

Abstract
Fanconi anemia (FA) is a chromosome fragility syndrome characterized by bone marrow failure and cancer susceptibility. The central FA protein FANCD2 is known to relocate to chromatin upon DNA damage in a poorly understood process. Here, we have induced subnuclear accumulation of DNA damage to prove that histone H2AX is a novel component of the FA/BRCA pathway in response to stalled replication forks. Analyses of cells from H2AX knockout mice or expressing a nonphosphorylable H2AX (H2AXS136A/S139A) indicate that phosphorylated H2AX (γH2AX) is required for recruiting FANCD2 to chromatin at stalled replication forks. FANCD2 binding to γH2AX is BRCA1-dependent and cells deficient or depleted of H2AX show an FA-like phenotype, including an excess of chromatid-type chromosomal aberrations and hypersensitivity to MMC. This MMC hypersensitivity of H2AX-deficient cells is not further increased by depleting FANCD2, indicating that H2AX and FANCD2 function in the same pathway in response to DNA damage-induced replication blockage. Consequently, histone H2AX is functionally connected to the FA/BRCA pathway to resolve stalled replication forks and prevent chromosome instability.
Keywords: BRCA, Fanconi anemia, genome stability, histone H2AX, stalled replication forks
Introduction
Fanconi anemia (FA) is a genetic syndrome characterized by chromosome fragility, congenital malformations, progressive pancytopenia and cancer susceptibility (Kutler et al, 2003). FA is a rare disease with a carrier frequency of 1/65 to 1/300 (Callén et al, 2005). There are at least 12 FA complementation groups (A, B, C, D1, D2, E, F, G, I, J, L and M), each connected with a distinct gene. All these genes, but FANCI, have been cloned (reviewed in Thompson, 2005). The FA proteins FANC-A, B, C, E, F, G, L and M assemble in a nuclear complex (FA complex), which is required for FANCD2 activation via monoubiquitination at the residue K561 during S phase and in response to DNA damage (García-Higuera et al, 2001; Taniguchi et al, 2002a). FANCL has been suggested to provide the monoubiquitin E3 ligase function for FANCD2 (Meetei et al, 2003a), although no direct evidence has been provided to date. The recently cloned FANCJ/BRIP1 and FANCM/Hef both have a helicase domain, but the molecular biology of the whole FA/BRCA pathway is far from being understood (reviewed in Thompson, 2005).
The FANCD2 gene is a key player in the FA pathway (reviewed in Bogliolo and Surrallés, 2005). Together with FANCL and FANCM/Hef, it is the only FA gene present in non-vertebrates and its functionally important features are highly conserved (Castillo et al, 2003). The FA complex is assembled in the absence of FANCD2 indicating that FANCD2 is downstream to this complex in the FA pathway (García-Higuera et al, 2001) but upstream to other components, resulting in a central position. Active FANCD2 colocalizes or interacts with the breast cancer susceptibility and the double-strand break (DSB) repair proteins BRCA1, Rad51 and BRCA2/FANCD1 in DNA damage-induced nuclear foci or during S phase (García-Higuera et al, 2001; Taniguchi et al, 2002a; Hussain et al, 2004; Wang et al, 2004).
Both BRCA1 and BRCA2/FANCD1 interact with the recombination protein Rad51 and are involved in DSB repair by homologous recombination (HR) (Patel et al, 1998; Venkitaraman, 2002). It has also been proposed that FA proteins mediate translesion DNA synthesis and, therefore, promote replication block bypass (Niedzwiedz et al, 2004; reviewed in Thompson, 2005). Repair by HR predominates during S/G2 phase of the cell cycle when sister chromatids are present. The fact that FANCD2 forms foci with BRCA1 and RAD51 during S phase suggests that the FA/BRCA pathway is involved in HR repair of DSB during S phase (Taniguchi et al, 2002a; D'Andrea, 2003; D'Andrea and Grompe, 2003; Nakanishi et al, 2005). Indeed, a number of recent publications indicate that FANCD2 participates in the resolution of DNA interstrand crosslinks (ICL)-induced stalled replication forks (Andreassen et al, 2004; Hussain et al, 2004; Pichierri et al, 2004; Rothfuss and Grompe 2004; Wang et al, 2004) possibly by ensuring correct replication fork repair by HR (reviewed by Pichierri and Rosselli, 2004a; Macé et al, 2005).
ATR but not ATM is present at stalled replication forks (Abraham, 2001; Lupardus et al, 2002; Tercero et al, 2003) and, consistently, the ICL-activated kinase is ATR, not ATM (Pichierri and Rosselli, 2004b). Once activated, ATR phosphorylates FANCD2, probably allowing subsequent monoubiquitination of FANCD2 (Andreassen et al, 2004; Pichierri and Rosselli, 2004b; Ho et al, 2006). Interestingly, ATR-mediated FANCD2 phosphorylation in response to ICLs depends on both an intact FA complex and nibrin, the product of the NBS1 gene, mutated in Nijmegen breakage syndrome patients (Pichierri and Rosselli, 2004b). NBS1 also interacts with FANCD2 after DNA damage (Nakanishi et al, 2002). In turn, RAD50-MRE11-NBS1 (RMN) foci formation after ICLs requires the FA complex (Pichierri et al, 2002). Consistent with a crosstalk between ATR, NBS1 and the FA/BRCA pathways, not only FA cells but also NBS and Seckel syndrome cells with leaky splicing mutations leading to low levels of ATR are hypersensitive to MMC (Nakanishi et al, 2002; O'Driscoll et al, 2003) and NBS; Seckel syndrome and FA patients share some clinical features (Andreassen et al, 2004). In addition, FA proteins crosstalk with other proteins involved in maintaining chromosome stability such as ATM and BLM (Taniguchi et al, 2002b; Meetei et al, 2003b; Pichierri et al, 2004), building an integrated network of genome stability pathways (Surrallés et al, 2004; Venkitaraman, 2004).
Exposure to ultraviolet radiation C (UVC) results in replication arrest as most of the DNA polymerases are unable to replicate templates containing UV-induced DNA lesions (Ward and Chen, 2001). Consistently, FANCD2 becomes monoubiquitinated and forms foci in response to not only MMC and X-rays, but also UVC (García-Higuera et al, 2001; Pichierri et al, 2004). ATR inactivation leads to the inhibition of histone H2AX phosphorylation and phosphorylated H2AX (γH2AX) foci formation upon treatment with UVC or hydroxyurea (HU), indicating that H2AX is phosphorylated by ATR in response to replication blockage (Ward and Chen, 2001). Conversely, ATM is not required for H2AX phosphorylation following UVC or HU replication block (Ward and Chen, 2001). Many components of the DNA damage response including BRCA1, the MRN complex, 53BP1, MDC1 and Rad51 form foci that colocalize with γH2AX foci (Fernández-Capetillo et al, 2004). As FANCD2 relocates to the chromatin fraction upon DNA damage in a poorly understood process (Montes de Oca et al, 2005), here we have locally irradiated cells of different genetic backgrounds with UVC to induce subnuclear accumulation of stalled replication forks and hence study the dynamics and regulation of FANCD2 relocation to damaged sites. These studies led us to uncover that histone H2AX is involved in the FA/BRCA pathway.
Results
FANCD2 dynamically relocates to UVC-induced stalled replication forks: a functional assay for the FA/BRCA pathway
In the first series of experiments, wild-type MRC5 fibroblasts were locally irradiated with 20, 40 or 60 J/m2 of UVC radiation through a filter with 5-μm-diameter pores and the locally irradiated nuclear areas were visualized by immunofluorescence with antibodies against cyclobutane pyrimidine dimers (CPD). At the pore size and density used, over 70% of the nuclei initially presented a UV spot. FANCD2 presented a diffuse or speckle nuclear pattern in the majority of non-irradiated cells or in cells immediately after irradiation but relocated to the site of UVC-induced damage only few hours after irradiation (Figure 1A). FANCD2 relocation followed a specific and consistent dynamics: immediately after irradiation, no FANCD2 signal was observed at the site of damage but the percentage of UV spots containing FANCD2 signal progressively increased until reaching a plateau 6–8 h after irradiation, when 70–80% of UV spots presented FANCD2 signal (Figure 1B; wild-type cells). FANCD2 signal was present at the site of irradiation as long as the CPD were detectable, up to 48 h after irradiation in few cells (data not shown). Six hours after irradiation, we observed a one-to-one correspondence between cells staining positive for both CPD at the irradiation site and FANCD2, and no FANCD2 spots were detected in the cells without UV-spots. These results indicate that FANCD2 relocation correlates with the presence of CPD.
Figure 1.

FANCD2 relocates to the sites of UVC radiation-induced stalled replication forks in S phase: a functional assay for the FA/BRCA pathway. Cells (human wild-type MRC5 fibroblasts) irradiated through a 5-μm-pore filter with UVC (20 J/m2) present FANCD2 signal (detected with anti-FANCD2 antibodies in red) at the site of damage (detected with anti-CPD antibodies in green) 6 h after irradiation but not immediately after irradiation (A). FANCD2 relocates to UV spots following a specific dynamics and in a manner dependent on ATR, FANCA, BRCA1 and FANCD2 K561 but independent of ATM (means and s.d. of three experiments are shown in the graph) (B). PCNA and FANCD2 (red) relocate to the site of irradiation (green) following the same dynamics (C). FANCD2 (red) relocates to the irradiated site (green) only in replicating but not in G1 synchronized (>95% of the cells in G1 phase) wild-type primary fibroblasts (D); FANCD2 relocates to UV spots (right green panel) only in S-phase cells, as shown by BrdU incorporation (red middle panel). Nuclear counterstaining in DAPI (left blue panel) (E).
It is known that UVC induces stalled replication forks (see Introduction). Consistent with this notion, the pattern of UVC-induced FANCD2 relocation correlates with the pattern of PCNA staining (Figure 1C), reflecting that FANCD2 and PCNA colocalize during DNA replication stress response (Howlett et al, 2005). In addition, FANCD2 relocated to the UV spot in exponentially growing primary human fibroblasts but not in the same cells synchronized in the G0/G1 phase of the cell cycle (>95% of the cells in G0/G1 phase) by serum starving (Figure 1D) or other methods (data not shown). Finally, when cells were grown in the presence of BrdU, 80 out of 80 cells with FANCD2 signal at the site of damage were positive for BrdU incorporation as detected with antibodies against BrdU (Figure 1E). Our unpublished confocal microscopy studies with cells expressing YFP-FANCD2 are also consistent with FANCD2 relocating to UV locally induced stalled forks during S phase (data not shown).
To determine whether this assay was a functional assay to study the FA/BRCA pathway, we analyzed the dynamics of FANCD2 relocation to the site of UVC-induced damage in cells deficient in well-known players of the FA/BRCA pathway (FANCA, BRCA1, ATR or ATM) and in FANCD2-deficient cells stably transduced with a non-ubiquitinable K561R-FANCD2 cDNA. As shown in Figure 1B, FANCD2 does not relocate in cells deficient in BRCA1, FANCA, ATR or expressing a non-monoubiquitinable K561R-FANCD2 but it normally relocates to the UV spots in wild-type cells (green), in FANCA-deficient cells functionally corrected by retrovirus-mediated FANCA cDNA transduction (blue), in ATM-deficient cells (orange), in BRCA2-deficient cells and in BRCA1-deficient cells functionally corrected by microcell-mediated chromosome transfer (data not shown). Thus, all known components required for FANCD2 monoubiquitination (the upstream FA proteins, ATR and FANCD2 K561) or FANCD2 foci formation (BRCA1) in response to DNA ICLs are also required for FANCD2 relocation to UVC-induced stalled replication forks. Therefore, all the data obtained by local irradiation and presented in Figure 1 are in keeping with all known functional requirements of the FA pathway and therefore validate our approach as a novel in vivo functional assay for dissecting the FA/BRCA pathway.
γH2AX is required for FANCD2 relocation to damaged sites but not for FANCD2 activation
As mentioned in Introduction, histone H2AX is phosphorylated by ATR but not by ATM on its S139 residue in response to UVC-induced stalled replication forks (Shiloh, 2001; Ward and Chen, 2001) and γH2AX is required for the accumulation of several DNA repair/damage response factors to the site of DNA damage (Fernández-Capetillo et al, 2004). Consequently, we applied our newly developed functional assay of local UVC-irradiation to check whether H2AX is required for FANCD2 relocation to the site of UVC-induced stalled replication forks. As shown in Figure 2, FANCD2 does not relocate to the site of UVC-induced damage in H2AX−/− mouse embryonic fibroblasts (MEFs) (Figure 2A) or H2AX−/− MEFs expressing a nonphosphorylable H2AX (H2AXS136A/S139A) (Figure 2B). However, normal FANCD2 relocation was observed in genetically matched wild-type MEF, indicating a critical role for H2AX and H2AX phosphorylation in FANCD2 relocation to UVC-induced stalled replication forks.
Figure 2.
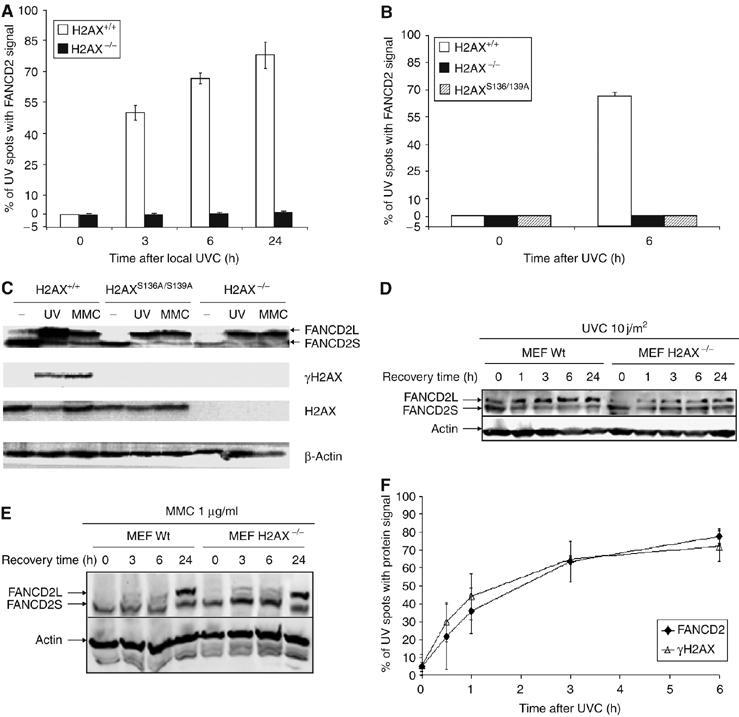
Relocation of FANCD2 to UVC-induced damage requires H2AX phosphorylation. FANCD2 does not relocate to locally induced stalled replication forks with UVC in H2AX−/− MEFs (A) or in H2AX−/− MEFs expressing a nonphosphorylable H2AX (H2AXS136A/S139A) (B). H2AX is not required for UVC- or MMC-induced FANCD2 monoubiquitination (C–E). H2AX-deficient MEF and their wild-type counterpart were treated with either UVC (10 J/m2; C, D) or with MMC (1 mg/ml; C, E). Cells were then harvested at the indicated recovery times and analyzed by Western blot for FANCD2 monoubiquitination. The equal loading of each slot was confirmed by probing the blot with anti-actin antibody (C–E). Local UVC irradiation results in concurrent H2AX phosphorylation and FANCD2 accumulation at the site of irradiation, with a dynamics of H2AX phosphorylation identical to the dynamics of FANCD2 relocation (F). (Means and s.d. of three experiments are shown in panels A, B and F.)
H2AX and γH2AX are not required for FANCD2 activation, as FANCD2 is normally monoubiquitinated in response to UVC or MMC, both in H2AX−/− and in nonfunctional H2AXS136A/S139A MEF cells (Figure 2C). In addition, the kinetics of FANCD2 monoubiquitination upon UVC (Figure 2D) or MMC (Figure 2E) treatments are not compromised in H2AX-deficient cells. The reduced amount of monoubiquitinated FANCD2 (FANCD2L) in H2AX−/− MEFs observable in Figure 2D is attributable to subtle differences in the proportion of cells in S phase between genetic backgrounds (data not shown). These results indicate that H2AX phosphorylation is not involved in FANCD2 activation but rather in FANCD2 recruitment or accumulation to stalled replication forks.
We then compared the dynamics of FANCD2 relocation and H2AX phosphorylation at the site of local irradiation using antibodies specific for H2AX phosphorylated at serine 139. Interestingly, the dynamics of FANCD2 relocation was identical to the dynamics of H2AX phosphorylation at the site of damage (Figure 2F), suggesting that FANCD2 relocates to the site of damage immediately after H2AX phosphorylation. As expected (Ward and Chen, 2001), H2AX phosphorylation at the site of UV irradiation was greatly reduced in Seckel syndrome cells expressing low levels of functional ATR (data not shown). As previously mentioned, ATR phosphorylates H2AX at stalled replication forks, thus further supporting the notion that stalled replication forks are the initiating signal for ATR-mediated H2AX phosphorylation and subsequent FANCD2 relocation to the damaged site.
To confirm our observations with H2AX-deficient cells and local UVC irradiation, we studied FANCD2 foci formation upon MMC treatment in the presence or absence of H2AX by RNA interference (RNAi) (Figure 3). Our data indicate that depletion of H2AX by RNAi results in normal FANCD2 monoubiquitination (Figure 3A) but impaired FANCD2 foci formation (Figure 3B–D) in response to MMC.
Figure 3.

H2AX depletion by RNAi disrupts FANCD2 foci formation ability after MMC treatment but does not impair FANCD2 monoubiquitination. HeLa cells were interfered with siRNA versus H2AX or versus GFP and treated with MMC for 0, 6 and 24 h. Western blot showing inhibition of H2AX phosphorylation after siRNA and normal FANCD2 monoubiquitination (A). FANCD2 foci in HeLa cells interfered with siRNA versus GFP (B). FANCD2 foci in HeLa cells interfered with siRNA versus H2AX (C). Graph showing percentage of inhibition of FANCD2 foci formation in GFP-interfered cells versus H2AX-depleted cells (D). Mean and s.d. of two independent experiments are reported. H2AX expression was knocked down by transfection with a SMARTpool® reagent (Dharmacon, USA) designed against H2AX. SiRNA was transfected using the Oligofectamine reagent (Invitrogen, USA) according to the manufacturer's indications. As a control, an siRNA duplex directed against GFP (Dharmacon, USA) was used. All the experiments were carried out 72 h after transfection when maximal inhibition was observed as analyzed by Western blot, (A). The equal loading of each slot was confirmed by probing the blot with anti-actin antibody.
As FANCD2 binds to chromatin upon DNA damage (Montes de Oca et al, 2005), we studied relocation of FANCD2 to the chromatin fraction in HeLa cells (Figure 4A) and in wild-type or H2AXS136A/S139A MEFs (Figure 4B) to probe that normal MMC-induced binding of FANCD2 to chromatin requires a functional H2AX. FANCD2 was normally detected in the chromatin pellet in HeLa cells upon ICL (8-methoxypsoralen (8-MOP)) or UVC treatment (Figure 4A) and in wild-type MEF upon MMC treatment (Figure 4B). However, MMC-induced chromatin loading of FANCD2 was strongly impaired in H2AXS136A/S139A MEFs, consistent with the notion that FANCD2 relocation to damaged chromatin requires a functional H2AX. However, we cannot disregard the possibility that this impairment, at least in part, is attributable to reduced overall levels of monoubiquitinated FANCD2 in H2AX-deficient cells.
Figure 4.
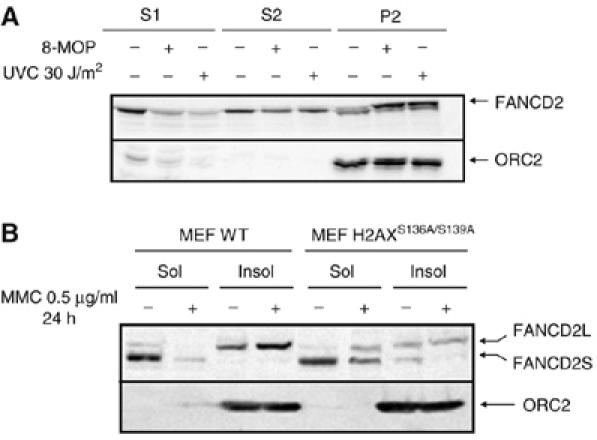
H2AX-dependent association of FANCD2 to chromatin after DNA damage. HeLa cells were left untreated or treated with 8-MOP+UVA or with 30 J/m2 of UVC, let to recover for 6 h and then fractionated and analyzed by Western blot (A). S1=cytoplasmic fraction, S2=nucleoplasmic fraction, P2=chromatin fraction. Wild type and H2AX−/− MEFs were treated with 1 μg/ml MMC for 24 h and then fractionated and analyzed by Western blot (B). S1 and S2 fractions were pooled (Sol) and 50 μg of total protein of the Sol fraction and of the chromatin fraction (Insol) were analyzed by Western blot. ORC2 was used as a marker of the chromatin fraction (A, B).
FANCD2 interacts with γH2AX upon DNA damage in a BRCA1-dependent manner
We then pursued the question of whether FANCD2 is recruited to the site of damage in a γH2AX-dependent manner by performing immunoprecipitation (IP) experiments in H2AX+/+, H2AX−/− or H2AXS136A/S139A MEF cell extracts as well as in BRCA1-deficient and genetically complemented cells. We were able to show that γH2AX and FANCD2 co-immunoprecipitate (Figure 5A). Importantly, FANCD2–γH2AX binding was significantly dependent on DNA damage (MMC or UVC) and was abolished in BRCA1-deficient cells (Figure 5B) and in H2AX−/− and H2AXS136A/S139A cells, suggesting that only phosphorylated form of H2AX is required, in concert with BRCA1, for recruitment of FANCD2 to damaged DNA. Consistently, we observed that MMC-induced chromatin loading of FANCD2L is also impaired in BRCA1-deficient cells (data not shown), again supporting the notion acquired in chicken cells that BRCA1 is dispensable for FANCD2 monoubiquitination but required for FANCD2 foci formation (Vandenberg et al, 2003).
Figure 5.
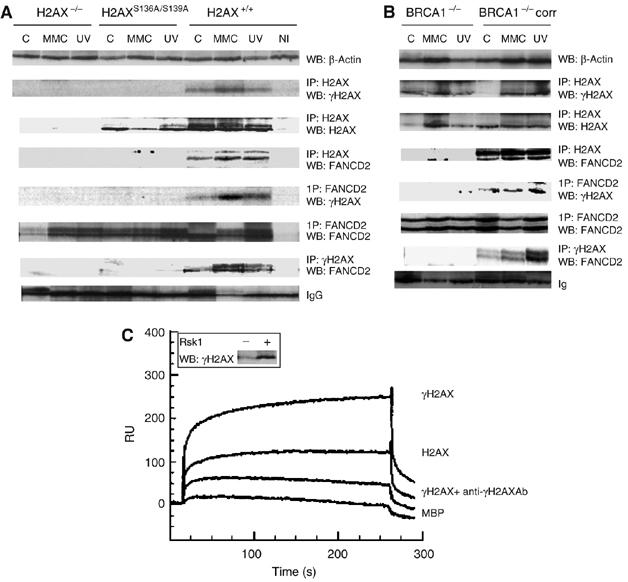
BRCA1-dependent binding of FANCD2 to γH2AX upon DNA damage. FANCD2 co-immunoprecipitates with γH2AX after DNA damage in a BRCA1-dependent manner. H2AX+/+, H2AXS136A/S139A, H2AX−/− MEF (A) or BRCA1−/− and BRCA−/− corrected cells (B) were not treated (−), MMC-treated (MMC) or UVC-irradiated (UV) as described in Materials and methods. Cells were lysed and immunoprecipitated with γH2AX, H2AX, FANCD2 or non-immune control (NI) antibodies followed by Western blot, and the protein bands were detected by probing with FANCD2, H2AX or γH2AX antibody, respectively. Loading control was monitored either by β-actin Western (A, B, upper row) or nonspecific binding to IgG (A, B, lowest row). Steady-state levels of γH2AX, FANCD2 or β-actin are maintained by Western blot of the corresponding samples before IP. In panels A and B, blots 2–4 and 5–7 represent H2AX IPs and FANCD2 IPs, respectively. Direct binding of FANCD2 to γH2AX by SPR (C). Equal aliquots of H2AX and H2AX phosphorylated by Rsk1 kinase were monitored by Western blot probed with γH2AX antibodies. Binding sensorgram of phosphorylated H2AX to the immobilized FANCD2. FANCD2 was immobilized (5 ng/mm2) on a dextran surface, as described in Materials and methods. Phosphorylated (A), nonphosphorylated (C) or phosphorylated H2AX premixed with anti-γH2AX antibody diluted at 1:500 (B) was injected over immobilized FANCD2. Control binding was performed on an unrelated (MBP) protein surface (C). The double bands of the FANCD2 WB panels in panels A and B correspond to FANCD2L (monoubiquitinated FANCD2; upper band) and FANCD2S (non-monoubiquitinated FANCD2).
FANCD2–γH2AX interaction was further studied by surface plasmon resonance (SPR) technique when comparing binding of recombinant human FANCD2 with γH2AX (Figure 5C). We recognized the increase of FANCD2 binding upon in vitro H2AX phosphorylation. This binding was abolished by coinjection of γH2AX with anti-γH2AX antibodies. Some binding was also observed for H2AX protein alone that could be due to the partial (less than 10%) H2AX phosphorylation in the sample. Thus, all these experiments allow us to conclude that in DNA-damaged cells, γH2AX is involved in the recruitment of FANCD2 to chromatin at stalled replication forks.
H2AX−/− cells are hypersensitive to MMC and both FANCD2 and H2AX cooperate in the same pathway in response to MMC
FA cells are phenotypically characterized by an increased sensitivity to the chromosome breaking ability of DNA crosslinkers such as MMC. In fact, the final diagnostic confirmation of FA is an excess of chromatid-type aberrations such as radial figures after treating the cells with crosslinking agents. Our data on the requirement of γH2AX for FANCD2 relocation but not for FANCD2 monoubiquitination strongly suggest that H2AX is important for the proper functioning of the FA/BRCA pathway. Consistent with this notion, H2AX−/− MEFs treated with MMC have an excess of chromatid-type aberrations, including radial figures, when compared to genetically matched wild-type MEF (Figure 6A and B). In addition, H2AX−/− MEF (Figure 6C) or H2AX-depleted wild-type MEF (Figure 6D) are hypersensitive to the cytotoxic effects of MMC, another hallmark of FA. A similar phenotype of an excess chromosome fragility was observed in H2AX KO MEFs reconstituted with a nonphosphorylable H2AX (Figure 6A and B). Thus, our data indicate that H2AX-deficient cells have FA-like cellular phenotype.
Figure 6.

H2AX deficiency leads to an excess of MMC-induced chromatid-type chromosomal aberrations and cytotoxicity. Excess of MMC-induced chromatid-type aberrations (A) and radial (B) in MEF derived from H2AX KO mice or H2AX−/− MEF expressing a nonphosphorylable H2AX (H2AXS136A/S139A). Means and s.d. of 2–4 experiments are shown. Differences between H2AX−/− and H2AXS136A/S139A are not statistically significant. Differences between wild-type and H2AX−/− or H2AXS136A/S139A cells are highly significant (P<0.001). H2AX-deficient cells are hypersensitive to the cytotoxic effect of MMC (C) (means and s.d. of 48 measures per dose and cell type are shown). Differences between wild-type and H2AX−/−cells are highly significant (P<0.001). Depletion of H2AX by RNAi in wild-type (but not in H2AX−/−) MEF increases cellular sensitivity to MMC (D). FANCD2 and H2AX function in the same pathway in response to MMC (E). FANCD2 depletion by RNAi does not further increase MMC sensibility in H2AX-deficient cells, suggesting that both FANCD2 and H2AX are in the same pathway. H2AX+/+, H2AX−/− and H2AXS136A/S139A MEFs were exposed to either FANCD2 RNAi or scramble RNAi (the same RNA sequence with five mismatches) and then exposed to MMC (200 nM for 24 h). FANCD2 immunoblot shows inhibition of FANCD2 synthesis by siRNA-FANCD2, not by scramble siRNA-FANCD2, irrespective of the H2AX genetic background (upper blot). The double bands of the FANCD2 WB panel in panel E correspond to FANCD2L (monoubiquitinated FANCD2; upper band) and FANCD2S (non-monoubiquitinated FANCD2). Percent of inhibition is calculated by TotaLab2.1 software as a ratio of FANCD2 RNAi and scRNA signals and normalized to the loading control signals (β-actin; upper blot, lowest row). The effect of FANCD2 depletion on MMC sensitivity in the three genetic backgrounds is shown in the graph. FANCD2 depletion by RNAi in wild-type cells increases MMC sensitivity. Similar and concurrent FANCD2 depletion in H2AX-deficient cells does not further increase MMC sensibility, suggesting that both FANCD2 and H2AX act in the same pathway in response to MMC.
Finally, we investigated whether H2AX and FANCD2 cooperate in response to DNA damage in the same pathway. For that purpose, MMC sensitivity was evaluated in H2AX+/+, H2AX−/− or H2AXS136A/S139A MEF after depleting FANCD2 by RNAi. As shown in Figure 6E, siRNA FANCD2 depletion led to an increased sensitivity to MMC in H2AX+/+cells, as expected. However, similar FANCD2 depletion did not further increase the sensitivity to MMC of H2AX−/− or H2AXS136A/S139A MEF. This indicates that FANCD2 and H2AX are likely to function in the same pathway in response to MMC.
Discussion
Understanding the role of the FA/BRCA pathway is essential not only for finding a cure for the disease but also for the general population as the FA pathway is a central node in a complex network of tumor suppressor and genome stability pathways (Surrallés et al, 2004; Venkitaraman, 2004), and its disruption leads to an increasing number of sporadic cancers (reviewed in Lyakhovich and Surrallés, 2006). However, the downstream function/s of the FA proteins is/are still a mystery. Here, we have focused our attention on the functional requirements of FANCD2, a key protein of this pathway as it is placed at the convergence point between ATM, ATR, BRCA1, BRCA2/FANCD1, NBS1, RAD51 and the upstream FA core complex proteins. In the present investigation, human or mouse fibroblasts with different genetic backgrounds and under various culture conditions were locally irradiated with UVC to induce subnuclear accumulation of stalled replication forks and the relocation of FANCD2 and other proteins to the site of damage was measured at different times after irradiation. This approach resulted to be a novel, practical and versatile functional assay for the FA/BRCA pathway as only when all known components of this pathway are fully functional, FANCD2 does normally relocate to the site of local irradiation.
Although FANCD2 relocates to UV-induced stalled replication forks, primary FA cells of complementation group FA-D2 (deficient in FANCD2) are only 20% more sensitive to UV than normal cells (Kalb et al, 2004). This sensitivity might be even stronger considering that all FA-D2 patients generally have hypomorphic mutations and express low levels of FANCD2 (Kalb et al, submitted). However, it is true but not surprising that, unlike XP cells, FA cells are not known for their hypersensitivity to UVC. The main reason is probably that the FA pathway is only the third line of defense, after nucleotide excision repair and translesion synthesis, against UV-induced DNA lesions. A similar picture occurs with ionizing radiation: FANCD2 gets phosphorylated by ATM and monoubiquitinated in response to ionizing radiation (Taniguchi et al, 2002b) but FA-D2 cells are not hypersensitive to ionizing radiation. Understanding the differential spectrum of mutagen sensitivities between genome instability syndromes that crosstalk at the molecular level is an important avenue for future research.
Here, we show that the ATR substrate histone H2AX is not required for FANCD2 monoubiquitination in response to UVC or MMC. However, we cannot disregard the possibility of reduced levels of FANCD2 monoubiquitination at longer times of mutagen exposure in H2AX-deficient cells as some differences in intensities have been observed between experiments. This might as well be explained by the variations in the proportion of cells in S phase between H2AX-proficient and -deficient genetic backgrounds (see Results). An alternative, yet speculative, possibility is that the active FANCD2 is more easily deubiquitinated in H2AX-deficient cells as it is not properly retained at damaged chromatin. Importantly, γH2AX is strictly required for proper accumulation of active FANCD2 to the site of damage. Thus, FANCD2 phosphorylation and monoubiquitination are necessary but not sufficient for FANCD2 relocation to stalled replication forks. The observations that (i) FANCD2 does not relocate to the site of damage in H2AX-deficient cells expressing a nonphosphorylable H2AX, (ii) FANCD2 relocates to UVC spots in concert with H2AX phosphorylation and (iii) ATR is required for proper H2AX phosphorylation suggest that ATR-mediated phosphorylation of H2AX at stalled replication forks is an additional requirement for FANCD2 relocation. Consistent with this model, γH2AX and FANCD2 co-immunoprecipitate in response to DNA damage, both H2AX-deficient cells and H2AX−/− cells reconstituted with a nonphosphorylable H2AX have a FA-like cellular phenotype, and FANCD2 and H2AX cooperate in the same pathway in response to MMC. In addition, H2AX depletion by RNAi disrupts FANCD2 foci formation after ICL treatment and results in an increased MMC sensitivity. Thus, all avenues lead to the conclusion that γH2AX is a novel functional requirement of the FA/BRCA pathway to resolve stalled replication forks. It is currently accepted that UV-induced photoproducts can be bypassed by the HR machinery when nucleotide excision repair is saturated and our data suggest that the FANCD2–H2AX interaction is probably functional for the repair of these types of lesions at stalled replication forks, not for the general response to fork stalling.
The way in which BRCA1 affects the accumulation of FANCD2 at sites of DNA damage is not yet precisely defined. BRCA1 has been reported to mediate FANCD2 foci formation and to colocalize with FANCD2 at damaged sites (García-Higuera et al, 2001), but it does not interact with FANCD2 (Folias et al, 2002) and is dispensable for FANCD2 monoubiquitination (Vandenberg et al, 2003). However, lack of BRCA1 partially disrupts this post-translational modification (García-Higuera et al, 2001; Bruun et al, 2003). Consistent with previous data (Foray et al, 2003), H2AX is normally phosphorylated in response to MMC or UV even in the absence of BRCA1, but normal FANCD2 binding to γH2AX requires BRCA1. Thus, our results suggest that BRCA1 mediates the recruitment of FANCD2 by γH2AX to damaged chromatin.
The role of H2AX in genome stability is controversial. γH2AX has been reported to be essential for the recruitment of repair/DNA damage response proteins to the site of DNA damage (Paull et al, 2000) or replication break sites (Furuta et al, 2003). However, it is dispensable for the initial recognition of DNA breaks, suggesting that it is more involved in the retention or accumulation of repair factors than in the actual recruitment (Celeste et al, 2003a; Fernández-Capetillo et al, 2003). γH2AX may keep the broken chromosome ends tethered together or prevent the premature separation of chromosome ends as a safeguard against exchanges (Bassing and Alt, 2004; Fernández-Capetillo et al, 2004; Xie et al, 2004). γH2AX is not essential for NHEJ or HR in mammalian cells but, resembling the FA pathway, it appears to modulate both pathways probably because it increases the likelihood of assembling a functional repair complex or prevents the premature separation of chromosome ends by reorganizing the chromatin structure surrounding the DSB (Fernández-Capetillo et al, 2004: Xie et al, 2004). It is known that FANCD2 binds to chromatin once activated by DNA damage and that monoubiquitinated FANCD2 then recruits BRCA2/FANCD1 into chromatin complexes suggesting that the FA/BRCA pathway and FANCD2 function to load BRCA2/FANCD1 on to chromatin, a process required for normal homology-directed DNA repair (Wang et al, 2004). It is possible that γH2AX plays a key role in this process, although further studies are needed to elucidate this issue.
Resembling FA patients and FA KO mice with mutations in either upstream or downstream FA genes (Navarro et al, 2006), H2AX−/− KO mice are viable and exhibit defects in DNA repair, growth defects, male sterility (small testes), modest radiosensitivity and hypersensitivity to MMC (Celeste et al, 2002; this study). In addition, H2AX deficiency increases tumor susceptibility in synergism with P53 deficiency (Bassing et al, 2003; Celeste et al, 2003b), again resembling FANCD2-deficient mice (Houghtaling et al, 2005). This phenotypic coincidence and the herein reported functional involvement of H2AX in the FA pathway downstream of FANCD2 monoubiquitination open the possibility that H2AX might be a candidate FA gene different from BRCA2/FANCD1 and FANCJ. In a collaborative multicenter study, we recently reported the existence of a 13th FA protein downstream of FANCD2 monoubiquitination (de Vries et al, 2005). However, the sensitivity of H2AX-deficient cells is lower than that observed in FA cells and we have been unable to find H2AX mutations in any FA patient with normal FANCD2 monoubiquitination and normal BRCA2/FANCD1 and FANCJ (data not shown). This suggests that, resembling ATR or NBS1, H2AX interacts with the FA pathway to prevent MMC-induced damage but that H2AX itself is not a FA gene.
The data reported here and in previously published studies can be integrated in the following model: UVC (or MMC, HU, 8-MOP) induces DNA lesions that block replication forks when the cell is (or once the cell enters) in S phase. The stalled replication forks activate ATR, which will then phosphorylate both FANCD2 and H2AX. Phosphorylated FANCD2 is then monoubiquitinated at K561 by FANCL in concert with the rest of the FA complex. Phosphorylated H2AX then allows recruitment and/or retention of active FANCD2 to chromatin at the site of stalled replication forks in concert with BRCA1. Once the DNA lesions are repaired, no signal for ATR activation is present, leading to inactivation of the FA pathway. Another possibility is that chromatin remodeling at the site of irradiation mediated by γH2AX allows FANCD2 accumulation at the damaged site and subsequent interaction of FANCD2, ATR and the rest of the upstream components of the FA/BRCA pathway. However, our observation that FANCD2 is normally activated in the absence of H2AX is at variance with this second possibility.
Materials and methods
Cell lines and culturing
The following human transformed fibroblasts cell lines were used in this study: MRC5 and HeLa (wild type); PD20 (FANCD2−/−); PD20 retrovirally corrected with pMMp-FANCD2 cDNA; PD20 transduced with pMMp-FANCD2/K561R; PD20 transduced with the empty pMMp vector; FANCA−/−-transformed fibroblasts; FANCA−/− retrovirally corrected with wild-type FANCA cDNA; CAPAN1 (BRCA2/FANCD1-deficient); HCC1937 (BRCA1-deficient); HCC1937 corrected for BRCA1 deficiency by microcell-mediated chromosome transfer; AT-7 (ATM−/−) and F02/98 (ATR−/−). In addition, we used primary wild-type human skin fibroblasts and MEFs derived from histone H2AX KO mice, wild-type genetically matched counterparts, as well as H2AX KO MEFs reconstituted with a cDNA coding for a nonphosphorylable H2AX (H2AXS136A/S139A). All the cell lines were cultured as described previously (Bogliolo et al, 2000; Callén et al, 2002; Surrallés et al, 2002).
Local UV irradiation
A discrete nuclear area of human fibroblasts of diverse genetic backgrounds and under various culture conditions was locally irradiated with UVC light by covering the cells during irradiation with a filter with 5-μm-diameter pores. The site of irradiation (UV spot) was visualized with antibodies against CPD and the relocation of FANCD2 or other proteins of interest to the site of damage was measured at different times using specific antibodies. All the relocation experiments were repeated at least three times and 200 locally irradiated nuclei per time point and experiment were analyzed to measure the percentage of UV spots containing the protein of interest.
The local UVC irradiation was performed essentially by the method of Volker et al (2001) with some minor modifications. Briefly, cells were seeded on 18 × 18 mm sterile coverslips and grown to near-confluency. Before irradiation, the medium was aspirated and the cells were washed with phosphate-buffered saline (PBS). The cells on the coverslips were then covered with an isopore polycarbonate filter with pores of 5-μm-diameter (Millipore, Bedford, MA, USA) and exposed to UVC from above with a Philips 15 W UVC lamp G15-T8. The UVC radiation dose was measured with a VLX-3W radiometer with a CX-254 sensor (Vilber Lourmat, France). Following this procedure, only the light passing through the pores locally irradiated a 5 μm diameter area of the nuclei. The size of the irradiated area was confirmed by confocal microscopy measurements (data not shown). Subsequently, the filter was removed, fresh prewarmed medium was added back to the cells, and cells were returned to culture conditions or immediately processed.
Cell cycle analysis and flow cytometry
Primary fibroblasts growing on coverslips or in normal tissue flasks (unsynchronized) or cells subjected to serum starvation were harvested for flow cytometric analysis of the cell cycle. The harvested cells were washed twice in PBS and fixed in cold 70% ethanol for at least 30 min at 4°C. The samples were then centrifuged at 2000 r.p.m. and the pellets washed with PBS and resuspended in 1 ml of PBS. The samples were then treated with RNAse (200 μg/ml final concentration), and 20 μg/ml of propidium iodide was added to each sample. The cell-cycle distributions were then analyzed with the flow cytometer FACScalibur (Becton Dickinson).
Immunohistochemistry and microscopy
Cells were washed with PBS and subsequently fixed by adding PBS containing 4% formaldehyde (Sigma-Aldrich, St Louis, MO, USA) for 10 min at room temperature (RT). Cells were washed with PBS and incubated with PBS and 0.5% Triton (Sigma-Aldrich) for 15 min at RT. To visualize the site of UV irradiation, cells were washed twice with PBS, treated with 1 M HCl for 5 min at RT to denature the DNA and washed once with PBS. Cells were subsequently rinsed with a washing buffer (WB) consisting of 3% bovine albumin (Sigma-Aldrich) and 0.05% Tween 20 (Sigma-Aldrich) in PBS, incubated with a mouse primary anti-CPD antibody at 1:200 dilution in WB for 45 min at 37°C, and washed for 15 min in WB with gentle agitation. In experiments requiring double labeling, the primary rabbit antibodies against the specific proteins were mixed in WB in the appropriate dilutions and incubated simultaneously with the anti-CPD antibody. The antibodies used and the corresponding working dilutions were as follows: anti-FANCD2 (Abcam, Cambridge, UK; 1:200; polyclonal); anti-FANCD2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:200; monoclonal); anti-γH2AX (R&D Systems, Minneapolis, MN, USA; 1:100; rabbit polyclonal); anti-PCNA (Becton Dickinson; 1:10 000). Incubation with secondary antibodies anti-mouse Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA) and anti-rabbit Alexa Fluor 546 (Molecular Probes) diluted in WB was performed at 37°C for 20 min followed by a 15 min washing step in WB with gentle agitation. After the last antibody labeling step, the preparations were mounted in anti-fading medium containing 4′-6′-diamidino-2-phenylindole (DAPI) as a DNA counter stain (Vector Laboratories, Burlingame, CA, USA). Using this color combination, nuclei were visualized in blue, the site of UV irradiation in green and the specific proteins in red. Microscopic analysis and quantification were performed using an Olympus BX-50 fluorescence microscope. Images were captured with either a laser confocal microscope TCS 4D (Leica Microsystems GmbH, Heidelberg, Germany) or a Leica DMRB epifluorescence microscope equipped with a DC 200 digital camera and the Leica DC Viewer capturing software. Images were finally computer-edited using the Adobe Photodeluxe 1.0 program.
FANCD2 monoubiquitination studies
Cell extracts were prepared basically by the method of Tanaka et al (1992) with some modifications. Briefly, harvested cells were washed three times with PBS and resuspended in buffer I (10 mM Tris–HCl pH 7.8 and 200 mM KCl) at 1 × 106 cells/10 μl. After the addition of an equal volume of buffer II (10 mM Tris–HCl, pH 7.8, 200 mM KCl, 2 mM EDTA, 40% glycerol, 0.2% Nonidet P-40, 2 mM dithiothreitol, 0.5 mM PMSF, 10 mg/ml aprotinin, 5 mg/ml leupeptin and 1 mg/ml pepstatin), the cell suspension was stirred for 1 h at 4°C and centrifuged at 13 000 r.p.m. for 15 min. The total protein concentration in the supernatant was then measured using the Bio-Rad Protein Assay (Biorad, Hercules, CA, USA) according to the manufacturer's instructions. Total proteins (50–100 μg) were then loaded on a 7% SDS–PAGE and subjected to standard Western blot procedure followed by immunodetection with monoclonal anti-human FANCD2 (1:250 Santa Cruz Biotechnology).
Cytogenetic and cell viability assays for MMC sensitivity
Exponentially growing MEFs with different H2AX genetic backgrounds were allowed to attach to coverslips for 24 h and subsequently treated with 50 nM MMC for 2 days. Concurrently growing cultures were left untreated. Cells were then harvested and Giemsa-stained following standard cytogenetic methods. A total of 20–60 well-spread metaphases were scored per treatment point and all the experiments were repeated 2–4 times. For cell growth/viability assay, cells were seeded in 96-well ELISA microtiter plates at a density of 2000 cells in a final volume of 100 μl culture medium per well. The cells were allowed to grow for 24 h and MMC was added at increasing concentrations for 1 week. Then the viability of the cells was assessed with Cell Proliferation KitII (XTT) (Roche, Basel, Switzerland) according to the manufacturer's instructions. The plates were then read with a microtiter plate ELISA reader. A total of 48 wells per dose and cell line were measured.
Immunoprecipitation experiments
IP was carried out as described previously (Lyakhovich and Shekhar, 2003). Briefly, 1 μg of anti-γH2AX, H2AX (Abcam, UK), FANCD2 (FI17, Santa Cruz Biotechnology Inc.) or unrelated (non-immune control) antibody was bound to protein A-agarose beads (Invitrogen) in the presence of ethidium bromide (20 μg/ml) and incubated with appropriate cell lysates overnight at 4°C. After extensive washing with PBS buffer, beads were eluted by boiling in SDS gel sample buffer and proteins were separated by SDS–PAGE and immunoblotted onto nylon membrane. Protein bands were visualized with anti-rabbit or anti-mouse IgG coupled to horseradish peroxidase using the ECL kit (Amersham, Arlington Heights, IL, USA).
RNAi and cytotoxicity
RNAi experiments were maintained as described by Nijman et al (2005). Shortly, cells were grown in RPMI medium and transfected with FANCD2 siRNA (Invitrogen, primer number 97480D01), set of H2AFX StealthTM RNAs (Invitrogen HSS142373; HSS142373; HSS142374) or corresponding scrambled RNA by Lipofectamin 2000 transfection (Invitrogen) two times with the an interval of 24 h. After second transfection, cells were either harvested to prepare lysates or submitted to MMT cytotoxicity assay. The sensitivity of siRNA-transfected MEF cells was evaluated using an MTT assay as described previously (Zhang et al, 2004). Immediately after second transfection, cells seeded into 96-well plates with 100 μl of culture medium were treated with indicated concentrations of MMC for 5 days and processed for viability assay as above.
Binding affinity measurements by SPR
Interaction between immobilized FANCD2 and phosphorylated and nonphosphorylated H2AX was determined using SPR technique on a BIACORe 2000 (BIAcore AB, Max Planc Institute, Munich). H2AX (80 μM) was phosphorylated by incubation with the Rsk1 kinase as described previously (Keogh et al, 2006). Immobilization was carried out to the activated carboxyl groups of CM5 sensor chip by coupling reagent 2-(2-pyridinyldithio)ethaneamine according to the manufacturer's procedures. Analytes were dissolved in 10 mM HEPES pH 7.4 containing 150 mM NaCl, 3 mM EDTA and 0.005% surfactant P-20, and were injected over the sensor chip at a flow rate of 10 μl/min at 25°C. Specificity of interaction was monitored by competition assay with analyte premixed with corresponding anti-phosphorylated H2AX antibody. Control experiments were performed on an unrelated (MBP) protein surface. The blank sensorgram was subtracted from the assay curve. All data were interpreted using the BioEvaluation 3.2 software (Biacore).
Chromatin fractionation experiments
All chromatin fractionation experiments are based on previous reports (Mendez and Stillman, 2000; Zou et al, 2002): briefly, 3 × 106 cells were washed twice with PBS (Ca2+- and Mg2+-free) and resuspended in solution A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol, 10 mM NaF, 1 mM Na2VO3, protease inhibitors (Roche)). Triton X-100 was added to a final concentration of 0.1%, and the cells were incubated for 5 min on ice. Cytosolic and soluble proteins (S1) were separated from nuclei by centrifugation (4 min, 1300 g). Nuclei were washed once in solution A, and then lysed in solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, protease inhibitors) for 30 min at 4°C. Insoluble chromatin was then separated from soluble nuclear proteins (S2) by centrifugation (4 min, 1700 g), washed once in solution B and collected by centrifugation (1 min, 10 000 g). The final chromatin pellet (P2) was resuspended in SDS sample buffer and sonicated. In the case of MEF cells analysis, the soluble fractions (S1 and S2) were pooled (‘Sol' in Figure 4B) and the equivalent of 50 μg of total proteins of the Sol fraction and of the chromatin fraction (‘Insol' in Figure 4B) was analyzed by Western blot. FANCD2 was detected with the Santa Cruz sc-28194 rabbit polyclonal antibody. 8-MetoxyPsoralen (Mop) treatment was as follows: cells were exposed to 10 μM 8-MOP (Sigma) for 20 min followed by 10 kJ/m2 of UVA and they were recovered for the indicated times and then fractionated.
Acknowledgments
We are grateful to Drs Barbara Cox (Portland, USA), Alan D D'Andrea (Boston, USA), Anna Genescà (Barcelona, Spain), Mien-Chie Hung (Houston, USA), Penny Jeggo (Sussex, UK), Andre Nussenzweig (Bethesda, USA), Tsukusa Matsunaga (Kanazawa, Japan), Jeff Parvin (Boston, USA), Filippo Rosselli (Villejuif, France) Joseph S Siino (Davis, USA), Margaret Z Zdzienicka (Leiden, The Netherlands) and Marina Zhuravleva (Bethesda, USA) for sharing materials; to Dr Nosov (Munich, Germany) for the help with SPR experiments; to Mr Jean Hugues Guervilly for technical help with the cell fractionation experiments; and to the Universitat Autònoma de Barcelona (UAB) Microscopy Service for image acquisition. FA research in JS laboratory is funded by the Generalitat de Catalunya (SGR-00197-2002), the La Caixa Fundation Oncology Program (BM05-67-0), Genoma España, the Spanish Ministry of Health and Consumption (projects FIS PI020145, FIS PI051205, FIS G03/073 and CIBER-ER CB06/07/0023), the Spanish Ministry of Science and Technology (projects SAF2001-5138, SAF2002-11833-E, SAF2003-020328, SAF2004-20372-E and SAF2006-3440), the Commission of the European Union (projects HPMF-CT-2001-01330, FIGH-CT-2002-00217 and FI6R-CT-2003-508842) and the European Regional Development Funds. MB was supported by a postdoctoral Marie Curie fellowship awarded by the Commission of the European Union (contract HPMF-CT-2001-01330).
References
- Abraham RT (2001) Cell cycle checkpoint signalling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Andreassen PR, D'Andrea AD, Taniguchi T (2004) ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 18: 1958–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW (2003) Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114: 359–370 [DOI] [PubMed] [Google Scholar]
- Bassing CH, Alt FW (2004) H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle 3: 149–153 [DOI] [PubMed] [Google Scholar]
- Bogliolo M, Taylor RM, Caldecott KW, Frosina G (2000) Reduced ligation during DNA base excision repair supported by BRCA2 mutant cells. Oncogene 19: 5781–5787 [DOI] [PubMed] [Google Scholar]
- Bogliolo M, Surrallés J (2005) The Fanconi anemia/BRCA pathway: FANCD2 at the crossroad between repair and checkpoint response to DNA damage. In: DNA Repair and Human Diseases, A Balajee (ed) New York: Ed. Landes Bioscience Ed. [Google Scholar]
- Bruun D, Folias A, Akkari Y, Cox Y, Olson S, Moses R (2003) siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair 2: 1007–1013 [DOI] [PubMed] [Google Scholar]
- Callén E, Samper E, Ramírez MJ, Creus A, Marcos R, Ortega JJ, Olivé T, Badell I, Blasco MA, Surrallés J (2002) Breaks at telomeres and TRF2-independent end fusions in Fanconi anemia. Hum Mol Genet 11: 439–444 [DOI] [PubMed] [Google Scholar]
- Callén E, Casado JA, Tischkowitz MD, Bueren JA, Creus A, Marcos R, Dasi A, Estella JM, Muñoz A, Ortega JJ, de Winter J, Joenje H, Schindler D, Hanenberg H, Hodgson SV, Mathew CG, Surrallés J (2005) A common founder mutation in FANCA underlies the world highest prevalence of Fanconi anemia in gypsy families from Spain. Blood 105: 1946–1949 [DOI] [PubMed] [Google Scholar]
- Castillo V, Cabré O, Marcos R, Surrallés J (2003) Molecular cloning of the Drosophila Fanconi anaemia gene FANCD2 cDNA. DNA Repair 2: 751–758 [DOI] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernández-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A (2003b) H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 114: 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernández-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A (2003a) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675–679 [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernández-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A (2002) Genomic instability in mice lacking histone H2AX. Science 296: 922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea AD (2003) The Fanconi road to cancer. Genes Dev 15: 1933–1936 [DOI] [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M (2003) The Fanconi anaemia/BRCA pathway. Nat Rev Cancer 3: 23–34 [DOI] [PubMed] [Google Scholar]
- De Vries Y, Levitus M, Steltenpool J, Rooimans MA, Thacker J, Surralles J, Arwert F, Joenje H, de Winter JP (2005) Evidence for a new complementation group with a defect downstream of FANCD2 in the Fanconi anemia pathway. Seventeenth Fanconi Anemia Research Fund Scientif Symposium, Abstract book, p 77
- Fernández-Capetillo O, Celeste A, Nussenzweig A (2003) Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle 2: 426–427 [PubMed] [Google Scholar]
- Fernández-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: the histone guardian of the genome. DNA Repair 3: 959–967 [DOI] [PubMed] [Google Scholar]
- Folias A, Matkovic M, Bruun D, Reid S, Hejna J, Grompe M, D'Andrea A, Moses R (2002) BRCA1 interacts directly with the Fanconi anemia protein FANCA. Hum Mol Genet 11: 2591–2597 [DOI] [PubMed] [Google Scholar]
- Foray N, Marot D, Gabriel A, Randrianarison V, Carr AM, Perricaudet M, Ashworth A, Jeggo P (2003) A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J 22: 2860–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y (2003) Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 278: 20303–20312 [DOI] [PubMed] [Google Scholar]
- García-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- Ho GP, Margossian S, Taniguchi T, D'Andrea AD (2006) Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol 26: 7005–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghtaling S, Granville L, Akkari Y, Torimaru Y, Olson S, Finegold M, Grompe M (2005) Heterozygosity of p53, Trp53−/−, accelerated epithelial tumor formation in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Cancer Res 65: 85–91 [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005) The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet 14: 693–701 [DOI] [PubMed] [Google Scholar]
- Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, de Winter JP, Ashworth A, Jones NJ, Mathew CG (2004) Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet 13: 1241–1248 [DOI] [PubMed] [Google Scholar]
- Kalb R, Duerr M, Wagner M, Herterich S, Gross M, Digweed M, Joenje H, Hoehn H, Schindler D (2004) Lack of sensitivity of primary Fanconi's anemia fibroblasts to UV and ionizing radiation. Radiat Res 161: 318–325 [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, Lieberman J, Shen X, Buratowski S, Haber JE, Durocher D, Greenblatt JF, Krogan NJ (2006) A phosphatase complex that dephosphorylates γH2AX regulates DNA damage checkpoint recovery. Nature 439: 497–501 [DOI] [PubMed] [Google Scholar]
- Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD (2003) A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 101: 1249–1256 [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, Cimprich KA (2002) A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev 16: 2327–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyakhovich A, Shekhar MP (2003) Supramolecular complex formation between Rad6 and proteins of p53 pathway during DNA damage-induced response. Mol Cell Biol 23: 2463–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyakhovich A, Surrallés J (2006) Disruption of the Fanconi anemia/BRCA pathway in sporadic cancer. Cancer Lett 232: 99–106 [DOI] [PubMed] [Google Scholar]
- Macé G, Bogliolo M, Guervilly JH, Dugas du Villard JA, Rosselli F (2005) 3R coordination by Fanconi anemia proteins. Biochimie 87: 647–658 [DOI] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003a) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35: 165–170 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003b) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23: 3417–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Stillman B (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca R, Andreassen PR, Margossian SP, Gregory RC, Taniguchi T, Wang X, Houghtaling S, Grompe M, D'Andrea AD (2005) Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood 105: 1003–1009 [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D'Andrea AD (2002) Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol 12: 913–920 [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yang Y-G, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang Z-Q, Jasin M (2005) Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci USA 102: 1110–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro S, Meza NW, Quintana-Bustamante O, Casado JA, Jacome A, McAllister K, Puerto S, Surralles J, Segovia JC, Bueren JA (2006) Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol Ther 14: 525–535 [DOI] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R (2005) The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 3: 331–339 [DOI] [PubMed] [Google Scholar]
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ (2004) The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell 15: 607–620 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR (1998) Involvement of Brca2 in DNA repair. Mol Cell 1: 347–357 [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10: 886–895 [DOI] [PubMed] [Google Scholar]
- Pichierri P, Rosselli F (2004a) Fanconi anemia proteins and the s phase checkpoint. Cell Cycle 3: 698–700 [PubMed] [Google Scholar]
- Pichierri P, Rosselli F (2004b) The DNA crosslink-induced S-phase checkpoint depends on ATR–CHK1 and ATR–NBS1–FANCD2 pathways. EMBO J 10: 1178–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Averbeck D, Rosselli F (2002) DNA cross-link-dependent RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C protein. Hum Mol Genet 11: 2531–2546 [DOI] [PubMed] [Google Scholar]
- Pichierri P, Franchitto A, Rosselli F (2004) BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J 23: 3154–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuss A, Grompe M (2004) Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol 24: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y (2001) ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 11: 71–77 [DOI] [PubMed] [Google Scholar]
- Surrallés J, Jackson SP, Jasin M, Kastan MB, West SC, Joenje H (2004) Molecular cross talk among chromosome fragility syndromes. Genes Dev 18: 1359–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrallés J, Ramírez MJ, Marcos R, Natarajan AT, Mullenders LHF (2002) Clusters of transcription-coupled repair in the human genome. Proc Natl Acad Sci USA 99: 10571–10574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Lai JS, Herr W (1992) Promoter-selective activation domains in Oct-1 and Oct-2 direct differential activation of an snRNA and mRNA promoter. Cell 68: 755–767 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD (2002a) S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100: 2414–2420 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D'Andrea AD (2002b) Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109: 459–472 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Thompson L (2005) Unraveling the Fanconi anemia–DNA repair connection. Nat Genet 37: 921–922 [DOI] [PubMed] [Google Scholar]
- Vandenberg CJ, Gergely F, Ong CY, Pace P, Mallery DL, Hiom K, Patel KJ (2003) BRCA1-independent ubiquitination of FANCD2. Mol Cell 12: 247–254 [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2002) Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108: 171–182 [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2004) Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer 4: 266–276 [DOI] [PubMed] [Google Scholar]
- Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8: 213–224 [DOI] [PubMed] [Google Scholar]
- Wang X, Andreassen PR, D'Andrea AD (2004) Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol 24: 5850–5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Chen J (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R (2004) Control of sister chromatid recombination by histone H2AX. Mol Cell 16: 1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang X, Bai CX, Chen J, Wei MQ (2004) Inhibition of epidermal growth factor receptor by RNA interference in A549 cells. Acta Pharmacol Sin 25: 61–67 [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ (2002) Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes and Dev 16: 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]