 20.Chinnery P F, Turnbull D M. Lancet. 1999;354, Suppl. 1:SI17–SI21. doi: 10.1016/s0140-6736(99)90244-1. [DOI] [PubMed] [Google Scholar]
- 21.Schapira A H. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- 22.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 23.Rizet G, Engelmann C. Rev Cytol Biol Veg. 1949;11:201–304. [Google Scholar]
- 24.Esser K. In: Podospora anserina. King R C, editor. Vol. 1. New York: Plenum; 1974. pp. 531–551. [Google Scholar]
- 25.Rizet G. Rev Cytol Biol Veg. 1952;13:51–92. [Google Scholar]
- 26.Belcour L, Begel O. J Gen Microbiol. 1980;119:505–515. [Google Scholar]
- 27.Lecellier G, Silar P. Curr Genet. 1994;25:122–123. doi: 10.1007/BF00309536. [DOI] [PubMed] [Google Scholar]
- 28.Cummings D J, Belcour L, Grandchamp C. Mol Gen Genet. 1979;171:229–238. doi: 10.1007/BF00267577. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 30.Sachs M S, Bertrand H, Metzenberg R L, RajBhandary U L. Mol Cell Biol. 1989;9:566–577. doi: 10.1128/mcb.9.2.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang S H, Hu Y Y, Wu C H, Holcenberg J. Nucleic Acids Res. 1990;18:1922. doi: 10.1093/nar/18.7.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calmels T, Parriche M, Durand H, Tiraby G. Curr Genet. 1991;20:309–314. doi: 10.1007/BF00318520. [DOI] [PubMed] [Google Scholar]
- 33.Bergès T, Barreau C. J Gen Microbiol. 1989;135:601–604. doi: 10.1099/00221287-135-3-601. [DOI] [PubMed] [Google Scholar]
- 34.Belcour L, Begel O. Mol Gen Genet. 1977;153:11–21. doi: 10.1007/BF01035991. [DOI] [PubMed] [Google Scholar]
- 35.Siedow J N, Berthold D A. Physiol Plant. 1986;66:569–573. [Google Scholar]
- 36.Henderson L M, Chappell J B. Eur J Biochem. 1993;217:973–980. doi: 10.1111/j.1432-1033.1993.tb18328.x. [DOI] [PubMed] [Google Scholar]
- 37.Cumsky M G, McEwen J E, Ko C, Poyton R O. J Biol Chem. 1983;258:13418–13421. [PubMed] [Google Scholar]
- 38.Sohal R S, Orr W C. Aging (Milano) 1998;10:149–151. [PubMed] [Google Scholar]
- 39.Belcour L, Begel O, Picard M. Proc Natl Acad Sci USA. 1991;88:3579–3583. doi: 10.1073/pnas.88.9.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borghouts C, Kimpel E, Osiewacz H D. Proc Natl Acad Sci USA. 1997;94:10768–10773. doi: 10.1073/pnas.94.20.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silar P, Koll F, Rossignol M. Genetics. 1997;145:697–705. doi: 10.1093/genetics/145.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frese D, Stahl U. Mech Ageing Dev. 1992;65:277–288. doi: 10.1016/0047-6374(92)90041-b. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Ritzel R G, McLean L L, McIntosh L, Ko T, Bertrand H, Nargang F E. Genetics. 1996;142:129–140. doi: 10.1093/genetics/142.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambowitz A M, Sabourin J R, Bertrand H, Nickels R, McIntosh L. Mol Cell Biol. 1989;9:1362–1364. doi: 10.1128/mcb.9.3.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirimura K, Yoda M, Usami S. Curr Genet. 1999;34:472–477. doi: 10.1007/s002940050422. [DOI] [PubMed] [Google Scholar]
- 46.Wagner A M, Moore A L. Biosci Rep. 1997;17:319–333. doi: 10.1023/a:1027388729586. [DOI] [PubMed] [Google Scholar]
- 47.McIntosh L. Plant Physiol. 1994;105:781–786. doi: 10.1104/pp.105.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lints F A. Gerontology. 1989;35:36–57. doi: 10.1159/000212998. [DOI] [PubMed] [Google Scholar]
- 49.Finch C. Longevity, Senescence and the Genome. Chicago: Univ. of Chicago Press; 1990. [Google Scholar]
- 50.Perez-Campo R, Lopez-Torres M, Cadenas S, Rojas C, Barja G. J Comp Physiol B. 1998;168:149–158. doi: 10.1007/s003600050131. [DOI] [PubMed] [Google Scholar]
- 51.Sluse F E, Jarmuszkiewicz W. Braz J Med Biol Res. 1998;31:733–747. doi: 10.1590/s0100-879x1998000600003. [DOI] [PubMed] [Google Scholar]
- 52.Popov V N, Simonian R A, Skulachev V P, Starkov A A. FEBS Lett. 1997;415:87–90. doi: 10.1016/s0014-5793(97)01099-5. [DOI] [PubMed] [Google Scholar]
- 53.Maxwell D P, Wang Y, McIntosh L. Proc Natl Acad Sci USA. 1999;96:8271–8276. doi: 10.1073/pnas.96.14.8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsuo M. Comp Biochem Physiol Comp Physiol. 1993;105:653–658. doi: 10.1016/0300-9629(93)90264-5. [DOI] [PubMed] [Google Scholar]
- 55.Belcour L, Begel O. Mol Gen Genet. 1978;163:113–123. [Google Scholar]
- 56.Rieck A, Griffiths A J, Bertrand H. Can J Genet Cytol. 1982;24:741–759. doi: 10.1139/g82-080. [DOI] [PubMed] [Google Scholar]
- 57.Poyton R O, McEwen J E. Annu Rev Biochem. 1996;65:563–607. doi: 10.1146/annurev.bi.65.070196.003023. [DOI] [PubMed] [Google Scholar]
- 58.Kirchman P A, Kim S, Lai C Y, Jazwinski S M. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Orr W C. Dev Genet. 1996;18:93–98. doi: 10.1002/(SICI)1520-6408(1996)18:2<93::AID-DVG1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]