

Abstract
Self-renewal of Bcr-Abl+ chronic myeloid leukemia (CML) cells is sustained by a nuclear activated serine/threonine-(S/T) unphosphorylated β-catenin. Although β-catenin can be tyrosine (Y)-phosphorylated, the occurrence and biological relevance of this covalent modification in Bcr-Abl-associated leukemogenesis is unknown. Here we show that Bcr-Abl levels control the degree of β-catenin protein stabilization by affecting its Y/S/T-phospho content in CML cells. Bcr-Abl physically interacts with β-catenin, and its oncogenic tyrosine kinase activity is required to phosphorylate β-catenin at Y86 and Y654 residues. This Y-phospho β-catenin binds to the TCF4 transcription factor, thus representing a transcriptionally active pool. Imatinib, a Bcr-Abl antagonist, impairs the β-catenin/TCF-related transcription causing a rapid cytosolic retention of Y-unphosphorylated β-catenin, which presents an increased binding affinity for the Axin/GSK3β complex. Although Bcr-Abl does not affect GSK3β autophosphorylation, it prevents, through its effect on β-catenin Y phosphorylation, Axin/GSK3β binding to β-catenin and its subsequent S/T phosphorylation. Silencing of β-catenin by small interfering RNA inhibited proliferation and clonogenicity of Bcr-Abl+ CML cells, in synergism with Imatinib. These findings indicate the Bcr-Abl triggered Y phosphorylation of β-catenin as a new mechanism responsible for its protein stabilization and nuclear signalling activation in CML.
Keywords: Bcr-Abl, β-catenin protein stability, CML
Introduction
The WNT/β-catenin signalling promotes stem cell renewal by coordinating changes in gene expression and cell adhesion (Reya and Clevers, 2005). The key player in this network is β-catenin, which acts as a nuclear coactivator of the TCF/LEF (T-cell factor/lymphoid enhancer factor) transcription factors or as a structural adaptor protein at cell adherens junctions (Nelson and Nusse, 2004; Harris and Peifer, 2005).
WNT factors are cysteine-rich lipid-modified proteins that bind to several Frizzled (FZD) receptors. Under physiological conditions, WNT proteins accumulate β-catenin by inhibiting its glycogen synthase kinase 3 (GSK3)-dependent serine/threonine (S/T) phosphorylation on specific N-terminal residues. As GSK3 targets β-catenin for ubiquitination and proteasome degradation (Klymkowsky, 2005), detection of a nuclear S/T-nonphospho β-catenin is a hallmark of its transcriptional activation. Expression of β-catenin/TCF-induced cell cycle regulators (such as c-Myc and cyclin D1) is crucial for maintaining cell homeostasis in normal proliferating tissues, such as colon and skin (Pinto and Clevers, 2005). The WNT/β-catenin cascade has also pivotal roles in the self-renewal of hematopoietic stem cells (HSC), as a forced expression of a nondegradable β-catenin (S33-mutant) is sufficient to perpetuate themselves in vitro and sustain bone marrow reconstitution in vivo (Reya et al, 2003). Whereas the loss of WNT responsiveness allows multilineage differentiation of HSC (Baba et al, 2005), this link appears uncoupled in several human malignancies as a result of an increased β-catenin expression and protein stabilization in committed myeloid and lymphoid progenitors (Staal and Clevers, 2005).
Chronic myeloid leukemia (CML) begins as an indolent disease when an HSC expresses the oncogenic tyrosine kinase Bcr-Abl, which confers a proliferative advantage to its progeny. At this phase, Bcr-Abl does not interfere with HSC differentiation, and its levels decrease in committed progenitors (Daley, 2004; Huntly et al, 2004). An Abl kinase-selective inhibitor, imatinib mesylate (CGP57148B, STI571, and Gleevec/Glivec), represents the treatment of choice for CML inducing remissions in most CML patients in chronic phase (CP) (Goldman and Melo, 2003; Deininger et al, 2005). However, mutations in the catalytic site of Bcr-Abl or bcr-abl gene amplification (le Coutre et al, 2000; Krause and Van Etten, 2005) can select imatinib-resistant clones during CML progression. Blast crisis (BC) represents the terminal outcome of CML and was recently correlated with the expansion of committed granulocyte–macrophage precursors with persistence of high Bcr-Abl mRNA levels and accumulation of a nuclear S/T-nonphospho β-catenin (Jamieson et al, 2004).
In this report, we investigated the molecular causes underlying β-catenin deregulation in CML and identified Bcr-Abl-mediated Y phosphorylation of β-catenin as a cause for its increased protein stability and transcriptional signalling activity.
Results
GSK3 inhibition promotes proliferation and nuclear accumulation of a Y-phospho β-catenin in Bcr-Abl+ BC-CML cells
Nuclear accumulation of β-catenin is a hallmark of WNT signalling activation. We tested whether Bcr-Abl+ CML cells could contain an intact WNT/GSK3 pathway by using SB-216763, a GSK3 inhibitor, which promotes β-catenin stabilization. Proliferation of a Bcr-Abl+ BC cell line (Ku812) and of fresh BMMC isolated from a BC-CML patient was increased by SB-216763 (Figure 1A), correlating with an impaired Y216 autophosphorylation of GSK3β (Cole et al, 2004) (Figure 1B, α p-Y GSK3β, b,d versus a,c). These data indicated the integrity of an APC/Axin/GSK3 pathway, suggesting that the rate of stabilized β-catenin was suboptimal in these Bcr-Abl+ cells or still exogenously inducible.
Figure 1.
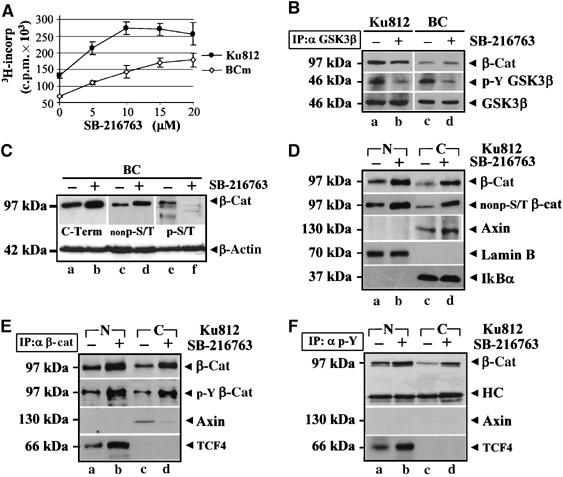
SB-216763 promotes nuclear accumulation of Y-phospho β-catenin and BC-CML cell growth. (A) Proliferation of Ku812 and fresh BC cells treated with the indicated doses (μM) of SB-216763 for 24 h. Errors bars indicate s.d. of triplicate experiments. (B) GSK3β (IP: αGSK3β) was immunoprecipitated from Ku812 (a and b) and BC (c and d) cells treated with DMSO (−) or 5 μM SB-216763 (+) for 2 h and immunoblotted with an anti-GSK3β antibody or an antibody against its phospho-Y216 residue. β-Catenin was also shown. (C) Upon 8 h of treatment with 5 μM SB-216763, whole lysates from BC cells were analyzed for total β-catenin levels with a C-terminal antibody (C-Term). β-Catenin was also probed with two antibodies recognizing its specific S/T-unphospho- (nonp-S/T) or phosphorylated (p-S/T) form. Total β-actin levels were indicated as a loading control. (D) Nuclear (N) and cytosolic (C) extracts from Ku812 cells cultured with DMSO (−) or 5 μM SB-216763 (+) for 8 h were analyzed with antibodies against β-catenin, its specific S/T-nonphospho form (nonp-S/T), nuclear Lamin B and cytoplasmic IkBα. (E, F) Nuclear (N) and cytosolic (C) extracts of Ku812 cells treated as described above were immunoprecipitated with an anti-β-catenin (IP: α β-cat, E) or an anti-phosphotyrosine (IP: α p-Y, F) antibody and then immunoblotted as indicated (HC=heavy chain of IgG antibody used for immunoprecipitation).
To verify the effect of SB-216763 on GSK3-mediated phosphorylation, β-catenin was probed with an antibody against its N-terminal S/T-phospho residues (Ser33, Ser37 and Thr41) (α p-S/T). We also used an antibody specific for β-catenin that is not phosphorylated on Ser33 and Ser37 (α nonp-S/T) and a pan C-terminal antibody (α C-term) that does not distinguish between phosphorylated and dephosphorylated β-catenin (Figure 1C). SB-216763 enhanced β-catenin accumulation in fresh BC cells (α C-term, b compared to a), increasing its S/T-nonphospho levels (α nonp-S/T, d versus c). Consistent with GSK3 kinase inhibition, the S/T-phospho pool of β-catenin was reduced by SB-216763 (α p-S/T, f compared to e). Figure 1D shows that the proliferative effect of SB-216763 correlated with increased nuclear and cytoplasmic levels of β-catenin (α β-cat, b,d versus a,c), further identified as transcriptionally active (α nonp-S/T, b,d versus a,c). β-Catenin could also be immunoprecipitated in a Y-phospho form from enriched nuclear and cytosolic Ku812 extracts (Figure 1E) (α p-Y β-cat, a and c), and the SB-216763 released β-catenin was highly phosphorylated on tyrosine residue(s) (α p-Y β-cat, b,d versus a,c). Interestingly, Y-phospho β-catenin was associated with the nuclear transcription factor TCF4 (Figure 1E and F, α TCF4, b versus a) and, to a lesser extent, to Axin in the cytosol (Figure 1E, α Axin, d versus c). As Axin was never immunoprecipitated using an antiphosphotyrosine antibody (Figure 1F, α Axin), as instead observed for β-catenin or TCF4, the Y-phospho β-catenin could be considered a true Axin-uncomplexed fraction in Bcr-Abl+ CML cells either in the presence or in absence of a WNT signal.
β-Catenin accumulates in CML as a Y-phosphoprotein coupled to Bcr-Abl
Our initial evidence that β-catenin accumulation might correlate with Bcr-Abl protein levels derived from an analysis of Bcr-Abl and β-catenin expression in BMMC from four CML patients in CP and six in BC. Figure 2A presents the results obtained in a representative BC patient and in the CP patient with the highest Bcr-Abl expression, the other CP patients being negative for Bcr-Abl in total cell lysates. Equal numbers of cell (5 × 105) were analyzed. As an expected feature of CML progression (Barnes et al, 2005), higher expression of Bcr-Abl (α Bcr-Abl) in BC-CML (lanes e and f) and Ku812 (lanes g and h) cells compared to CP-CML (lanes c and d) was detected and correlated with higher levels of total (α β-cat) and transcriptionally active (α nonp-S/T β-cat) β-catenin. Whereas β-catenin accumulation in BC cells could be accounted for by restored mRNA transcription (Jamieson et al, 2004), we observed that imatinib reduced the active S/T-nonphospho pool of β-catenin, pointing to a Bcr-Abl-mediated β-catenin stabilization and transcriptional activation.
Figure 2.
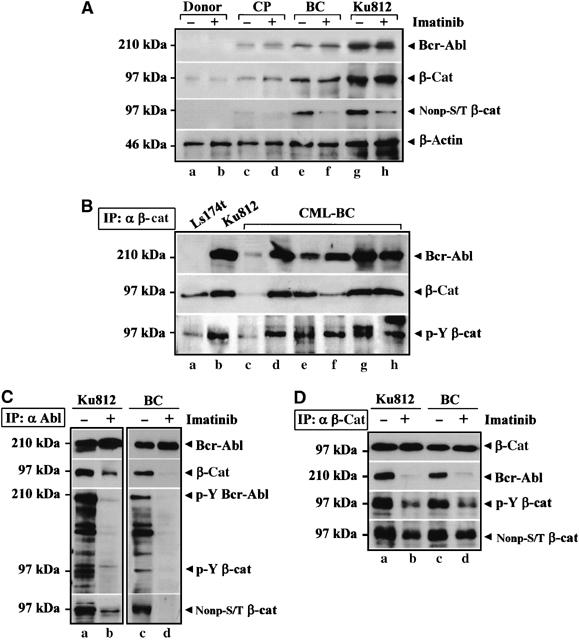
β-Catenin is coupled to Bcr-Abl and accumulates in a Y-phosphoform in CML. (A) A representative sample of normal (donor) or CML-BMMC isolated from a CP-CML (CP) or BC-CML (BC) patient was compared to Ku812 cells. Total cell lysates (±1 μM Imatinib for 2 h) were immunoblotted with the indicated antibodies. (B) Ls174T (a), Ku812 (b) and fresh BC-CML (c–h) cells were immunoprecipitated with an anti-β-catenin antibody (IP: α β-cat). Immunocomplexes were analyzed by SDS–PAGE for Bcr-Abl and β-catenin. The same blot was stripped and analyzed for Y-phospho β-catenin (p-Y β-cat). (C) Anti-Abl immunoprecipitates (IP: α Abl) from Ku812 (a and b) and BC-CML (c and d) cells treated with DMSO (−) or 1 μM Imatinib (+) for 2 h were analyzed for Bcr-Abl and β-catenin levels. The same blot was reprobed by using an anti-phosphotyrosine antibody indicating two proteins of 210 kDa (p-Y Bcr-Abl) and 97 kDa (p-Y β-cat) and with a nonphospho-S/T β-catenin antibody (nonp-S/T β-cat). (D) Ku812 (a and b) and BC-CML (c and d) lysates were immunoprecipitated with an anti-β-catenin antibody (IP: α β-cat) and probed with the indicated antibodies.
As β-catenin can interact directly with oncogenic tyrosine kinases such as c-MET (Hiscox and Jiang, 1999), RON (Danilkovitch-Miagkova et al, 2001) and c-erbB-2 (Kanai et al, 1995), we investigated a potential association of Bcr-Abl with β-catenin (Figure 2B) in Ku812 (b) and BC-CML (lanes c–h) cells. The CRC Ls174T cells (a), which contain high β-catenin levels, were included as negative controls for Bcr-Abl expression. Cells were immunoprecipitated with an anti-β-catenin antibody (IP: α β-cat). Bcr-Abl (α Bcr-Abl) co-precipitated with β-catenin (α β-cat), which was Y phosphorylated (α p-Y β-cat). As shown in Figure 2C, total lysates from Ku812 and fresh BC-CML (BC) cells treated with dimethyl sulfoxide (DMSO) (−) or imatinib (+) were also immunoprecipitated with an anti-Abl antibody (IP: α Abl). β-Catenin was detected in the anti-Abl immunoprecipitates and imatinib prevented both Bcr-Abl (α p-Y Bcr-Abl) and β-catenin Y activation (α p-Y β-cat), decreasing their physical interaction (α β-cat, a,c versus b,d). The finding that an S/T-nonphospho β-catenin is coupled to Bcr-Abl (α nonp-S/T, a and c) suggests that Bcr-Abl recruits a signalling competent pool of β-catenin. In Figure 2D, we performed reciprocal anti-β-catenin immunoprecipitates (IP: α β-cat). Whereas a comparable amount of β-catenin was detected in all samples (α β-cat), the co-precipitation of Bcr-Abl and β-catenin was impaired in cells treated with imatinib (α Bcr-Abl, b,d compared to a,c) as well as Y phosphorylation of β-catenin (α p-Y β-cat, b,d versus a,c) and its nonphospho-S/T levels (α nonp-S/T, b,d versus a,c).
These data show a functional link between the increased expression of Bcr-Abl and the accumulation of a nuclear Y-phospho β-catenin in the BC phase of CML.
Bcr-Abl kinase activity is required to trigger Y phosphorylation of β-catenin
β-Catenin is a target for several members of the Src family tyrosine kinase (Piedra et al, 2001; Coluccia et al, 2006), which are known to contribute to Bcr-Abl+ leukemogenesis (Tipping et al, 2004). Therefore, β-catenin might be phosphorylated by either Bcr-Abl itself and/or its proximal Src effectors in human CML cells.
A search for Src kinase inhibitors not active against Bcr-Abl identified SU6656 (IC50 of 20 nM for c-Src). In Figure 3A, Ku812 cells treated with DMSO (a), 1 μM SKI-606 (b), 1 μM imatinib (c) or 5 μM SU6656 (d) were immunoprecipitated with an anti-Abl antibody (IP: α Abl). Whereas SKI-606 and imatinib inhibited Bcr-Abl (α p-Y Bcr-Abl, b and c) and Src (α p-Y Src, b and c) Y phosphorylation, SU6656 selectively reduced the activation of Src kinases bound to Bcr-Abl (α p-Y Src, d) without affecting Bcr-Abl autoactivation (α p-Y Bcr-Abl, d). The ability of SKI-606, imatinib or SU6656 kinase inhibitors to prevent β-catenin Y phosphorylation was also analyzed by immunoprecipitating Ku812 cells with a C-terminal β-catenin antibody (Figure 3B). β-Catenin Y activation observed in the untreated control (α pY β-cat, a) was inhibited by SKI-606 (b), a dual Src/Abl inhibitor, or Imatinib (c), a specific Abl antagonist, whereas SU6656 (d), a Src family kinase inhibitor, had a minor effect. Interestingly, SU6656 was unable to disrupt the Bcr-Abl/β-catenin association, as instead observed for SKI-606 and Imatinib (α Bcr-Abl, d versus c and b). Imatinib reduced the levels of Y-phospho Src associated with Bcr-Abl to a greater extent than SU6656, suggesting a downstream effect of Bcr-Abl on c-Src activation.
Figure 3.
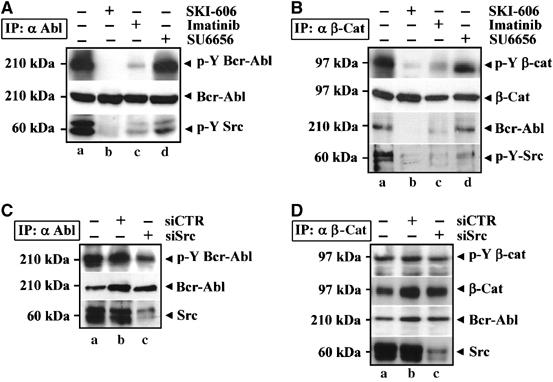
Tyrosine kinase activity of Bcr-Abl is required to trigger β-catenin Y phosphorylation in BC-CML cells. (A) Ku812 cells were treated with DMSO (a), SKI-606 (b), Imatinib (c) or SU6656 (d) for 2 h and then immunoprecipitated with an anti-Abl antibody (IP: α Abl). Protein levels and Y phosphorylation of Bcr-Abl (p-Y Bcr-Abl) were analyzed by immunoblotting. Activation of c-Src was also probed by using a specific anti-phospho-Y418 antibody (p-Y Src). (B) Anti-β-catenin immunoprecipitates (IP: α β-cat) from Ku812 cells treated with DMSO (a), SKI-606 (b), Imatinib (c) or SU6656 (d) for 2 h were immunoblotted with the indicated antibodies. (C) Ku812 cells were transiently transfected with siRNAs for c-Src (c), a control siRNAs pool (b) or oligofectine alone (a). After 48 h, cells were immunoprecipitated with an anti-Abl antibody (IP: α Abl) and analyzed for c-Src content and Y-phospho-Bcr-Abl (p-Y Bcr-Abl). Levels of Bcr-Abl were shown as loading control for the immunoprecipitation. (D) Lysates obtained from Ku812 described were immunoprecipitated with an anti-β-catenin antibody (IP: α β-cat) and probed with the indicated antibodies.
To confirm these data further, we targeted Src expression by using a mixture of four selected double-stranded small interfering RNA (siRNA) directed against Src. As shown in Figure 3C, this procedure inhibited 80% of Src associated with Bcr-Abl (α Src, c versus b). Although the autoactivation of Bcr-Abl (α p-Y Bcr-Abl, c) appeared slightly decreased by silencing of Src, this did not inhibit the binding of Bcr-Abl to β-catenin and its Y phosphorylation (Figure 3D).
These findings cannot exclude the contribution of other Src-related kinases, which can immunoprecipitate with the Bcr-Abl/β-catenin complex, but they indicate that an active Bcr-Abl tyrosine kinase is required to trigger Y phosphorylation of β-catenin in CML cells. They also indicate that Src phosphorylation in CML cells is dependent on Bcr-Abl kinase activity, but not vice versa.
Bcr-Abl phosphorylates β-catenin at Y86 and Y654 and promotes its protein stabilization
Tyrosine residues of β-catenin that can be phosphorylated by Src kinases were identified as Y86, Y142 and Y654 (Roura et al, 1999; Piedra et al, 2003). To validate the effects of Bcr-Abl on β-catenin Y phosphorylation and stability, human embryonic kidney (HEK293) T cells were transiently cotransfected with Bcr-Abl and histidine (His)-tagged plasmids encoding for wild-type (WT) β-catenin or its specific Y-to-F mutants Y86F, Y142F, Y654F and the double-mutant Y86F-Y654F (Figure 4A). Analysis of anti-His immunoprecipitates (IP: α His) confirmed the Bcr-Abl/β-catenin association also in these cells (α Bcr-Abl, b–f) and showed that β-catenin was differently modified on its Y/S/T residues in the presence of the oncogene. In fact, the exogenous WT β-catenin resulted phosphorylated on S/T (α p-S/T β-cat: a), but not on Y-residues (α p-Y β-cat: a) when transfected alone. The coexpression of the Bcr-Abl (α Bcr-Abl, b–f) prevented the S/T phosphorylation of β-catenin (α p-S/T β-cat: b) by triggering its Y-modification (α p-Y β-cat: b). Both Y86F (α p-Y β-cat: c) and Y654F (α pY β-cat: e) mutants were less Y phosphorylated (approximately 50%) in presence of Bcr-Abl than the Y142F (α p-Y β-cat: d) or WT β-catenin (α p-Y β-cat: b), whereas the Y phosphorylation of the double-mutant Y86F-Y654F (α p-Y β-cat: f) was completely inhibited. Interestingly, a lower degree of S/T phosphorylation of the Y654F mutant compared to Y86F (α p-S/T β-cat: e versus c) correlated with a decreased amount of Axin detectable in Y654F-immunoprecipitates (α Axin: e compared to c), indicating that the Bcr-Abl-mediated phosphorylation of β-catenin Y86 could be more efficient than Y654 in impairing its binding affinity to Axin.
Figure 4.
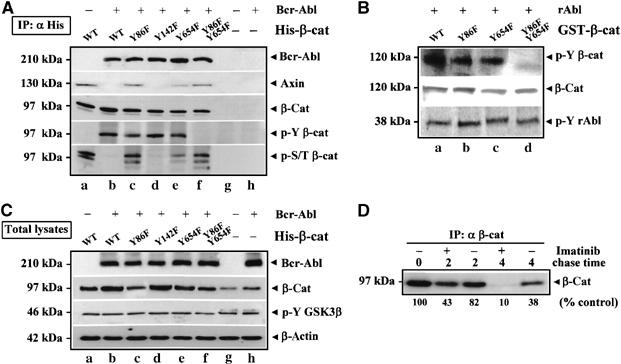
Bcr-Abl promotes β-catenin Y86-Y654 phosphorylation and stability. (A) HEK293T cells were transiently transfected with Bcr-Abl (b and h) or an empty vector (g). A histidine (His)-tagged plasmid encoding for WT β-catenin (A) and its tyrosine-to-phenylalanine (Y-to-F) mutants Y86F (c), Y142F (d), Y654F (e) and Y86F-Y654F (f) were coexpressed with Bcr-Abl (c–f). After 48 h, transfected cells were immunoprecipitated with an anti-His antibody (IP: α His) and blotted for Bcr-Abl, Axin and β-catenin. The same blot was stripped and assessed for Y-phospho β-catenin (p-Y-β-cat). Levels of phospho-S/T β-catenin (p-S/T β-cat) are also shown. (B) GST-purified WT β-catenin (A,B) and the indicated Y-to-F fusion proteins Y86F (C), Y654F (D) and Y86F-Y654F (E) were phosphorylated by recombinant Abl kinase in vitro. Samples were analyzed by immunoblotting with the indicated antibodies. (C) Total lysates from cells transfected as described in (A) were blotted as indicated. (D) Ku812 cells were labeled with [35S]methionine for 1 h and then chased with nonradioactive medium without or with Imatinib for the indicated time points. Cells were then lysed and immunoprecipitated for β-catenin (IP: α β-cat). The results were analyzed by densitometry and expressed as a percentage of the intensity value at time 0.
The ability of recombinant Abl (rAbl) kinase to Y phosphorylate purified GST-β-catenin fusion proteins in vitro indicated that β-catenin is a direct substrate of Abl (Figure 4B).
As shown in Figure 4C, expression of Bcr-Abl (α Bcr-Abl, b–f and lane h) in HEK293T cells increased the total protein levels of either endogenous (α β-cat: h versus g) or ectopically induced WT β-catenin (α β-cat, b versus a). Also, different total levels of Y-to-F β-catenin mutants (α β-cat, c–f) correlated proportionately to their degree of Y phosphorylation (α p-Y β-cat: c–f). These effects were not accompanied by changes in GSK3β Y216 phosphorylation, as also detected in empty vector (−)-transfected sample (α p-Y GSK3β, a–h). The effect of Bcr-Abl on β-catenin protein turnover was analyzed by performing a pulse-chase analysis of Bcr-Abl+ CML cells cultured with or without Imatinib. Autoradiography of anti-β-catenin immunoprecipitates prepared at different times during a chase showed that the estimated half-life of β-catenin was decreased from 3.1 to 1.5 h in the presence of Imatinib compared with untreated cells (Figure 4D).
In conclusion, these data indicate that the delayed degradation of β-catenin correlated with its Bcr-Abl-mediated Y phosphorylation on Y86 and Y654. This evidence further supports a causal role for Bcr-Abl in promoting β-catenin stabilization without affecting GSK3β autophosphorylation.
Tyrosine-phosphorylated β-catenin does not interact with the Axin/GSK3β complex
Total β-catenin levels are tightly regulated by a regulatory multi-protein complex involving Axin, APC and GSK3 (Harris and Peifer, 2005; Klymkowsky, 2005). In Figure 5A, APC (α APC, b) and Axin (α Axin, b) were immunoprecipitated with β-catenin (IP: α β-cat) from BC-CML patient cells. Although Imanitib did not change the amount of APC coupled to β-catenin (α APC, c versus b), it significantly increased β-catenin/Axin association (α Axin, c compared to b) and binding of β-catenin to the Y-activated GSK3β kinase (α p-Y GSK3β, c versus b). By analyzing reciprocal anti-Axin immunoprecipitates obtained from the same BC-CML sample (Figure 5B, IP: α Axin), we observed that the amount of β-catenin captured by Axin was higher (α β-cat, b versus a) in the presence of Imatinib, justifying the increases on its S/T phosphorylation levels (α p-S/T β-cat, b versus a). A similar analysis was carried out in Ku812 cells (Figure 5C and D) obtaining comparable results. In addition, as Imatinib did not alter the Axin/GSK3β interaction (Figure 5C and D, α GSK3β, b versus a), these findings indicate that the Bcr-Abl-induced Y-phospho pool of β-catenin has a reduced binding affinity to Axin. In this view (Figure 5E), Ku812 cells were cultured in the absence (−) or presence (+) of Imatinib and then immunoprecipitated with either an anti-β-catenin (IP: α β-cat, a and b) or an anti-Axin antibody (IP: α Axin, c and d). After removal of Axin-immunocomplexes from total cell lysates, the supernatants were further immunoprecipitated by using an anti-phosphotyrosine antibody (IP: α supPY, e and f). The immunoprecipitation with an anti-Axin antibody showed that the β-catenin/Axin interaction was enhanced upon Imatinib treatment (α Axin: d versus c). Interestingly, the analysis of the Axin-coupled and Axin-uncoupled fractions for β-catenin (α β-cat, c–f) revealed that Y-phospho β-catenin could be immunoprecipitated only from the Axin-free cell lysate supernatants (α p-Y-β-cat: e versus c).
Figure 5.
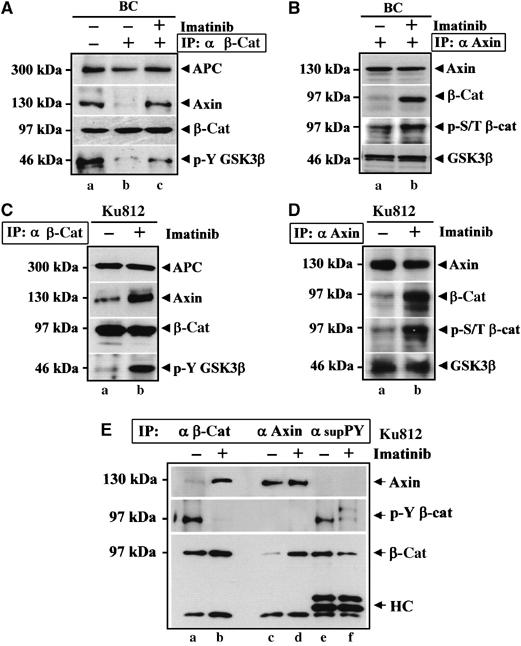
Tyrosine-phosphorylated β-catenin does not bind Axin. (A) BC cells (5 × 106) treated with DMSO (b) or 1 μM Imatinib (c) for 2 h were immunoprecipitated for β-catenin (IP: α β-cat). A lysate from 5 × 106 BC cells not immunoprecipitated was also prepared (a). The protein lysates were immunoblotted with the indicated antibodies. (B) BC cells (5 × 106) cultured with DMSO (−) or 1 μM Imatinib (+) for 2 h were immunoprecipitated with an anti-Axin antibody (IP: α Axin) and immunoblotted with the indicated antibodies. (C, D) Ku812 cells incubated with DMSO (−) or 1 μM Imatinib (+) for 2 h were immunoprecipitated for β-catenin (IP: α β-cat, C) or Axin (IP: α Axin, D) and immunoblotted with the indicated antibodies. (E) Ku812 cells cultured with DMSO (−) or 1 μM Imatinib (+) for 2 h were immunoprecipitated with an anti-β-catenin (IP: α β-cat) (a and b) or an anti-Axin antibody (IP: α Axin) (c and d). After removal of Axin immunocomplexes from cell lysates, Y phosphorylated proteins were immunoprecipitated by anti-phosphotyrosine antibody (IP: α sup-PY) and analyzed in Western blot for Axin and β-catenin. The same blot was stripped and assessed for Y phosphorylation of β-catenin (p-Y β-cat) with an antiphosphotyrosine antibody.
In conclusion, these data indicate that Bcr-Abl-induced Y phosphorylation of β-catenin could sterically modify the protein, preventing its recruitment by the Axin/GSK3β.
Effect of Bcr-Abl kinase inhibition on β-catenin cellular distribution and nuclear signalling
We tested if the β-catenin/TCF signalling could be impaired by inhibition of Bcr-Abl kinase activity (Figure 6A). In Ku812 cells treated for 2 h with DMSO or Imatinib, β-catenin protein levels were unchanged (α β-cat: a versus b). Cleavage products of β-catenin became detectable after 16 h of exposure to Imatinib (α Δβ-cat: c), whereas total levels of Bcr-Abl (α Bcr-Abl), Axin (α Axin) and TCF4 (α TCF4) were unaffected. Imatinib-induced β-catenin cleavage was associated with reduced levels of pro-caspase-3 (α pro-caspase-3) and blocked by the irreversible caspase-3 inhibitor Z-DEVD-fmk (α Δβ-cat: e compared to c).
Figure 6.
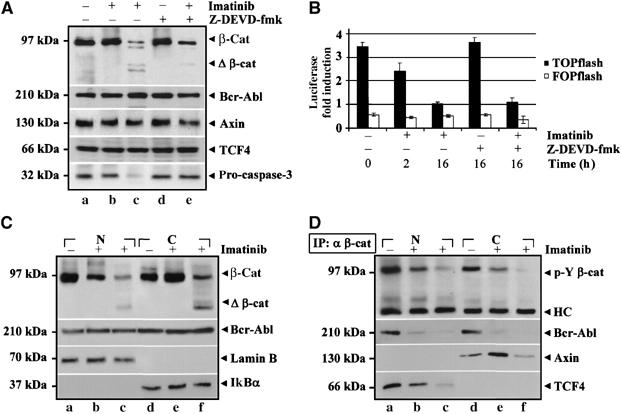
Imatinib reduces nuclear levels of Y-phospho β-catenin by impairing its TCF4-related transcription. (A) Ku812 cells were treated with DMSO (a) or with 1 μM Imatinib for 2 h (b) or 16 h (c), respectively. Cells were also incubated with 20 μM caspase-3 inhibitor Z-DEVD-fmk alone (d) or with 1 μM Imatinib (e) for 16 h. Total cell lysates were then immunoblotted as indicated. (B) TOP and FOP-flash plasmids were transfected into Ku812 cells. After 24 h of transfection, cells were treated for an additional 24 h and processed for determining the luciferase gene-reporter activity. Data indicate the mean+s.d. of three independent experiments. (C) Ku812 cells were treated with DMSO (a and d) or Imatinib for 2 h (b and e) or 16 h (c and f). Nuclear (N) and cytosolic (C) cell extracts were then prepared and probed as indicated. (D) Nuclear (N) and cytosolic (C) fractions described in (C) were also immunoprecipitated for β-catenin (IP: α β-cat) and then subjected to immunoblotting with the indicated antibodies.
Luciferase activity of a β-catenin-responsive TOPflash plasmid decreased rapidly in response to Imatinib, whereas the activity of the control FOPflash remained constant (Figure 6B). Treatment of cells with Z-DEVD-fmk alone had no effect on β-catenin-related transcription and, interestingly, the Z-DEVD-fmk maintained integrity of β-catenin (Figure 6A: α β-cat: e versus c) and did not impair the decrease in the TOPflash reporter activity induced by Imatinib.
The rapidity of the loss of β-catenin nuclear signalling suggested a mechanism different from β-catenin proteolysis. Thus, in Figure 6C, we analyzed nuclear (N) and cytosolic (C) extracts from Ku812 cells treated with DMSO (a,d) and Imatinib for 2 h (b,e) or 16 h (c,f) showing that Imatinib promoted a rapid cytosolic retention of β-catenin (α β-cat: e versus b), which was Y-dephosphorylated (Figure 6D: α p-Y β-cat: e versus b). The nuclear interaction of Y-phospho β-catenin, TCF4 and Bcr-Abl (α TCF4 and α Bcr-Abl b versus a) was inhibited by Imatinib, which also promoted a cytosolic Axin/β-catenin binding (α Axin: e compared to d). The presence of Bcr-Abl was also confirmed in anti-TCF4 immunoprecipitates (Figure 7A): this complex was disrupted by Imatinib but not by SU6656.
Figure 7.
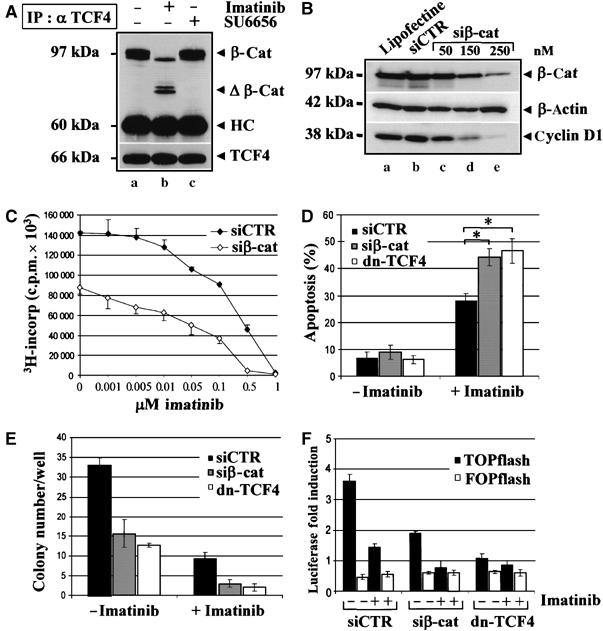
β-Catenin downregulation cooperates with Imatinib in reducing Bcr-Abl+ BC-CML cell growth and clonogenicity. (A) Ku812 cells were treated with DMSO (−) or 1 μM Imatinib (+) for 16 h. TCF4 and β-catenin were immunoprecipitated using an anti-TCF4 antibody (IP: α TCF4). β-Catenin and Bcr-Abl protein levels were analyzed by immunoblotting. The blot was reprobed to assess TCF4 protein content. (B) Ku812 cells were transfected with 50 (c), 150 (d) or 250 nM (e) of siRNA for β-catenin, 250 nM of a control siRNA (b) or carrier alone (a). Total lysates prepared 48 h after transfection were analyzed for β-catenin and cyclin D1 expression. The amounts of β-actin were reported as loading control. (C) Proliferation assay of Ku812 cells transfected with a control siRNA (siCTR) or β-catenin siRNA (siβ-cat) for 24 h and then cultured in the presence of decreasing concentrations of Imatinib for an additional 24 h. A labeling with [3H]thymidine was carried out for the last 8 h. (D) Quantitative analysis of apoptosis in Ku812 cells transfected with siCTR or siβ-cat for 24 h and then cultured in the absence or presence of 1 μM Imatinib for additional 24 h was performed by flow cytometry. Values represent the mean±s.d. of three independent experiments (*P<0.003 for siβcat and <0.05 for dn-TCF4). (E) Ku812 cells were cotransfected with a pEGFP plasmid and siCTR, siβ-cat or a dominant-negative TCF4 construct (dn-TCF4). Total 1000 GFP+ sorted cells by FACS analysis were transferred to 0.5% soft agar in the absence or presence of 1 μM Imatinib. Colonies were scored on an inverted Leica microscope on day 15. (F) Ku812 cells were transfected with either siCTR, siβ-cat or dn-TCF4. After 24 h, cells were cotransfected with TOP-flash or FOP-flash plasmid and cultured in the absence or presence of Imatinib for an additional 24 h when luciferase reporter activity was determined. Data represent the mean±s.d. of three independent experiments.
β-Catenin downregulation synergizes with Imatinib in reducing Bcr-Abl+ cell proliferation and clonogenicity
To validate the biological role of β-catenin in Bcr-Abl+ leukemogenesis, we tested if siRNA-reduced β-catenin levels could affect the expression of cyclin D1, a crucial mediator of Bcr-Abl activity (de Groot et al, 2000) (Figure 7B). Whereas a scrambled siCTR oligo had no effect on β-catenin (α β-cat, b), a substantial downregulation was observed at 250 nM siβ-cat (α β-cat, e) correlating with lower cyclin D1 levels (α cyclin D1). In Figure 7C, siβ-cat alone decreased Ku812 proliferation by 40% compared with siCTR. The combined use of Imatinib and siβ-cat further inhibited cell growth with a reduction of IC50 value for Imatinib from 0.3 to 0.1 μM. Although survival of Ku812 cells was not apparently affected by siβ-cat, β-catenin downregulation increased the apoptogenic effect of Imatinib (Figure 7D). Similar results were observed upon expression of a dominant-negative TCF4 (dnTCF4), used to test a more specific inhibition of β-catenin/TCF signalling. We also observed a synergistic effects of Imatinib and siβ-cat in reducing CML clonogenicity, which was also decreased by dnTCF4 (Figure 7E). To further corroborate these data, we measured β-catenin/TCF-dependent transcription with a TOP/FOPflash reporter assay (Figure 7F). Luciferase induction was reduced by expression of siβ-cat and, to a greater extent, when siβ-cat and Imatinib were combined, likely reflecting a suboptimal β-catenin downregulation (Figure 7B, α β-cat, e). The inhibition of TOPflash activity caused by dnTCF4 was comparable to that obtained with Imatinib alone, and the combination of the two treatments did not exert any synergism, thus excluding more downstream effects of Imatinib.
In conclusion, targeting of β-catenin interferes with transforming ability of Bcr-Abl and enhances CML sensibility to Imatinib.
Discussion
The relevance of regulating β-catenin protein stability is supported by the mutations of APC and Axin genes as well as of GSK3 phosphorylation sites of β-catenin in many human malignancies (Kikuchi, 2003; Staal and Clevers, 2005). In this report, we show that Bcr-Abl+ CML cells contain a S/T-phospho pool of β-catenin that can be reduced by using a GSK3 inhibitor (SB-216763). This implies that an intact APC/Axin/GSK3-destruction complex retains the ability to recruit and promote β-catenin degradation in Bcr-Abl+ CML, rendering therefore unlikely the β-catenin activating mutations identified in many solid human cancers (Kikuchi, 2003). Evidence was also provided that Bcr-Abl-mediated Y phosphorylation of β-catenin impairs its association with Axin and promotes its nuclear translocation and TCF4 binding. The WNT-activated Frizzled-LRP5/LRP6 receptors were shown to provide additional mechanisms by which Axin can be seized from cytosolic β-catenin thereby blocking its proteasome degradation (Davidson et al, 2005; Zeng et al, 2005). As a GSK3-mediated S/T phosphorylation of LRP5/6 is required for subsequent binding of Axin (Zeng et al, 2005), we observed that SB-216763 could slightly increase the cytosolic amount of Axin. Nevertheless, the finding that β-catenin/Axin interaction was rather impaired in SB-216763-treated cells, presumably because of the enhanced Y-phospho pool of β-catenin, point to a dominant role of Bcr-Abl in causing β-catenin protein stabilization and transcriptional activation. To our knowledge, these data represent the first evidence that β-catenin is coupled to the TCF4 transcription factor in a constitutively Y phosphorylated form in Bcr-Abl+ CML cells. A physical interaction between Y-phospho β-catenin and Bcr-Abl was shown by their reciprocal co-immunoprecipitation in leukemic cells as well as by the ability of purified recombinant Abl kinase to Y phosphorylate β-catenin in vitro. Ress and Moelling (2005) recently reported that Bcr-Abl was not immunoprecipitated from CML cells by using a different β-catenin C-terminal antibody. As we also failed to reproduce our results by using this antibody, its epitope on β-catenin (residues 680–781) could contain the binding site to Abl.
A site-directed mutagenesis of β-catenin at Y86 and Y654 was sufficient to prevent the Bcr-Abl-mediated accumulation of β-catenin in HEK293T cells as well as the Y phosphorylation of purified GST-β-catenin by rAbl in vitro. These findings identified these two Y-residues as targets of Bcr-Abl. The weak inhibitory effect of SU6656, a pan Src family kinase inhibitor, on β-catenin Y phosphorylation cannot exclude a minor contribution of other Src-related kinases, which are also activated by Bcr-Abl in CML cells (Wilson et al, 2002). However, Src-mediated phosphorylation seems to be dispensable, as the β-catenin/TCF4 binding was not affected by SU6656. The notion that Y86 and Y654 are located respectively within the N- and C-terminal transcriptional domains of β-catenin suggests that one or both residues might regulate the transactivating function of β-catenin. In this regard, phosphorylation of Y654 was reported to strengthen β-catenin association with the basal transcription factor TATA-binding protein (TBP) (Hecht et al, 1999; Piedra et al, 2001).
Inhibition of β-catenin Y phosphorylation by Imatinib rapidly increased β-catenin protein turnover and its binding affinity to the APC/Axin/GSK3 degradation machinery in CML cells. A lower degree of S/T phosphorylation of the Y654F mutant compared to Y86F in HEK293T cells correlated with a reduced amount of Axin detectable in Y654F-immunoprecipitates, thus suggesting that the Bcr-Abl-mediated phosphorylation of Y86 could induce a conformational change of β-catenin impairing its binding to Axin.
Although cleavage of β-catenin occurred in apoptotic CML cells treated with Imatinib for 16 h, the rapidity of the loss of TOPflash transcription induced by Imatinib upon 2 h suggested a mechanism different from β-catenin proteolysis. We observed that Imatinib caused a rapid nuclear to cytosolic shift of β-catenin, with decrease in Y-phospho β-catenin, thus indicating that Bcr-Abl could control both β-catenin nuclear import/export and its cytoplasmic degradation. In this regard, many cytosolic factors such as E-cadherins (Roura et al, 1999; Coluccia et al, 2006), MUC1 (Huang et al, 2005) or ICAT (Gottardi and Gumbiner, 2004) can recruit β-catenin, keeping the protein in the cytosol in a transcriptionally inactive form. Although there is no definitive evidence that β-catenin has to be Y-dephosphorylated when it is not coupled to E-cadherins, it is likely that this covalent modification (Y phosphorylation) could be a general mechanism to disrupt these cytosolic protein–protein interactions as shown recently for β-catenin/BCR (Ress and Moelling, 2005). On the other hand, the effects of Bcr-Abl on the nuclear–cytoplasmic shuttling of β-catenin, with regard to the role of APC (Henderson and Fagotto, 2002), require further studies.
The analysis of Bcr-Abl and β-catenin expression in BMMC from CML-patients supports a model in which increasing expression of Bcr-Abl in BC over CP can progressively achieve β-catenin stabilization (Figure 8). As only Axin-coupled β-catenin is targeted to proteosome, it is likely that the relative levels of Bcr-Abl and Axin can determine the equilibrium between the Y-phospho (not accessible to Axin) and Y-nonphospho (accessible to Axin) pool of β-catenin. In accordance with our findings, forced overexpression of Axin was reported to increase β-catenin degradation reducing the self-renewal potential of leukemic blasts (Jamieson et al, 2004). Although further studies are needed to test whether β-catenin accumulation in BC cells could be accounted for by restored transcription of its mRNA in committed granulocytes and macrophages precursors, quantitative other than qualitative (i.e. presence or absence) differences in Bcr-Abl oncogenic signalling (le Coutre et al, 2000; Perrotti et al, 2002; Barnes et al, 2005) seem to be responsible for the degree of β-catenin stabilization and response to Imatinib in BC versus CP CML cells. The synergistic effect of β-catenin siRNA and Imatinib in reducing Bcr-Abl+ cell growth and clonogenicity indicates that targeting β-catenin could represent a potential ‘loss-of-function' approach and may offer a therapeutic value in patients with CML.
Figure 8.
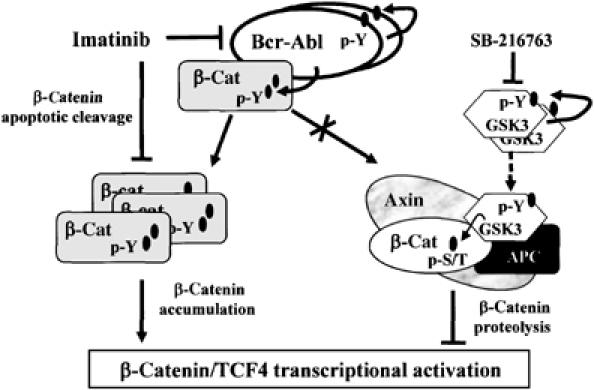
A model of β-catenin/TCF4 transcriptional activation in Bcr-Abl+ CML cells. The figure summarizes the effects of Bcr-Abl, Imatinib and SB-216763 on β-catenin protein stabilization and nuclear signaling.
Materials and methods
Cell cultures
Bcr-Abl+ Ku812 cell line was derived from a CML patient in BC (le Coutre et al, 1999). Colorectal cancer cells (Ls174T) and HEK293T were from ATCC (Rockville, MD) and cultured in RPMI-1640 (Life Technologies, Gaithersburg, MD) containing 10% fetal bovine serum (Cambrex, Baltimore, MD), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (GibcoBRL, Paisley, UK) at 37°C. Bone marrow (BM) samples were obtained after informed consent from two healthy donors (donor), four CML patients in CP and six in BC. BM mononuclear cells (BMMC) were purified by standard Ficoll–Hypaque density gradient centrifugation (Amersham Biosciences, Uppsala, Sweden). BC samples with at least 70% of blasts were used.
Kinase inhibitors
Imatinib was synthesized by Dr Alfonso Zambon (University of Venezia, Italy). Stock solutions of Imatinib at 1 or 10 mM in sterile water were filtered and stored at −20°C. The dual Src/Abl inhibitor SKI-606 (a kind gift from Drs Diane and Frank Boschelli, Wyeth Research, NY) (Golas et al, 2003) was dissolved in DMSO (Sigma Chemical Co., St Louis, MO). SU6656, an Src family kinase inhibitor, was from Calbiochem (Darmstadt, Germany). The GSK3 inhibitor SB-216763 was from Sigma Chemicals Co., and was prepared in DMSO at 10 mM and stored at 4°C.
Plasmids and transfections
The pcDNA3.1-Bcr-Abl cDNA was a kind gift from Dr Brian Druker (OSHU, Portland, OR, USA). A mutant form of TCF4 (dnTCF4) lacking the β-catenin-binding domain, WT or specific tyrosine-to phenylalanine (Y-to-F) point mutant (Y86F, Y142F, Y654Fand Y86F-Y654F), β-catenin cDNAs cloned in the pcDNA3.1His(C) vector or the pGEX-6P2 plasmid to express them as GST-fusion proteins were described elsewhere (Roura et al, 1999; Piedra et al, 2003). HEK293T cells were transfected by Lipofectamine2000 (Invitrogen, Carlsbad, CA) with 5 μg of pEGFP vector (Clontech, Palo Alto, CA, USA) and the indicated His-β-catenin plasmids (15 μg total/well). At 48 h, GFP+ cells were counted using a Zeiss fluorescence microscope and the efficiency of transfection was expressed as a percentage of total cells counted. Cultures containing at least 60% of transfected cells were lysed and subjected to immunoprecipitation or directly analyzed by Western blotting.
In vitro phosphorylation assay
Purification of GST-β-catenin (WT, Y86F, Y654F and Y86F-Y654F) fusion proteins was reported previously (Roura et al, 1999). Recombinant Abl protein, including its kinase domain, was expressed in Sf9 insect cells using the baculovirus expression system (MaxBac 2.0, Invitrogen) and purified using a Q-sepharose Fast Flow anion exchange column (Amersham) as described recently for ALK (Gunby et al, 2005). β-Catenin WT and mutants, 10 pmoles each, were incubated with 150 ng of recombinant Abl kinase in a buffer containing 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 1 mM EGTA and 100 μM γ32ATP (specific activity 4000 c.p.m./pmol). The reactions were stopped with Laemmli buffer and the samples were analyzed by SDS–PAGE.
Small interfering RNA
Ku812 cells (5 × 105/well) were transiently transfected with 150 nM of c-Src siRNA or of scrambled siRNA (SMARTpool; Dharmacon, Lafayette, CO) by using Oligofectine (Qiagen, Crawley, UK). Ku812 cells were also transiently transfected with a β-catenin siRNA (siβ-cat, GUGGGUGGUAUAGAGGCUC) (van de Wetering et al, 2003) or a control siRNA against luciferase (siCTR, CUUACGCUGAGUA CUUCGA), both synthesized and purified by MWG-Biotech, AG.
Proliferation assay
Ku812 cells (104/well) were treated with the indicated drug in a total volume of 200 μl and harvested as reported previously (le Coutre et al, 2000). The IC50 inhibitory drug concentration (producing a 50% decrease in proliferation compared to control) was calculated.
Apoptosis analysis
Cells were cotransfected with the indicated constructs and a green fluorescent pEGFP vector. After 24 h cells were treated with Imatinib for further 24 h, fixed in 70% cold ethanol and stained with 50 μg/ml propidium iodide (Sigma Chemicals Co.) A FACScalibur flow cytometry analysis was confined to GFP+ cells and the amount of apoptotic cells in sub-G1 phase of the cell cycle was determined with a Cell-Quest software (Becton Dickinson, San Josè, CA).
Clonogenic assay
Ku812 cells were cotransfected with pEGFP vector and a dominant-negative TCF4, siβ-cat or a control siCTR. GFP+-sorted cells were resuspended in complete medium containing 0.5% agar in the absence or presence of Imatinib and plated in six-well plates over a basal layer of complete medium containing 1% agar. The plates were cultured at 37°C and the clonogenicity was determined in three independent experiments by counting the number of colonies per well on day 15.
Luciferase assay
Ku812 cells (2 × 105/well) were transfected by Lipofectamine2000 (Invitrogen) with 5 μg of TCF-reporter construct TOPflash or FOPflash (Upstate Biotechnology, Lake Placid, NY) and 25 ng of pSV-βgal construct (Promega, WI), as internal control, to normalize luciferase activity for transfection efficiency. After 24 h, cells were treated with Imatinib for an additional 24 h. Luciferase activity was measured using a luminometer (Turner Designs TD-20/20) and a luciferase assay system from Promega.
Cell fractionation
Ku812 cells were lysed in a digitonin buffer (1% digitonin, 150 mM NaCl, 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 10 μM benzamidine–HCl, 10 μg each of aprotinin, leupeptin and pepstatin A per ml). The lysates were centrifuged at 13 000 r.p.m. for 10 min, and supernatants representing cytosolic fractions were saved. The pellets representing nuclear components were lysed in RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS and 50 mM Tris (pH 7.5)).
Immunoprecipitation and Western blotting
Cell lysates were immunoprecipitated and immunoblotted as described previously (Coluccia et al, 2006). Antibodies against C-terminal β-catenin (immunogenic epitope within amino acids 560–781), GSK3β and p-Y216 GSK3β were from BD Transduction Laboratories (Heidelberg, Germany). N-terminal anti-β-catenin S/T-phosphorylated was from Cell Signaling Technology (Beverly, MA), whereas N-terminal anti-β-catenin S/T-nonphosphorylated (clone 8E4), anti-phosphotyrosine (clone 4G10), anti-Axin, anti-APC (clone ALI 12-28), anti-TCF4 (clone 6H5-3), anti-cyclinD1 and anti-Src antibody (clone GD11) were from Upstate Biotechnology. Antibody to p-Y418 Src was from Biosource (Camarillo, CA). Anti-β-Actin, anti-Abl (clone K-12), anti-Lamin B (C-20) and anti-IkBα (H4) antibodies were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA), whereas the anti-His-tag (Xpress) antibody was from Invitrogen.
Pulse-chase assay
Ku812 cells (5 × 106) were washed in cold PBS and kept in RPMI-1640 medium without cysteine/methionine (Sigma Chemical Co.) for 1 h at 37°C. Cells were pulsed for 30 min with [35S]methionine Promix (10 μCi/106 cells; Amersham Pharmacia), chased in RPMI-1640 (±1 μM Imatinib). Lysates were then immunoprecipitated with a C-terminal anti-β-catenin antibody. Immunocomplexes were separated by SDS–PAGE, stained with Coomassie blue as loading control, destained and incubated with Amplify fluorographic reagent (Amersham), dried and developed by autoradiography. Densitometric analysis (Eagle Eye II Photo-densitometer, Stratagene) was performed with Image analysis software from Scion Corporation (MD, USA).
Statistical analysis
Results were statistically compared by simple t-test and differences were considered significant if P<0.05.
Acknowledgments
We are grateful to Drs Diane and Frank Boschelli (Wyeth Research, NY) for kindly providing SKI-606 and to Dr Brian Druker for the pcDNA3.1-neo vector expressing the full-length cDNA for Bcr-Abl. We warmly thank Dr Antonio de Herreros (Unitat de Biologia Cellular I Molecular, IMIM-UPF, Barcelona, Spain) for dominant-negative TCF4 (dnTCF4), WT and point mutants (Y86F, Y142F and Y654F) β-catenin cDNAs. We thank G Lagna for technical and graphic help. This work was supported by the Italian Association for Cancer Research (AIRC), MIUR-COFIN and PRIN programs (2003–2004), EU (Prokinase Research #503467), CFI and NCI-C. AMLC is recipient of a fellowship from FIRC (Fondazione Italiana per la Ricerca sul Cancro).
References
- Baba Y, Garrett KP, Kincade PW (2005) Constitutively active β-catenin confers multilineage differentiation potential on lymphoid and myeloid progenitors. Immunity 23: 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DJ, Palaiologou D, Panousopoulou E, Schultheis B, Yong A, Wong A, Pattacini L, Goldman JM, Melo JV (2005) Bcr-Abl expression levels determinate the rate of development of resistance to Imatinib mesylate in chronic myeloid leukaemia. Cancer Res 65: 8912–8919 [DOI] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P (2004) Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J 377: 249–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccia AM, Benati D, Dekhil H, De Filippo A, Lan C, Gambacorti Passerini C (2006) SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of β-catenin and its nuclear signalling. Cancer Res 66: 2279–2286 [DOI] [PubMed] [Google Scholar]
- Daley GQ (2004) Chronic myeloid leukemia: proving ground for cancer stem cells. Cell 119: 314–316 [DOI] [PubMed] [Google Scholar]
- Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ (2001) Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the β-catenin pathway. Mol Cell Biol 21: 5857–5868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C (2005) Casein kinase 1γ couples Wnt receptor activation to cytoplasmic signal transduction. Nature 438: 867–872 [DOI] [PubMed] [Google Scholar]
- de Groot RP, Raaijmakers JA, Lammers JW, Koenderman L (2000) STAT5-dependent cyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol Cell Biol Res Commun 3: 299–305 [DOI] [PubMed] [Google Scholar]
- Deininger M, Buchdunger E, Druker BJ (2005) The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 105: 2640–2653 [DOI] [PubMed] [Google Scholar]
- Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, Frost P, Ye F, Boschelli DH, Boschelli F (2003) SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res 63: 375–381 [PubMed] [Google Scholar]
- Goldman JM, Melo JV (2003) Chronic myeloid leukemia: advances in biology and new approaches to treatment. N Engl J Med 349: 1451–1464 [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, Gumbiner BM (2004) Role for ICAT in beta-catenin-dependent nuclear signaling and cadherin functions. Am J Physiol Cell Physiol 286: C747–C756 [DOI] [PubMed] [Google Scholar]
- Gunby RH, Tartari CJ, Porchia F, Donella-Deana A, Scapozza L, Gambacorti-Passerini C (2005) An enzyme-linked immunosorbent assay to screen for inhibitors of the oncogenic anaplastic lymphoma kinase. Haematologica 90: 988–990 [PubMed] [Google Scholar]
- Harris TJ, Peifer M (2005) Decisions, decisions: beta-catenin chooses between adhesion and transcription. Trends Cell Biol 15: 234–237 [DOI] [PubMed] [Google Scholar]
- Hecht A, Litterst CM, Huber O, Kemler R (1999) Functional characterization of multiple transactivating elements in beta-catenin, some of which interact with the TATA-binding protein in vitro. J Biol Chem 274: 18017–18025 [DOI] [PubMed] [Google Scholar]
- Henderson BR, Fagotto F (2002) The ins and outs of APC and β-catenin nuclear transport. EMBO Rep 3: 834–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscox S, Jiang WG (1999) Association of the HGF/SF receptor, c-Met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun 261: 406–411 [DOI] [PubMed] [Google Scholar]
- Huang L, Chen D, Liu D, Yin L, Kharbanda S, Kufe D (2005) MUC1 oncoprotein blocks glycogen synthase kinase 3β-mediated phosphorylation and degradation of β-catenin. Cancer Res 65: 10413–10422 [DOI] [PubMed] [Google Scholar]
- Huntly BJ, Shigematsu H, Deguchi K, lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG (2004) MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6: 587–596 [DOI] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtiens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL (2004) Granulocyte–macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351: 657–667 [DOI] [PubMed] [Google Scholar]
- Kanai Y, Ochiai A, Shibata T, Oyama T, Ushijima S, Akimoto S, Hirohashi S (1995) c-erbB-2 gene product directly associates with beta-catenin and plakoglobin. Biochem Biophys Res Commun 208: 1067–1072 [DOI] [PubMed] [Google Scholar]
- Kikuchi A (2003) Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci 94: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymkowsky MW (2005) beta-catenin and its regulatory network. Hum Pathol 36: 225–227 [DOI] [PubMed] [Google Scholar]
- Krause DS, Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N Engl J Med 353: 172–187 [DOI] [PubMed] [Google Scholar]
- le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E, Giardini R, Formelli F, Gambacorti Passerini C (1999) In vivo eradication of human BCR/ABL-Positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst 2: 163–168 [DOI] [PubMed] [Google Scholar]
- le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L, Cabrita G, Marchesi E, Supino R, Gambacorti Passerini C (2000) Induction of resistance to the Abelson inhibitor STI571 in human leukemc cells through gene amplification. Blood 95: 1758–1766 [PubMed] [Google Scholar]
- Nelson WJ, Nusse R (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, Iervolino A, Condorelli F, Gambacorti Passerini C, Caligiuri MA, Calabretta B (2002) BCR/ABL suppresses C/EBP expression via the translation inhibitory activity of the poly(rC)-binding protein hnRNP E2. Nat Genet 30: 48–58 [DOI] [PubMed] [Google Scholar]
- Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG (2001) Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem 276: 20436–20443 [DOI] [PubMed] [Google Scholar]
- Piedra J, Miravet S, Pálmer HG, Heisterkamp N, García de Herreros A, Duñach M (2003) p120 catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin–α-catenin interaction. Mol Cell Biol 23: 2287–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Clevers H (2005) Wnt, stem cells and cancer in the intestine. Biol Cell 97: 185–196 [DOI] [PubMed] [Google Scholar]
- Ress A, Moelling K (2005) Bcr is a negative regulator of the Wnt signalling pathway. EMBO Rep 6: 1095–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423: 409–414 [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434: 843–850 [DOI] [PubMed] [Google Scholar]
- Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M (1999) Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J Biol Chem 274: 36734–36740 [DOI] [PubMed] [Google Scholar]
- Staal FJ, Clevers HC (2005) WNT signalling and haematopoiesis: a WNT–WNT situation. Nat Rev Immunol 5: 21–30 [DOI] [PubMed] [Google Scholar]
- Tipping AJ, Baluch S, Barnes DJ, Veach DR, Clarkson BM, Bornmann WG, Mahon FX, Goldman JM, Melo JV (2004) Efficacy of dual-specific Bcr-Abl and Src-family kinase inhibitors in cells sensitive and resistant to imatinib mesylate. Leukemia 18: 1352–1356 [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Oving I, Muncan V, Pong Fong MT, Brantjes H, van Leenen D, Holstege FC, Brummelkamp TR, Agami R, Clevers H (2003) Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep 4: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MB, Schreiner SJ, Choi HJ, Kamens J, Smithgall TE (2002) Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene 21: 8057–8088 [DOI] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X (2005) A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 438: 873–877 [DOI] [PMC free article] [PubMed] [Google Scholar]