

Abstract
Actin polymerization plays a critical role in clathrin-mediated endocytosis in many cell types, but how polymerization is regulated is not known. Hip1R may negatively regulate actin assembly during endocytosis because its depletion increases actin assembly at endocytic sites. Here, we show that the C-terminal proline-rich domain of Hip1R binds to the SH3 domain of cortactin, a protein that binds to dynamin, actin filaments and the Arp2/3 complex. We demonstrate that Hip1R deleted for the cortactin-binding site loses its ability to rescue fully the formation of abnormal actin structures at endocytic sites induced by Hip1R siRNA. To determine when this complex might function during endocytosis, we performed live cell imaging. The maximum in vivo recruitment of Hip1R, clathrin and cortactin to endocytic sites was coincident, and all three proteins disappeared together upon formation of a clathrin-coated vesicle. Finally, we showed that Hip1R inhibits actin assembly by forming a complex with cortactin that blocks actin filament barbed end elongation.
Keywords: actin, cortactin, endocytosis, Hip1R, huntingtin
Introduction
Several lines of evidence support a role for actin polymerization in clathrin-mediated endocytosis (CME). In yeast and in many mammalian cells, actin assembly is transiently nucleated at coated pits (Merrifield et al, 2002; Kaksonen et al, 2003), and the impairment of actin polymerization affects vesicle scission and many aspects of clathrin-coated pit (CCP) dynamics (Engqvist-Goldstein and Drubin, 2003; Merrifield et al, 2005; Yarar et al, 2005). On the basis of protein interactions, several pathways could link the actin nucleating Arp2/3 complex to dynamin, a large GTPase that is required for vesicle scission in mammals. The proline-rich domain (PRD) of dynamin is capable of binding to intersectin, endophilin A and syndapin 1, which might recruit N-WASP (Hussain et al, 2001; Kessels and Qualmann, 2002, 2006; Da Costa et al, 2003; Otsuki et al, 2003). N-WASP, in turn, activates the Arp2/3 complex to initiate actin assembly associated with CME (Merrifield et al, 2004; Benesch et al, 2005; Innocenti et al, 2005). Dynamin also binds to cortactin (McNiven et al, 2000), another Arp2/3 complex activator (Uruno et al, 2001; Weaver et al, 2001). Cortactin is localized at CCPs (Cao et al, 2003; Engqvist-Goldstein et al, 2004) and, interestingly, the timing of cortactin recruitment coincides with vesicle scission (Merrifield et al, 2005). Cortactin is also required for the clathrin-dependent entry of Listeria into host cells (Veiga and Cossart, 2005).
Currently, how Arp2/3 activators are regulated at endocytic sites is not clear. Recent data suggested that Hip1R, an F-actin and clathrin-binding protein (Engqvist-Goldstein et al, 1999, 2001), and its yeast homologue, Sla2p, negatively regulate actin assembly associated with CME (Kaksonen et al, 2003; Engqvist-Goldstein et al, 2004) and clathrin-coated vesicle (CCV) budding from the trans-Golgi network (Carreno et al, 2004). However, the mechanism by which Hip1R inhibits actin assembly is currently not known. In addition, Hip1R binds to CIN85 (Cbl-interacting protein of 85 kDa), an SH3 domain-containing adaptor protein that controls trafficking of EGF receptors along the endocytic and recycling pathways (Kowanetz et al, 2004). Here, we provide evidence for a Hip1R–cortactin complex in vitro and in vivo. We show that the complex contributes to the regulation of actin assembly in HeLa cells and we establish the relative timing of recruitment of these proteins to endocytic sites. Specifically, we show that the Hip1R–cortactin complex inhibits actin assembly by capping filament barbed ends.
Results
Hip1R interacts directly with cortactin
Because Hip1R depletion leads to cortactin-dependent actin polymerization at coated pits (Engqvist-Goldstein et al, 2004), we tested for an interaction between these proteins. Cortactin was immunoprecipitated from clathrin coat extracts, and the immunoprecipitates were probed with an anti-Hip1R antibody (see Supplementary Materials and methods). The coprecipitation of Hip1R with cortactin suggests that a Hip1R-cortactin complex is present at the surface of CCVs (Figure 1A). The comparison between fractions of Hip1R bound and unbound to cortactin indicated that the totality of cortactin was immunoprecipitated but only a small amount of Hip1R is coprecipitated, suggesting that cortactin is not a structural protein of CCVs (Figure 1A). To test whether this interaction is direct, recombinant GST-tagged cortactin was used in attempts to pull down recombinant His-tagged Hip1R. We discovered that these two proteins interact directly (Figure 1B). To map precisely the Hip1R-binding domain of cortactin, we used GST fusions of cortactin to pull down His-tagged Hip1R (see Supplementary Materials and methods). We identified the SH3 domain (amino acids 491–546) of cortactin as the Hip1R-binding site (Figures 1B and D). We also quantified the affinity of the interaction between Hip1R and cortactin using a pull-down assay, and found a high affinity (Kd=20 nM, Figure 1C, see Supplementary Materials and methods). To map the cortactin-binding site of Hip1R, we used two constructs of Hip1R truncated from the N- and C-terminal domains, which contain, respectively, the ANTH domain that binds phosphatidylinositol-4,5-bisphosphate (PIP2), and the talin-like domain that binds F-actin. Although deletion of the N-terminal domain results in a lower affinity, deletion of the C-terminal domain had a much more severe effect (Figure 2A). To confirm the importance of the C-terminal domain, we used several GST fusions of Hip1R domains to pull down His-tagged cortactin. Only the C-terminal domain binds to cortactin (Figure 2B), albeit with a lower apparent affinity compared to full-length Hip1R. Interestingly, the C-terminal PRD of Hip1R contains several known and putative SH3-binding motifs within the last 51 amino acids (1018–1068) (Kowanetz et al, 2004). To characterize further this cortactin-binding domain, we deleted the last 51 amino acids of Hip1R. We found that this deleted version of Hip1R (1–1017) did not bind to the SH3 domain of cortactin or to full-length cortactin (Figure 2C). In total, these data show that the SH3 domain of cortactin binds to the C-terminal PRD of Hip1R, but that high-affinity binding is dependent upon the presence of the N-terminal domain of Hip1R (Figures 1D and 2D).
Figure 1.

Hip1R interacts with the SH3 domain of cortactin. (A) co-immunoprecipitation of Hip1R with cortactin from a clathrin coat extract. Cortactin immunoprecipitates (IP) and controls lacking antibody (C) were blotted with anti-Hip1R and anti-cortactin antibodies. (B) His-tagged Hip1R (40 nM) pull-down assays using GST and GST-cortactin 1–546; 1–81; 1–326; 324–546; 491–546 (10 μM). Bound (P) and unbound (S) fractions were analyzed by Western blotting (WB) using an anti-His tag antibody (upper panel) and by Coomassie blue staining (lower panel). (C) His-tagged Hip1R (1 nM) pull-down assays using the indicated concentrations of GST-cortactin. Total input (T) and bound fractions were probed with an anti-His tag antibody. The results were quantified to obtain the binding curve. The best fit of the curve gave a Kd=20 nM. (D) Summary of the binding assay results. All the experiments were performed three times with similar results.
Figure 2.

The C-terminal proline-rich region of Hip1R interacts with cortactin. (A) His-tagged Hip1R 1–1068; 346–1068 and 1–655 (400 nM) pull-down assays using GST and GST-cortactin (800 nM). Bound and unbound fractions were analyzed by Coomassie blue staining (upper panel), and by Western blotting using an anti-Hip1R antibody (lower panel). (B) His-tagged cortactin (400 nM) pull-down assays using GST and GST-Hip1R 1–312; 346–655; 766–1068 (10 μM). Bound and unbound fractions were analyzed by Coomassie blue staining (upper panel), and by Western blotting using an anti-cortactin antibody (lower panel). (C) His-tagged Hip1R 1–1017 (400 nM) pull-down assays using GST, GST-cortactin (1–546) and GST-SH3-cortactin (491–546) (800 nM). Bound and unbound fractions were analyzed by Western blotting using an anti-His tag antibody (upper panel) and by Coomassie blue staining (middle panel). The same experiment was carried out in parallel with His-tagged Hip1R 1–1068 (full length) as a positive control, bound and unbound fractions were analyzed by Western blotting using an anti-His tag antibody (lower panel). (D) Summary of the binding assay results. All experiments were performed three times with similar results.
A Hip1R–cortactin complex contributes to the regulation of actin assembly at endocytic sites
To investigate further the involvement of the Hip1R–cortactin complex in the regulation of actin dynamics in vivo, we used the mutant of Hip1R deleted for the last 51 amino acids (Hip1R 1–1017) that no longer binds to cortactin in vitro (Figure 2C). To test the effect of this Hip1R mutant on actin dynamics in vivo, we treated HeLa cells with RNAi to knock down endogenous Hip1R protein levels. This was shown to lead to the formation of abnormal actin structures at endocytic sites (Engqvist-Goldstein et al, 2004). These structures were also dependent on the presence of cortactin as double knockdown of Hip1R and cortactin led to a decrease in the number of these structures (Engqvist-Goldstein et al, 2004). Expression of wild-type Hip1R protein (Hip1R 1–1068) resulted in loss of these abnormal actin structures, indicating that Hip1R is necessary to negatively regulate actin dynamics at endocytic sites (Engqvist-Goldstein et al, 2004). To determine if the binding of cortactin to Hip1R contributes to this inhibition of actin assembly, we compared the ability of wild-type Hip1R (Hip1R 1–1068) and Hip1R 1–1017 to rescue the siRNA-dependent actin phenotype. When we reduced Hip1R levels in HeLa cells using the siRNA duplex A3 (described in Engqvist-Goldstein et al, 2004), 72.6±6.6% of the cells formed abnormal actin structures (Figure 3A and C), whereas only 7.5±1.2% of the control-treated cells (InvC1) contained these structures (Figure 3A and B). Expression of wild-type Hip1R in A3-treated cells abolished this phenotype and decreased the number of the structures to the level of the control (8.8±1.3%) (Figure 3A and D). In contrast, expression of Hip1R-1–1017 in A3-treated cells reproducibly failed to suppress the phenotype to the extent of the full-length protein because a significant portion of the cells (21.5±3.6%) showed abnormal actin structures (Figure 3A and E). Importantly, the A3-treated cells that were not rescued by the expression of Hip1R 1–1017 showed more and longer tails compared to the A3-treated cells that were not rescued by the wild-type Hip1R (Figure 3D and E). These results indicate that a functional Hip1R–cortactin complex contributes to normal actin turnover at endocytic sites.
Figure 3.
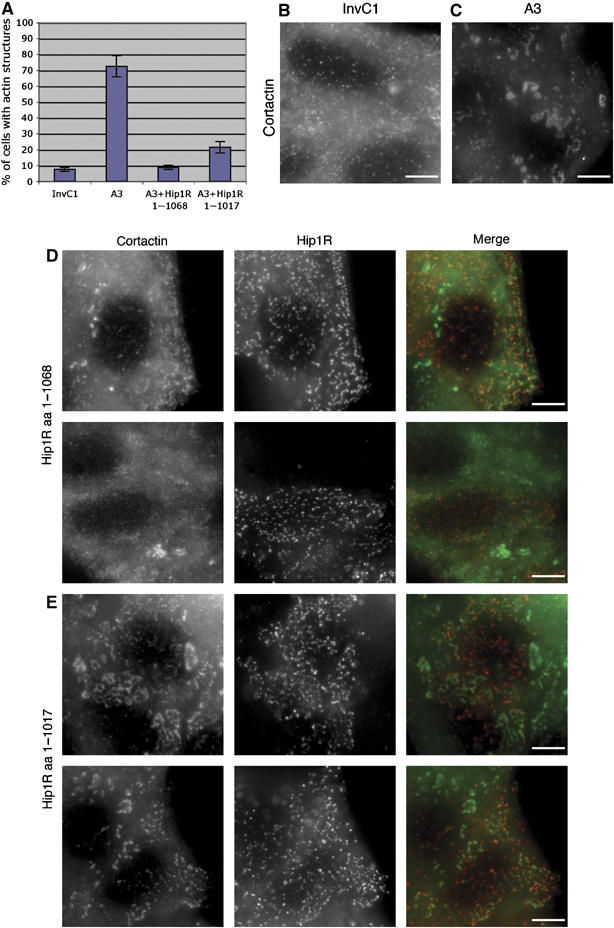
Hip1R–cortactin contributes to the regulation of actin assembly at endocytic sites. (A) The bar graph shows the percentage of HeLa cells showing abnormal actin structures after treatment with Hip1R siRNA duplex (A3), a control siRNA (InvC1), Hip1R siRNA and expression of wild-type Hip1R (A3+Hip1R 1–1068), Hip1R siRNA and expression of Hip1R 1–1017 (A3+Hip1R 1–1017). (B–E) Representative pictures of cells. (B) Cortactin staining in control siRNA (InvC1)-treated cells. (C) Cortactin staining in Hip1R siRNA (A3)-treated cells. (D–E) Representative pictures of non-rescued cells with remaining abnormal actin structures. (D) Hip1R siRNA-treated cells expressing wild-type Hip1R (1–1068). (E) Hip1R siRNA-treated cells expressing mutant Hip1R 1–1017. Cortactin staining (left panels), Hip1R staining (middle panels), merge (right panels). Scale bars are 10 μm.
Real-time imaging of Hip1R and cortactin at CCPs
To determine when this Hip1R–cortactin complex might function during endocytosis, we quantitatively analyzed the temporal appearance of Hip1R and cortactin at endocytic sites. Previous immuno-EM studies revealed that both proteins are present during all stages of CCP formation (Engqvist-Goldstein et al, 2001; Cao et al, 2003). In addition, Hip1R appeared to link actin filaments to CCPs, whereas cortactin was found on actin filaments and actin filament branches throughout the plasma membrane (Engqvist-Goldstein et al, 2001; Cao et al, 2003). Total internal reflection fluorescence microscopy (TIR-FM) was performed to image Hip1R-GFP or cortactin-GFP on the surfaces of cells stably expressing DsRed-clathrin (Figure 4). Consistent with previous reports, Hip1R appeared at essentially all of the clathrin patches, whereas cortactin appeared at about one-third of the clathrin patches (Engqvist-Goldstein et al, 2004; Merrifield et al, 2005). We found that Hip1R recruitment follows the course of CCP growth, albeit with a small delay compared to clathrin (Figure 4A, C and E). When a CCP started to disappear, Hip1R behavior was essentially the same as that of clathrin. These observations are consistent with previous observations, but with greater time resolution (Engqvist-Goldstein et al, 2001; Keyel et al, 2004). In contrast, cortactin recruitment was more precipitous, occurring close to the time of CCV formation, and cortactin fluorescence lingered slightly longer after clathrin disappeared (Figure 4B, D and E).
Figure 4.
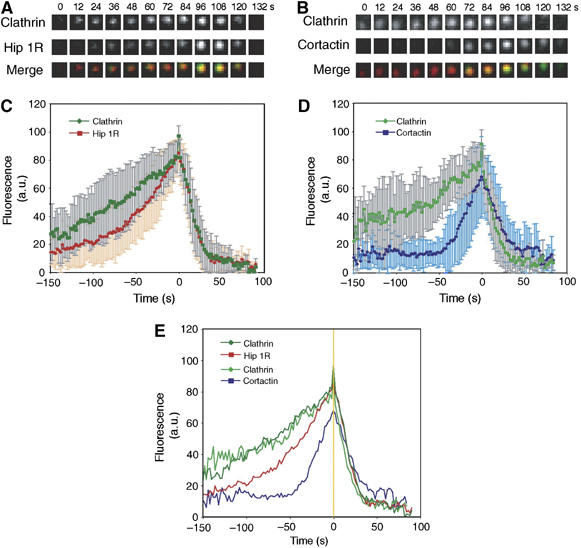
Timing of Hip1R and cortactin recruitment to CCPs. (A) Time series showing DsRed-clathrin and Hip1R-GFP recruitment to the same CCP. (B) Time series showing DsRed-clathrin and cortactin-GFP recruitment to the same CCP. (C) Average fluorescence of clathrin (dark green) and Hip1R (red) plotted against time from 30 CCPs in eight cells. The error bars represent the s.d. from 30 events. Time 0 corresponds to the moment at which the clathrin signal started to dim. All data were normalized (see Materials and methods) before averaging. (D) Average fluorescence for clathrin (light green) and cortactin (blue) plotted against time from 30 CCPs in 14 cells. (E) Summary of panels C and D without the error bars. Time 0 is marked by a yellow line.
A recent study by Merrifield et al (2005) showed a similar pattern for cortactin accumulation and correlated the cortactin peak with vesicle scission. As Hip1R and cortactin levels peaked concomitantly, it is plausible that the Hip1R–cortactin complex may also regulate actin polymerization during vesicle invagination, neck constriction and scission.
The Hip1R–cortactin complex inhibits actin assembly
In previous studies, we showed that the coiled-coil domain of Hip1R (346–655) is responsible for localization of Hip1R to CCPs (Engqvist-Goldstein et al, 1999), whereas the C-terminal domain (656–1068) is responsible for the inhibition of actin assembly in vivo (Engqvist-Goldstein et al, 2004). Interestingly, the C-terminal domain is known to bind F-actin, and we showed here that it also contains a cortactin-binding activity. Hence, we speculated that a Hip1R–cortactin complex might inhibit actin assembly.
To test for activities of the Hip1R–cortactin complex on actin dynamics, we performed actin polymerization kinetics assays using pyrenyl-labeled actin (Kouyama and Mihashi, 1981). Increasing concentrations of Hip1R alone up to 1 μM had only a very small effect on actin assembly (Figure 5A). However, in the presence of 0.4 μM cortactin, the same concentrations of Hip1R inhibited actin assembly in a dose-dependent fashion, whereas cortactin alone had only a very small effect (Figure 5B). A decrease in final polymerized actin in the presence of Hip1R and cortactin was also confirmed by an actin pelleting assay (Figure 5C). Finally, actin filaments from the same experiment were observed by fluorescence microscopy after dilution and stabilization by rhodamine-labeled phalloidin. The observation of single actin filaments and quantification of polymerized actin showed a decrease in actin assembly in the presence of cortactin and Hip1R (Figure 5D and E).
Figure 5.
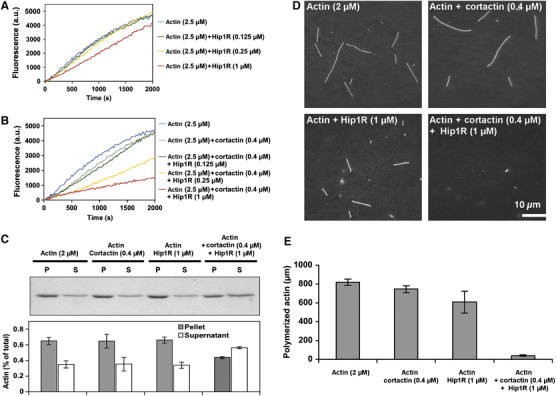
The Hip1R–cortactin complex inhibits actin assembly. (A, B) Effect of increasing amounts of Hip1R on assembly of 2.5 μM MgATP–G-actin, 10% pyrenyl-labeled, in the absence (A) or presence (B) of 0.4 μM cortactin. (C) Actin was polymerized 30 min at RT under the indicated conditions, polymerized (P) and unpolymerized (S) actin were separated by ultracentrifugation, resolved by 10% SDS–PAGE and detected by Coomassie blue (upper panel). The bar graph shows the percentage of actin in the pellets and supernatants (lower panel). The error bars represent the s.d. from three independent experiments. (D, E) Actin was polymerized in the presence of the indicated concentration of Hip1R and cortactin and the filaments were observed after 30 min by rhodamine phalloidin staining; (D) representative field for each condition; (E) quantification of the total length of polymerized actin (μm). The error bars represent the s.d. from three independent experiments.
The Hip1R–cortactin complex inhibits actin assembly at the barbed end specifically
Several mechanisms may account for the inhibition of actin assembly by the Hip1R–cortactin complex: sequestration of actin monomers, actin filament depolymerization or capping of actin filament barbed ends. To determine the mechanism by which the Hip1R–cortactin complex inhibits actin assembly, we first tested whether this complex caps the barbed end of growing actin filaments. First, we studied the elongation of spectrin–actin seeds, which are short actin filaments capped at their pointed ends. Barbed end growth was inhibited with a delay and a complete inhibition was observed after 600 s, with a half-time of 150 s (Figure 6A). In contrast, elongation of gelsolin–actin complexes, which are actin nuclei capped at their barbed ends, was not affected by the Hip1R–cortactin complex (Figure 6B). Therefore, we conclude that the Hip1R–cortactin complex inhibits barbed end elongation specifically, and is not a sequestering protein or a depolymerizing factor. To quantify this inhibition further, we calculated the concentration of free barbed ends over time from the curves presented in Figure 6A (Figure 6C; see Materials and methods). It is important to note that a total inhibition of barbed end elongation is observed in the presence of 0.5 μM Hip1R and 0.5 μM cortactin, demonstrating that the Hip1R–cortactin complex not only decreases the rate constant for actin assembly–disassembly at barbed ends like a ‘leaky capper' (Romero et al, 2004), but blocks actin assembly like a barbed end capping protein. The plateau of each curve was plotted as a function of the Hip1R–cortactin complex concentration using the Kd we calculated previously in Figure 1C. The best fit of this curve gave a Kd=85 nM, which represents the affinity of the Hip1R–cortactin complex for the barbed end (inset, Figure 6C).
Figure 6.
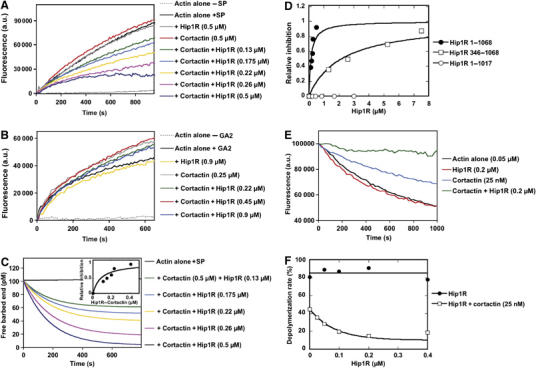
The Hip1R–cortactin complex inhibits actin filament barbed end assembly. (A) Barbed end growth was measured in the presence of 100 pM spectrin–actin seeds (SP), 1 μM MgATP–G-actin, 10% pyrenyl-labeled and the indicated concentrations of Hip1R and cortactin. (B) Pointed end growth was measured in the presence of 4 nM gelsolin–actin (GA2) complex, 1.5 μM MgATP–G-actin, 10% pyrenyl-labeled and the indicated concentrations of Hip1R and cortactin. (C) The concentration of free barbed ends over time in the presence of the indicated concentrations of Hip1R and cortactin are derived from the corresponding kinetics of barbed end growth shown in panel A (Pantaloni et al, 2000) (see also Materials and methods). Inset, the relative inhibition (fraction of inhibited barbed ends corresponding to the plateau of each kinetics curve) was plotted versus the concentration of the Hip1R–cortactin complex. The binding curve for the interaction of the complex with the actin barbed end was calculated using the equation described in the Materials and methods with the best-fit value of Kd=85 nM. (D) The fraction of barbed ends inhibited was plotted versus the total concentration of Hip1R 1–1068 (black circles), Hip1R 1–1017 (open circles) and Hip1R 346–1068 (open squares) in the presence of 0.5 μM cortactin. (E) Hip1R–cortactin inhibits actin depolymerization induced by dilution. Depolymerization of actin filaments (2 μM, 35% pyrenyl-labeled) was induced by a 40-fold dilution into polymerization buffer in the presence of indicated concentrations of Hip1R and cortactin. (F) Depolymerization rates in the presence of the indicated concentrations of Hip1R with (open squares) and without (closed circles) cortactin (25 nM). All the experiments were performed three times with the same results.
We compared the activity of Hip1R 1–1068 (full length) and Hip1R 1–1017. We found that increasing concentrations of Hip1R 1–1017 in the presence of 0.5 μM cortactin did not cap actin filament barbed ends, whereas similar concentrations of Hip1R 1–1068 in the presence of 0.5 μM cortactin blocked actin filament barbed end elongation, demonstrating that this activity requires a physical interaction between Hip1R and cortactin (Figure 6D). Hip1R 346–1068, deleted for the N-terminal domain, was also tested for its ability to inhibit barbed end growth in the presence of cortactin. Increasing concentrations of Hip1R 346–1068 inhibited the elongation of spectrin–actin seeds in the presence of cortactin (Figure 6D). Hip1R 346–1068 in conjunction with cortactin displayed a very low activity in agreement with our data showing that its affinity for cortactin is markedly reduced (Figures 2A and 6D). Barbed end capping was also tested by dilution-induced depolymerization of actin filaments (Figure 6E and F). Although Hip1R alone did not affect actin depolymerization significantly, 25 nM cortactin alone partially stabilized actin filaments from depolymerization (Figure 6E). This effect likely reflects the ability of cortactin to stabilize actin contacts along the filament and it would also explain why cortactin slightly enhances the elongation of gelsolin–actin complexes (Figure 6B). In the presence of Hip1R and cortactin, actin filament depolymerization was completely inhibited, confirming that the barbed end dynamics are inhibited (Figure 6E and F).
Binding of cortactin to Hip1R, N-WASP or dynamin is mutually exclusive
The N-terminal half of cortactin is known to bind to actin filaments and to activate the Arp2/3 complex (Uruno et al, 2001; Weaver et al, 2001), whereas the C-terminal SH3 domain of cortactin has been implicated in multiple functions. First, it binds and stimulates the ability of N-WASP to activate the Arp2/3 complex (Martinez-Quiles et al, 2004; Kowalski et al, 2005). Second, the SH3 domain of cortactin can bind to the PRD of dynamin, which is proposed to allow cortactin to activate the Arp2/3 complex at endocytic sites (Zhu et al, 2005). Therefore, it was important to determine how formation of the Hip1R–cortactin complex affects cortactin's activities on endocytosis-associated components of the actin nucleation machinery such as Arp2/3, N-WASP and dynamin.
Because it is thought that a major function of cortactin is to activate the Arp2/3 complex (Uruno et al, 2001; Weaver et al, 2001), we first tested whether Hip1R affects the ability of a high concentration of cortactin (1 μM) to activate the Arp2/3 complex. Increasing Hip1R concentrations up to 1.5 μM in the presence of 1 μM cortactin and 350 nM Arp2/3 complex inhibited actin assembly in a Hip1R dose-dependent fashion (Figure 7A).
Figure 7.
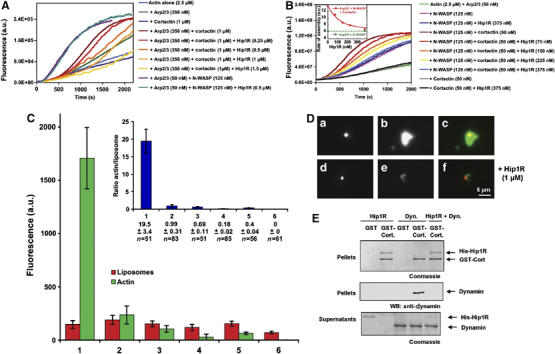
Hip1R inhibits Arp2/3-dependent polymerization pathways. (A) Effect of Hip1R levels on assembly of 2.5 μM MgATP–G-actin, 10% pyrenyl-labeled, in the presence of 1 μM cortactin and 350 nM Arp2/3 complex or 125 nM N-WASP and 50 nM Arp2/3 complex. (B) Effect of Hip1R levels on assembly of 2.5 μM MgATP–G-actin, 10% pyrenyl-labeled, in the presence of 50 nM cortactin, 50 nM Arp2/3 complex and 125 nM N-WASP. Inset; the maximum rate of actin assembly in the presence of Arp2/3, N-WASP and cortactin (red) or Arp2/3 and N-WASP (green) was plotted versus the concentration of Hip1R. (C, D) Reconstitution of actin assembly associated with lipid bilayers. DOPS (10 μM) liposomes containing 0.1% Texas Red DOPE were incubated with 1 μM dynamin, 1 μM cortactin, 0.5 μM Arp2/3 complex, 3 μM actin, 0.3 μM Alexa488-labeled actin and the following modifications: none (1), with 1 μM Hip1R (2), without Arp2/3 (3), without cortactin (4), without Arp2/3 and cortactin (5), without dynamin (6). Samples were observed by fluorescence microscopy. (C) Histograms showing the average fluorescence per Texas Red-labeled liposome (red) and the associated Alexa488-labeled actin cloud (green) for conditions 1–6 detailed above. The inset shows the actin/liposome ratio for the same conditions (blue). Data shown are average±s.e.m. (D) Representative images obtained for conditions (1) and (2). (a–c) Representative images obtained for liposomes (10 μM DOPS+0.1% Texas Red DOPE) incubated with 1 μM dynamin, 1 μM cortactin, 0.5 μM Arp2/3 complex, 3 μM actin, 0.3 μM Alexa488-labeled actin (condition 1). (d–f) Representative images obtained for liposomes (10 μM DOPS+0.1% Texas Red DOPE) incubated with 1 μM dynamin, 1 μM cortactin, 0.5 μM Arp2/3 complex, 3 μM actin, 0.3 μM Alexa488-labeled actin and 1 μM Hip1R (condition 2). (a, d) Liposomes; (b, e) actin; (c, f) merged. (E) His-tagged Hip1R (1 μM) pull downs using GST (1 μM) and GST-cortactin (1 μM) in the presence of dynamin (1.5 μM). Bound (P) and unbound (S) fractions were analyzed by Coomassie blue staining (upper and lower panels), bound fractions were also analyzed by Western blotting using a Hudy1 anti-dynamin antibody (middle panel). All the experiments were performed three times with the same results.
We also tested the ability of Hip1R to inhibit the activation of N-WASP by cortactin in an actin polymerization assay in vitro. We worked at a low concentration of cortactin (50 nM) at which the activation of Arp2/3 and the capping in the presence of high concentrations of Hip1R were both negligible (Figure 7B). Under these conditions, cortactin was still a potent activator of N-WASP. However, we found that Hip1R inhibited actin assembly in the presence of Arp2/3, N-WASP and cortactin in a dose-dependent fashion, whereas it did not affect actin assembly in the presence of Arp2/3 and N-WASP (Figure 7B). The maximum inhibition corresponds to the activity of N-WASP in the absence of cortactin, demonstrating that Hip1R inhibits the activation of N-WASP by cortactin independent of the capping activity of the Hip1R–cortactin complex (Figure 7B, inset).
In cells, dynamin assembles at the plasma membrane and is thought to recruit activators of the Arp2/3 complex such as cortactin to initiate actin assembly in a spatiotemporally regulated fashion. Previously, it has been shown that dynamin-coated liposomes recruit phalloidin-stabilized actin bundles in the presence of cortactin and the Arp2/3 complex (Schafer et al, 2002). We wished to establish the activity of the Hip1R–cortactin complex in a similar assay using non-stabilized material. Therefore, we observed the spatial organization of actin structures generated by dynamin-coated liposomes in the presence of cortactin and the Arp2/3 complex to determine the effect of Hip1R on the formation of these structures. We used an assay, in which dynamin assembles at the surface of Texas Red-labeled dioleyl phosphatidylserine (DOPS) liposomes (Sweitzer and Hinshaw, 1998) in the presence of cortactin, the Arp2/3 complex and Alexa488-labeled actin. In contrast to results from the previous study (Schafer et al, 2002), samples were observed directly by fluorescence microscopy without phalloidin and without subsequent dilution. We found that actin clouds formed on the liposome surfaces. However, essentially no actin structures were observed on liposomes in the absence of dynamin, cortactin or the Arp2/3 complex, demonstrating that the presence of liposome-associated actin clouds is the result of nucleation of filaments by the pathway leading from DOPS to dynamin to cortactin to Arp2/3 (Figure 7C and D). Importantly, in the presence of Hip1R, the ability of liposomes to generate actin clouds was greatly reduced. To determine whether in this assay Hip1R binds to cortactin to cap actin filaments or prevent the SH3 domain of cortactin from interacting with dynamin, we tested the ability of dynamin to bind GST-cortactin in the presence and absence of Hip1R. We found that Hip1R prevented the association of cortactin with dynamin (Figure 7E).
Discussion
Here, we reported the discovery and functional characterization of a novel endocytic complex composed of Hip1R and cortactin. We presented evidence that this complex has a novel actin filament capping activity, and explored when and how this complex may function during endocytosis. As both proteins are also present in actin-rich ruffles at the cell periphery (Wu and Parsons, 1993; Engqvist-Goldstein et al, 1999), and on the surface of the Golgi (Carreno et al, 2004; Cao et al, 2005), we speculate Hip1R–cortactin complexes may function to regulate a variety of actin-mediated processes.
Importance of Hip1R and cortactin in endocytosis
The depletion of Hip1R by siRNA induced the formation of abnormal actin tails suggesting that this protein plays a critical role in the inhibition of actin assembly at endocytic sites. The depletion of cortactin by siRNA abolished the formation of these actin tails, suggesting that the formation of these structures involves the activation of the Arp2/3 complex by cortactin (Engqvist-Goldstein et al, 2004). In addition, our data indicate that cortactin binding to Hip1R contributes to the inhibition of actin assembly at endocytic sites. We also showed that Hip1R and cortactin levels peak concomitantly during vesicle internalization, further pointing towards a role for the Hip1R–cortactin complex in the regulation of actin assembly during endocytosis. These observations clearly show the importance of Hip1R and cortactin in the regulation of actin assembly during CME.
However, the specific roles of these two proteins during endocytosis are still unclear. The depletion of Hip1R caused a mild defect in transferrin uptake, and we showed in a previous study that cortactin siRNA did not affect transferrin uptake (Barroso et al, 2006). Although Sauvonnet et al (2005) reported that the depletion of cortactin by siRNA decreased the amount of transferrin internalized, they did not take into consideration the possibility that there might be less transferrin receptor at the cell surface of siRNA-treated cells. By contrast, the importance of actin assembly in vesicle invagination and scission has been clarified recently (Merrifield et al, 2005; Yarar et al, 2005). One possibility to accommodate these various observations is that several actin nucleation machineries are involved in the process of vesicle internalization, and hence the impact of impairing the function of a single pathway may be hard to detect. Moreover, we found that a truncation mutant of Hip1R defective in cortactin binding, while being reduced relative to the full-length protein in the ability to rescue the actin tail formation phenotype of cells treated with a Hip1R siRNA, nevertheless, partially rescued the phenotype. These results point to complexity in the pathways that regulate actin assembly at endocytic sites, consistent to what has been observed in yeast cells (Sun et al, 2006).
Interestingly, the C-terminal domain of Hip1R contains two PXXXPR SH3-binding motifs (1025–1030 and 1044–1049). The three SH3 domains of the protein CIN85 bind to the first motif (Kowanetz et al, 2004). We showed that mutations of the two arginines in alanine that abolish the binding of CIN85 (Kowanetz et al, 2004) did not affect cortactin binding and fully rescue the Hip1R siRNA in HeLa cells, suggesting that the binding of CIN85 to Hip1R does not contribute to the regulation of actin assembly at endocytic sites (data not shown).
The Hip1R–cortactin complex caps actin filament barbed ends
In a previous study, we showed that RNAi depletion of Hip1R in HeLa cells increases actin assembly associated with endocytic sites and reduces the rate of CME, suggesting that Hip1R's inhibitory effect on actin assembly facilitates efficient endocytosis (Engqvist-Goldstein et al, 2004). Here, we showed that a Hip1R–cortactin complex inhibits actin filament barbed end growth in vitro. Interestingly, the barbed end growth inhibition was delayed, similar to what was reported for the heterodimeric capping protein (Schafer et al, 1996; Wear et al, 2003). A variety of capping proteins like CP, Eps8 or twinfilin are thought to block growth of actin filament barbed ends to favor elongation of uncapped barbed ends, in a process known as funneling (Schafer et al, 1996; Disanza et al, 2004; Helfer et al, 2006). This funneling mechanism might facilitate a variety of actin-based motility processes like lamellipodium protrusion and Listeria propulsion (Loisel et al, 1999; Pantaloni et al, 2001).
It is therefore tempting to speculate that the Hip1R–cortactin complex mimics capping proteins to facilitate steps in endocytic vesicle formation. Our in vivo data support the conclusion that the interaction of cortactin with Hip1R contributes to the inhibition of actin assembly at endocytic sites and show that Hip1R and cortactin concomitantly peak at the moment of vesicle internalization. In addition to our findings, the requirement of actin filament barbed end capping for endocytosis is supported by recent findings. In yeast, capping protein is important for the initial movement of endocytic vesicles away from the plasma membrane (Kaksonen et al, 2005; Kim et al, 2006). In addition to the funneling of actin monomers, local capping could limit the growth of the actin network to the neck of the vesicle to prevent the network from surrounding the vesicle and impairing its inward movement.
The roles of cortactin in Arp2/3-dependent actin nucleation and in capping with Hip1R are mutually exclusive
Previously, dynamin, cortactin and the Arp2/3 complex were shown to be capable of recruiting actin bundles to liposomes (Schafer et al, 2002). Here, we extended these observations by the discovery using liposomes in vitro that dynamin recruits cortactin, which in turn activates the Arp2/3 complex to initiate actin assembly. In mammalian cells, actin nucleation at endocytic sites involves the Arp2/3 complex and at least two activators, N-WASP and cortactin (Engqvist-Goldstein et al, 2004; Merrifield et al, 2004, 2005). Although the relative contribution of N-WASP and cortactin to Arp2/3 activation at endocytic sites is not known, we showed in a previous study that assembly of exaggerated actin ‘tail' structures at endocytic sites in the absence of Hip1R depends on cortactin (Engqvist-Goldstein et al, 2004). Recent studies also showed that N-WASP regulates actin assembly at CCPs (Benesch et al, 2005; Innocenti et al, 2005). Together, these data support the view that N-WASP and cortactin activate the Arp2/3 complex in a sequential or synergistic fashion to generate new barbed ends concentrated near the plasma membrane at the vesicle neck where dynamin is localized (Weaver et al, 2001, 2002; Uruno et al, 2003).
Interestingly, we found that the C-terminal PRD of Hip1R interacts with the SH3 domain of cortactin. The SH3 domain of cortactin activates N-WASP by binding to its PRD (Martinez-Quiles et al, 2004; Kowalski et al, 2005). This SH3 domain is thought to be responsible for the targeting of cortactin to the PRD of dynamin at endocytic sites, where cortactin directly stimulates the Arp2/3 complex (Zhu et al, 2005). Our data show that Hip1R inhibits the activation of N-WASP by cortactin and prevents cortactin from binding to dynamin-associated liposomes to stimulate the Arp2/3 complex. These results reflect the competition of Hip1R, N-WASP and dynamin PRDs for the SH3 domain of cortactin. Therefore, the binding of cortactin to N-WASP and dynamin-associated membranes leading to the localized activation of the Arp2/3 complex and the formation of a Hip1R–cortactin complex that caps actin filaments are mutually exclusive. Because dynamin is localized at the neck of the vesicle (Kosaka and Ikeda, 1983; Hinshaw and Schmid, 1995; Sweitzer and Hinshaw, 1998; Takei et al, 1998) and Hip1R is localized at the surface of the CCV (Engqvist-Goldstein et al, 1999), it is tempting to suggest a spatial separation of the capping and nucleating activities associated with cortactin (Figure 8).
Figure 8.
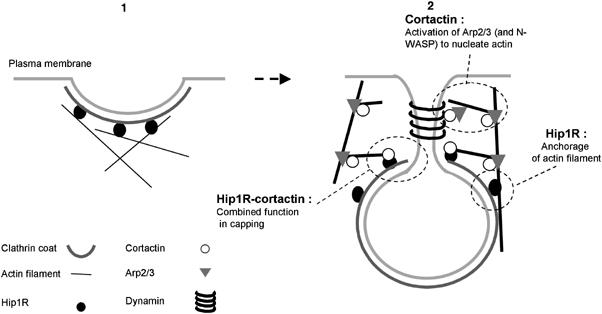
Model for the endocytic roles of actin, Hip1R and cortactin in endocytosis. (1) Hip1R binds to the clathrin coat, possibly stimulating or stabilizing its assembly on the inner face of the plasma membrane, and binds to actin filaments. (2) Dynamin assembles around the neck of the nascent CCV. Cortactin interacts with dynamin, stimulates the Arp2/3 complex to form branched actin filaments, and stabilizes the branched junctions. The actin meshwork grows via new filament assembly at the vesicle neck. The actin network is anchored to the clathrin coat by Hip1R, so that the vesicle is pushed away from the plasma membrane. Hip1R forms a complex with cortactin to cap actin filament barbed ends near the clathrin coat.
How do the individual and combined functions of Hip1R and cortactin cooperate to facilitate endocytosis?
The timing of cortactin and Hip1R appearance we reported here, together with similar recruitment kinetics for dynamin, actin, Arp3 and N-WASP, and the impairment of CCP dynamics after actin drug treatments, all point to an important role for carefully regulated actin polymerization during the late stages of CME (Merrifield et al, 2002, 2004, 2005; Yarar et al, 2005). FRAP analysis that addressed the polarity of this growing actin network demonstrated that actin nucleation occurs continuously at endocytic sites in both yeast and mammals (Kaksonen et al, 2003; Engqvist-Goldstein et al, 2004). Actin assembly in yeast occurred proximal to the plasma membrane, and actin filaments were then moved off the membrane into the cytoplasm (Kaksonen et al, 2003).
In this context, we propose that actin filaments are continuously generated at the vesicle neck by a mechanism that involves activation of the Arp2/3 complex by cortactin, N-WASP and possibly other proteins. The resulting filaments continuously move off the plasma membrane into the cytoplasm. Hip1R, by binding simultaneously to clathrin and actin filaments, anchors the CCV to the actin meshwork engaged in retrograde flow and facilitates movement of the vesicle into the cytoplasm. The studies presented here demonstrate that Hip1R binds to cortactin to form a complex that inhibits actin filament barbed end elongation. We propose that this complex funnels actin monomers to newly formed, uncapped barbed ends, to accelerate the growth of the actin network, hence facilitating the formation of CCVs.
Our model suggests versatility of Hip1R and cortactin function. Each provides distinct functions, in attaching the actin meshwork to the clathrin coat and promoting clathrin assembly (Engqvist-Goldstein et al, 2001; Chen and Brodsky, 2005; Legendre-Guillemin et al, 2005), and in activating the Arp2/3 complex and stabilizing filament branches (Uruno et al, 2001; Weaver et al, 2001), respectively, as well as a combined function, in capping filaments near the site of new filament assembly (Figure 8).
Materials and methods
Polymerization assays
Actin polymerization was monitored by the increase in fluorescence of 10% pyrenyl-labeled actin. Actin polymerization was induced by addition of 100 mM KCl, 1 mM MgCl2 and 0.2 mM EGTA to a solution of Ca-ATP-G-actin containing the desired proteins. Fluorescence measurements were carried out at 25°C in a Spex Fluorolog 2 spectrophotometer.
The concentration of free barbed end as a function of time was derived from spectrin actin seed elongation kinetics, and calculated for each time point using the following equation:
where [F] is the concentration of free barbed ends in μM, dC/dt the rate of actin assembly in μM s−1, k+ the association constant for monomer to the barbed end of actin filaments (10 μM−1 s−1) and [G] the concentration of polymerizable actin in μM.
The affinity of the Hip1R–cortactin complex for barbed ends was calculated as follows. The concentration of free barbed ends at the assembly plateau calculated as described above was taken as a measure of free barbed ends. The following equation was used to fit the data. The fraction R of Hip1R–cortactin bound to barbed ends is R=([A0]+[B0]+K−(([A0]+[B0]+K)2−4[A0][B0])0.5)/2[A0], where [A0] is the concentration of spectrin–actin seeds, [B0] the concentration of Hip1R–cortactin complex and K is the equilibrium dissociation constant (Kd) of the complex with the barbed end.
Live cell imaging
TIR-FM was performed using an Olympus IX81 microscope equipped with a × 60/NA1.45. lens and 488 nm argon ion laser (Melles Griot). The temperature was maintained at 27.5°C using a Bioptechs chamber. The 488 nm laser was used to excite both GFP and DsRed. Simultaneous two-color imaging was performed using an image splitter (Optical Insight) to separate the red and green emission signals to two sides of the camera sensor using a 565 nm dichroic mirror, and 530/30 and 630/50 nm emission filters. No bleed-through between the red and the green channel was detected under our conditions. Swiss 3T3 cells that stably express DsRed-mLCa (mouse clathrin light chain a) were transiently transfected with Hip1R-GFP or cortactin-GFP using Lipofectamine reagent (Invitrogen) 48 h before imaging. Each cell was imaged every 2 s for 200–300 frames. After each experiment, images of immobilized microbeads that fluoresce at both green and red wavelengths were captured. These images were used to align the cell images.
Image analysis
We used similar criteria to select single CCPs for analysis as described by Merrifield et al (2002). We only counted the CCPs, which had either Hip1R or cortactin as well. The maximum fluorescence intensity of GFP or DsRed at each CCP was measured using the ImageJ program, and was plotted against time. Data from 10 frames before the appearance of the CCP, and 10 frames after the disappearance of the CCP, were also obtained, and the smallest value was set as the background. After background correction, the fluorescence intensity was normalized with the maximum value set at 100. The last point of the clathrin fluorescence peak before dimming was set as the 0 time. An average of 30 CCPs that underwent a complete cycle of appearing and disappearing was analyzed for each protein pair.
Treatment of HeLa cells with siRNA
For knockdown of endogenous Hip1R, the siRNA-A3 was prepared as described in Engqvist-Goldstein et al (2004). This sequence is designed to target nucleotides 184–204 of human Hip1R (GenBank accession no. BAA31630). The siRNA-InvC1 was used as a control and does not target a gene. HeLa cells were plated 1 day before transfection in 24-well plates on coverslips at a density of 1 × 104 cells/well. On the day of transfection, the cell density was ∼30%. For transfection, 1.5 μl of siRNA duplex (20 μM) was diluted into 50 μl OptiMem (Invitrogen Corp., Carlsbad, CA) in tube 1. In tube 2, 3 μl OligoFectamine (Invitrogen Corp., Carlsbad, CA) was diluted into 12 μl OptiMem. Tubes 1 and 2 were incubated for 10 min at room temperature (RT) before being combined. This mixture was incubated at RT for 20 min. OptiMem (38 μl) was then added to the mixture and the solution was added to the cells grown in 0.5 ml of DMEM with 10% FBS.
Rescue experiments
One day after transfection of the siRNA, cells were transfected with mouse Hip1R (amino acids 1–1068)-6myc or mouse Hip1R (amino acids 1–1017)-6myc. Details of the cloning procedure and vector construction can be found in Engqvist-Goldstein et al (1999). Transfections were carried out using Fugene 6 (Roche Diagnostics) according to the manufacturer's instructions. Forty-eight hours after transfection, cells were fixed and processed for immunofluorescence as described previously (Engqvist-Goldstein et al, 1999). The following primary antibodies were used: anti-cortactin antibody (4F11; Upstate Biotechnology) at a dilution of 1:100 and anti-myc antibody (A-14; Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:400. Secondary antibodies were FITC donkey anti-mouse (Jackson ImmunoResearch Laboratories) for anti-cortactin, and rhodamine donkey anti-rabbit (Jackson ImmunoResearch Laboratories) for anti-myc, both used at a dilution of 1:200.
For scoring the A3 phenotype, cells were stained for cortactin (Engqvist-Goldstein et al, 2004). Cells were scored as positive if they had three or more ring- or tail-like structures containing cortactin. Two independent experiments were performed scoring 200 cells from each experiment. In the rescue experiments, scoring was performed as described above, except that in these experiments only cells expressing the rescue construct (as judged by myc staining) were scored.
Preparation of liposomes
A mixture of DOPS (Avanti Polar) (2 mM) and dioleyl phosphatidylethanolamine (DOPE) (2 μM) in chloroform/methanol (19:1) was dried in a rotary evaporator for 45 min at 37°C. After gently adding degassed 0.3 M sucrose, the flask was flushed with argon, sealed and left undisturbed for 2 h at 37°C to allow spontaneous formation of liposomes. The liposomes were recovered by centrifugation at 12 000 g for 20 min at RT, and resuspended in 25 mM HEPES–KOH, pH 7.2, 25 mM KCl, 2.5 mM magnesium acetate and 100 mM potassium acetate. Finally, the liposomes were extruded using a 5 μm filter (Avanti Polar).
Reconstitution of actin assembly on liposomes
DOPS liposomes (10 μM) were incubated with 1 μM dynamin, 1 μM cortactin, 0.5 μM Arp2/3 complex, 3 μM actin, 1 μM Hip1R, 0.3 μM Alexa488-labeled actin, 2 mM ATP, 2 mM MgCl2, 5 mM DTT, 0.1 mM 1,4-diazabicyclo(2.2.2)octan and 0.1% BSA, for 30 min at RT (proteins purifications are described in the Supplementary Materials and methods). Although the ANTH domain of Hip1R binds to PIP2 specifically, these conditions ensure that a possible nonspecific interaction with the negatively charged DOPS does not affect dynamin binding. The solution (3 μl) was pressed between a slide and a coverslip, sealed and imaged using a Nikon TE300 microscope equipped with a 100/NA 1.4 objective and Orca-100-cooled CCD camera (Hamamatsu). We analyzed all the liposomes and calculated the liposome/actin ratio for every liposome including the ones that did not form actin clouds.
Supplementary Material
Supplementary materials and methods
Supplementary materials and methods
Acknowledgments
We thank Takehito Uruno and Xi Zhan for the His-tagged cortactin plasmid, Marko Kaksonen for the cortactin-GFP plasmid, John Cooper for the GST-tagged cortactin plasmid, Henry Ho and Marc Kirschner for the pFastBac-N-WASP plasmid, Harvey MacMahon for the pGEX-SH3-Amph2 plasmid (used in dynamin purification), R Dyche Mullins for the pGEX-N-WASP-WA plasmid (used in Arp2/3 purification), Wolfhard Almers for the Swiss 3T3 cells that stably express DsRed-mLCa and Marie-France Carlier for the spectrin–actin seeds and the gelsolin. We are very grateful to Adam Martin for his help with actin and labeled actin preparations, Eugene Futai and the Schekman laboratory for help with liposome preparations, Marko Kaksonen for help with imaging, and members of the Drubin and Barnes laboratory for helpful discussions. This work was supported by Fondation pour la Recherche Médicale (France) and a European Molecular Biology Organization fellowship to CLC, a postdoctoral fellowship of the Deutsche Forschungsgemeinschaft (DFG) to BSP, an American Cancer Society Postdoctoral Fellowship PF-02-096-01-CSM to CXZ, and a National Institutes of Health grant GM65462 to DGD.
References
- Barroso C, Rodenbusch SE, Welch MD, Drubin DG (2006) A role for cortactin in Listeria monocytogenes invasion of NIH 3T3 cells, but not in its intracellular motility. Cell Motil Cytoskeleton 63: 231–243 [DOI] [PubMed] [Google Scholar]
- Benesch S, Polo S, Lai FP, Anderson KI, Stradal TE, Wehland J, Rottner K (2005) N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. J Cell Sci 118: 3103–3115 [DOI] [PubMed] [Google Scholar]
- Cao H, Orth JD, Chen J, Weller SG, Heuser JE, McNiven MA (2003) Cortactin is a component of clathrin-coated pits and participates in receptor-mediated endocytosis. Mol Cell Biol 23: 2162–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Weller S, Orth JD, Chen J, Huang B, Chen JL, Stamnes M, McNiven MA (2005) Actin and Arf1-dependent recruitment of a cortactin-dynamin complex to the Golgi regulates post-Golgi transport. Nat Cell Biol 7: 483–492 [DOI] [PubMed] [Google Scholar]
- Carreno S, Engqvist-Goldstein AE, Zhang CX, McDonald KL, Drubin DG (2004) Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J Cell Biol 165: 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Brodsky FM (2005) Huntingtin-interacting protein 1 (Hip1) and Hip1-related protein (Hip1R) bind the conserved sequence of clathrin light chains and thereby influence clathrin assembly in vitro and actin distribution in vivo. J Biol Chem 280: 6109–6117 [DOI] [PubMed] [Google Scholar]
- Da Costa SR, Sou E, Xie J, Yarber FA, Okamoto CT, Pidgeon M, Kessels MM, Mircheff AK, Schechter JE, Qualmann B, Hamm-Alvarez SF (2003) Impairing actin filament or syndapin functions promotes accumulation of clathrin-coated vesicles at the apical plasma membrane of acinar epithelial cells. Mol Biol Cell 14: 4397–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disanza A, Carlier MF, Stradal TE, Didry D, Frittoli E, Confalonieri S, Croce A, Wehland J, Di Fiore PP, Scita G (2004) Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nat Cell Biol 6: 1180–1188 [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Drubin DG (2003) Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol 19: 287–332 [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Kessels MM, Chopra VS, Hayden MR, Drubin DG (1999) An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J Cell Biol 147: 1503–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Warren RA, Kessels MM, Keen JH, Heuser J, Drubin DG (2001) The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J Cell Biol 154: 1209–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Zhang CX, Carreno S, Barroso C, Heuser JE, Drubin DG (2004) RNAi-mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol Biol Cell 15: 1666–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfer E, Nevalainen EM, Naumanen P, Romero S, Didry D, Pantaloni D, Lappalainen P, Carlier MF (2006) Mammalian twinfilin sequesters ADP-G-actin and caps filament barbed ends: implications in motility. EMBO J 25: 1184–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw JE, Schmid SL (1995) Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature 374: 190–192 [DOI] [PubMed] [Google Scholar]
- Hussain NK, Jenna S, Glogauer M, Quinn CC, Wasiak S, Guipponi M, Antonarakis SE, Kay BK, Stossel TP, Lamarche-Vane N, McPherson PS (2001) Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nat Cell Biol 3: 927–932 [DOI] [PubMed] [Google Scholar]
- Innocenti M, Gerboth S, Rottner K, Lai FP, Hertzog M, Stradal TE, Frittoli E, Didry D, Polo S, Disanza A, Benesch S, Di Fiore PP, Carlier MF, Scita G (2005) Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat Cell Biol 7: 969–976 [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Sun Y, Drubin DG (2003) A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115: 475–487 [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG (2005) A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell 123: 305–320 [DOI] [PubMed] [Google Scholar]
- Kessels MM, Qualmann B (2002) Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J 21: 6083–6094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels MM, Qualmann B (2006) Syndapin oligomers interconnect the machineries for endocytic vesicle formation and actin polymerization. J Biol Chem 281: 13285–13299 [DOI] [PubMed] [Google Scholar]
- Keyel PA, Watkins SC, Traub LM (2004) Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J Biol Chem 279: 13190–13204 [DOI] [PubMed] [Google Scholar]
- Kim K, Galletta BJ, Schmidt KO, Chang FS, Blumer KJ, Cooper JA (2006) Actin-based motility during endocytosis in budding yeast. Mol Biol Cell 17: 1354–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Ikeda K (1983) Reversible blockage of membrane retrieval and endocytosis in the garland cell of the temperature-sensitive mutant of Drosophila melanogaster, shibirets1. J Cell Biol 97: 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem 114: 33–38 [PubMed] [Google Scholar]
- Kowalski JR, Egile C, Gil S, Snapper SB, Li R, Thomas SM (2005) Cortactin regulates cell migration through activation of N-WASP. J Cell Sci 118: 79–87 [DOI] [PubMed] [Google Scholar]
- Kowanetz K, Husnjak K, Holler D, Kowanetz M, Soubeyran P, Hirsch D, Schmidt MH, Pavelic K, De Camilli P, Randazzo PA, Dikic I (2004) CIN85 associates with multiple effectors controlling intracellular trafficking of epidermal growth factor receptors. Mol Biol Cell 15: 3155–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Lemaire JF, Philie J, Gan L, Hayden MR, McPherson PS (2005) Huntingtin interacting protein 1 (HIP1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain. J Biol Chem 280: 6101–6108 [DOI] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF (1999) Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401: 613–616 [DOI] [PubMed] [Google Scholar]
- Martinez-Quiles N, Ho HY, Kirschner MW, Ramesh N, Geha RS (2004) Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol Cell Biol 24: 5269–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiven MA, Kim L, Krueger EW, Orth JD, Cao H, Wong TW (2000) Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol 151: 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Feldman ME, Wan L, Almers W (2002) Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol 4: 691–698 [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Perrais D, Zenisek D (2005) Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells. Cell 121: 593–606 [DOI] [PubMed] [Google Scholar]
- Merrifield CJ, Qualmann B, Kessels MM, Almers W (2004) Neural Wiskott–Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur J Cell Biol 83: 13–18 [DOI] [PubMed] [Google Scholar]
- Otsuki M, Itoh T, Takenawa T (2003) Neural Wiskott–Aldrich syndrome protein is recruited to rafts and associates with endophilin A in response to epidermal growth factor. J Biol Chem 278: 6461–6469 [DOI] [PubMed] [Google Scholar]
- Pantaloni D, Boujemaa R, Didry D, Gounon P, Carlier MF (2000) The Arp2/3 complex branches filament barbed ends: functional antagonism with capping proteins. Nat Cell Biol 2: 385–391 [DOI] [PubMed] [Google Scholar]
- Pantaloni D, Le Clainche C, Carlier MF (2001) Mechanism of actin-based motility. Science 292: 1502–1506 [DOI] [PubMed] [Google Scholar]
- Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, Carlier MF (2004) Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 119: 419–429 [DOI] [PubMed] [Google Scholar]
- Sauvonnet N, Dujeancourt A, Dautry-Varsat A (2005) Cortactin and dynamin are required for the clathrin-independent endocytosis of gammac cytokine receptor. J Cell Biol 168: 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Jennings PB, Cooper JA (1996) Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J Cell Biol 135: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Weed SA, Binns D, Karginov AV, Parsons JT, Cooper JA (2002) Dynamin2 and cortactin regulate actin assembly and filament organization. Curr Biol 12: 1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Martin AC, Drubin DG (2006) Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev Cell 11: 33–46 [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Hinshaw JE (1998) Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell 93: 1021–1029 [DOI] [PubMed] [Google Scholar]
- Takei K, Haucke V, Slepnev V, Farsad K, Salazar M, Chen H, De Camilli P (1998) Generation of coated intermediates of clathrin-mediated endocytosis on protein-free liposomes. Cell 94: 131–141 [DOI] [PubMed] [Google Scholar]
- Uruno T, Liu J, Li Y, Smith N, Zhan X (2003) Sequential interaction of actin-related proteins 2 and 3 (Arp2/3) complex with neural Wiscott-Aldrich syndrome protein (N-WASP) and cortactin during branched actin filament network formation. J Biol Chem 278: 26086–26093 [DOI] [PubMed] [Google Scholar]
- Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R, Mueller SC, Zhan X (2001) Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol 3: 259–266 [DOI] [PubMed] [Google Scholar]
- Veiga E, Cossart P (2005) Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol 7: 894–900 [DOI] [PubMed] [Google Scholar]
- Wear MA, Yamashita A, Kim K, Maeda Y, Cooper JA (2003) How capping protein binds the barbed end of the actin filament. Curr Biol 13: 1531–1537 [DOI] [PubMed] [Google Scholar]
- Weaver AM, Heuser JE, Karginov AV, Lee WL, Parsons JT, Cooper JA (2002) Interaction of cortactin and N-WASp with Arp2/3 complex. Curr Biol 12: 1270–1278 [DOI] [PubMed] [Google Scholar]
- Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA (2001) Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol 11: 370–374 [DOI] [PubMed] [Google Scholar]
- Wu H, Parsons JT (1993) Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol 120: 1417–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, Schmid SL (2005) A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell 16: 964–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhou K, Hao JJ, Liu J, Smith N, Zhan X (2005) Regulation of cortactin/dynamin interaction by actin polymerization during the fission of clathrin-coated pits. J Cell Sci 118: 807–817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Supplementary materials and methods