

Abstract
The epithelial isoform of fibroblast growth factor receptor 2 (Fgfr2b) is essential for embryogenesis, and Fgfr2b-null mice die at birth. Using Cre-Lox transgenics to delete Fgfr2b in cells expressing keratin 5, we show that mice lacking epidermal Fgfr2b survive into adulthood but display striking abnormalities in hair and sebaceous gland development. Epidermal hyperthickening develops with age, and 10% of mutant mice develop spontaneous papillomas, demonstrating the role of Fgfr2b in post-natal skin development and in adult skin homeostasis. Mice lacking epithelial Fgfr2b show great sensitivity to chemical carcinogenic insult, displaying several oncogenic ha-ras mutations with dramatic development of papillomas and squamous cell carcinomas. Mutant mice have increased inflammation in the skin, with increased numbers of macrophages and γδT cells with abnormal morphology. Mutant skin shows several changes in gene expression, including enhanced expression of the pro-inflammatory cytokine interleukin 18 and decreased expression of Serpin a3b, a potential tumor suppressor. Thus we describe a novel role of Fgfr2b and provide the first evidence of a tyrosine kinase receptor playing a tumor suppressive role in the skin.
Keywords: cancer, epidermis, Fgf, inflammation, sebaceous gland
Introduction
Reciprocal intercellular signalling between epithelium and mesenchyme is a fundamental process in the induction and patterning of many organs. Fibroblast growth factors (Fgfs) participate in this process by instructing cells to proliferate, survive, migrate or differentiate. In mammals, there are 22 Fgf family members that signal via one or more receptor tyrosine kinases encoded by four Fgf receptor genes (Fgfr1–4), which are expressed as alternatively spliced variants (Ornitz and Itoh, 2001). Germline knockout of the IIIb exon of the Fgfr2 gene results in mice that die at birth from multiple developmental defects, identifying Fgfr2b as a critical mediator of organogenesis (De Moerlooze et al, 2000; Revest et al, 2001). Similar results were obtained by overexpressing a soluble dominant-negative version of the Fgfr2b isoform (Celli et al, 1998), and studies in which Fgf-10 was knocked out showed this to be the key ligand for Fgfr2b during development (Min et al, 1998; Sekine et al, 1999).
A role of Fgfr2b in post-natal skin development was established by studying full-thickness skin grafts from late-stage Fgfr2b-null and wild-type fetuses, grown on the back of nude mice. When the first wave of hair morphogenesis was complete, after 21 days, histological analysis showed Fgfr2b signalling was crucial for normal epidermal growth and development as well as for subsequent hair follicle morphogenesis (Petiot et al, 2003). However, the grafting approach was not suitable for following long-term skin homeostasis. Thus, for this study, we adopted a Cre-Lox strategy to knock out Fgfr2b in the epidermis.
The expression of Cre recombinase under the control of the bovine keratin 5 (K5) promoter mediates extremely efficient excision of floxed target genes in the epidermis (Brakebusch et al, 2000) when inherited paternally (Ramirez et al, 2004). This approach is different from germline deletion, in that the skin develops normally during embryogenesis, as Cre is not active until embryonic day 15.5 (Ramirez et al, 2004), with Fgfr2b not being excised until late in fetal development. Therefore, our studies address the role of Fgfr2b in post-natal epidermal homeostasis rather than in skin development.
Various mouse models have been used to study Fgfr2b signalling in adult skin. Mice expressing a membrane-bound, dominant-negative Fgfr2b, lacking tyrosine kinase activity, under the control of the keratin 14 promoter, displayed epidermal atrophy, hair follicle abnormalities and dermal hyperthickening with severely delayed re-epithelialization of excisional wounds (Werner et al, 1994). Expression of Fgf-7, which binds to Fgfr2b exclusively, has been modulated by germline knockout (Guo et al, 1996) and by keratinocyte-specific overexpression, again under the control of the keratin 14 promoter (Guo et al, 1993). The only phenotype in the skin of Fgf-7 knockout mice was matting of the fur in aging male mice (Guo et al, 1996), whereas Fgf-7 overexpression resulted in neonatal epidermal hyperthickening, followed by progressive epidermal thinning and loss of adipose tissue as the mice aged (Guo et al, 1993).
None of the above studies address the role of Fgfr2b specifically; the dominant-negative receptor is able to outcompete any receptor that shares ligands with Fgfr2b, for example Fgfr1b, and the Fgf-7 ligand knockout fails to address the possibility of compensation by an alternative ligand, such as Fgf-10. Thus, we wished to address the role of Fgfr2b in the skin, both in terms of post-natal development and in terms of tissue homeostasis.
The FGFRs are not involved solely in the regulation of normal skin development. There is a growing body of evidence implicating FGFRs as causative and suppressive factors in cancer (Grose and Dickson, 2005), with activating mutations in FGFR2 having been described in human gastric cancer (Jang et al, 2001). FGFR2b has been suggested to act as a tumor suppressor in the urothelium, with decreased expression of FGFR2b correlating with poor prognosis in transitional cell carcinoma (Diez de Medina et al, 1997; Ricol et al, 1999). Further studies revealed that this tumor-suppressive effect was not dependent on the tyrosine kinase activity of FGFR2b, but suggested that the C-terminus of the receptor was modulating IGF-II signalling (Bernard-Pierrot et al, 2004). Thus, in the absence of FGFR2b, IGFII was overexpressed, resulting in increased tumor cell proliferation and decreased apoptosis. In human salivary adenocarcinoma, FGFR2b acts to induce differentiation and apoptosis of cancer cells (Zhang et al, 2001), whereas in prostate carcinoma, in both mouse and human, decreased expression of FGFR2 results in overexpression of FGFR1, which in turn drives cancer progression (Jin et al, 2003; Yasumoto et al, 2004). Our study provides the first evidence of a protective role of Fgfr2b in cancer development in the skin.
Results
Defective pelage growth in K5-R2b-null mice
K5-R2b-null (Fgfr2bflox/flox; K5-Cre positive) mice could be distinguished phenotypically as soon as their pelage hair begins to grow, a few days after birth, with their coat appearing thin and silky compared with control (Fgfr2bflox/flox; K5-Cre-negative) littermates. This difference in appearance persisted throughout adulthood, as illustrated in the comparison of two 5-week-old male littermates (Figure 1A). No difference in overall growth, behavior or fertility was observed, although K5-R2b-null mice developed abnormally long claws compared with control littermates (data not shown). RNase protection assays with a riboprobe specific for the IIIb variant of Fgfr2 and a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) riboprobe as a control confirmed that Fgfr2b mRNA was no longer expressed in the skin of 8-week-old K5-R2b-null mice (Figure 1B). Although Fgf receptors are expressed at low levels, a band corresponding to the protected fragment for Fgfr2b was clearly discernible in control samples and not in skin samples from K5-R2b mice (Figure 1B).
Figure 1.
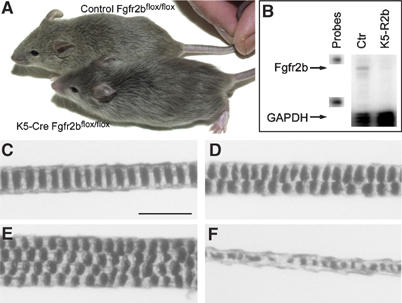
Skin-specific deletion of Fgfr2b leads to defective hair formation. (A) Five-week-old male littermates. Both mice are homozygous for the floxed Fgfr2b allele—the lower mouse also carries the K5 Cre transgene. Mice lacking Fgfr2b in the epidermis are normal size compared with wild-type siblings, but show clear coat differences, with their fur appearing sleek and silky compared with wild types. (B) RNase protection assay. Expression of Fgfr2b mRNA in skin from 8-week-old control and K5-R2b-null littermates. Levels of GAPDH mRNA were assayed in the same samples to serve as a control of both loading levels and RNA quality. Each hybridization probe (1000 c.p.m.) was loaded in the lanes labelled ‘probes' and used as size markers. Arrows indicate the protected fragments for Fgfr2b and control (GAPDH) mRNA. (C–F) Pelage hair structure. Light microscopy of the three main hair subtypes from control mice: (C) Zigzag, (D) Guard and (E) Awl hairs. (F) Disorganized zigzag hair from a K5-R2b-null mouse. Scale bar, 20 μm.
Light microscopy of hairs plucked from five 8-week-old wild-type mice (n=100 hairs) revealed all the usual hair subtypes present in the correct ratios; 70% Zigzag (Figure 1C), 2% Guard (Figure 1D) and 28% Awl/Auchene (Figure 1E). Hairs were classified by virtue of their length and classical patterns of air spaces within the hair (Sundberg and Hogan, 1994). In contrast, hairs from mice lacking Fgfr2b in the skin (n=100 hairs) showed a grossly disorganized zigzag structure (Figure 1F), as well as being of a thinner diameter (ranging from 5 to 7 μm when measured midway along the hair shaft, compared with 9 to 12 μm in control zigzag hairs).
Hair follicle abnormalities and cutaneous inflammation in the absence of Fgfr2b
Defects in hair follicle cycling were apparent, with immunostaining confirming the asynchronous nature of the first morphogenetic wave in K5-R2b-null mice. Longitudinal sections through back skin of 16 days post partum (d.p.p.) mice stained for keratin 6 permitted clear visualization of hair follicles and revealed no misexpression of keratin 6 in the interfollicular epidermis (Supplementary Figure 1A and B). Whereas the follicles of wild-type mice all were situated within the dermis in the resting phase (telogen) by 16 d.p.p. (Figure 2A), many follicles in knockout mice at the same stage still extended down beneath the dermis, with some reaching to the muscle layer at the base of the adipose tissue (mean number of follicles below dermis=3.2±2.6 (s.d.) per microscopic field (× 20); average of 10 fields from sections from three mice). Additionally, follicles of K5-R2b-null mice often appeared disorganized in structure, with more than one follicle coalescing to emerge at the same site in the epidermis (Supplementary Figure 1B). As with mice lacking Fgfr2b in the germ line (Petiot et al, 2003), no defects were observed in the expression of markers of epidermal differentiation (data not shown).
Figure 2.
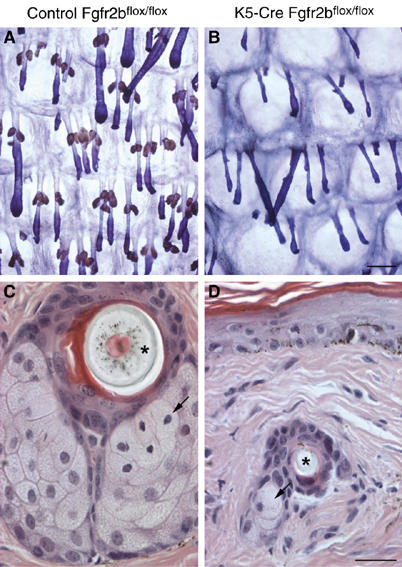
Sebaceous gland atrophy in K5-R2b-null mice. By 3 months of age, epidermal whole-mount peroxidase staining (sebaceous glands appear brown) combined with Mayer's hemalum counterstain revealed that sebaceous glands in the tail of K5-R2b-null mice were virtually absent (compare (A) with (B)). This absence was confirmed by scanning through H&E-stained serial transverse sections of tail skin, looking for sebocytes (arrows in (C) and (D); asterisks indicate transversely sectioned hair shafts). (C) and (D) represent sections showing the largest proportion of sebaceous tissue encountered in serial transverse sections from tails of wild-type and K5-R2b-null mice, respectively. Scale bar, 200 μm.
Concomitant with the hair defects, an increased level of macrophage recruitment was observed in K5-R2b-null skin. F4/80 immunostaining for macrophages and monocytes in control 16 d.p.p. skin revealed some resident tissue macrophages present in the dermis (Supplementary Figure 1C), but similar staining of K5-R2b-null skin revealed markedly increased macrophage recruitment to both the dermis and the adipose tissue (Supplementary Figure 1D and E).
Sebaceous glands develop in the skin of K5-R2b-null mice, but subsequently atrophy
Male mice lacking Fgf-7 were reported as having greasy, matted fur (Guo et al, 1996). As the coats of K5-R2b-null mice have a silky appearance that might be attributable to an oily coat, we investigated sebaceous gland development in these mice. Whereas the relatively small sebaceous glands that develop on pelage follicles appeared unaffected in mutant mice (data not shown), whole-mount staining of tail skin epidermis revealed dramatic differences in mutant mice. By 6 d.p.p., the tail hair follicles are already midway through the first anagen growth phase, and nascent sebaceous glands can be seen as rudimentary buds near the top of the follicle in both control and K5-R2b-null mice (Supplementary Figure 2). From day 6 onwards, the differences in the rate of sebaceous gland growth between the two genotypes became more evident, with K5-R2b glands barely increasing in size after 2 weeks of age, whereas control glands continued to grow (Supplementary Figure 2).
By virtue of their high endogenous peroxidase activity, mature sebocytes are readily apparent in epidermal whole-mount preparations (Figure 2A). By 3 months of age, in contrast to the abundance of sebaceous glands in control tail skin, in the null mice, there was a virtual absence of sebaceous glands (Figure 2B). This was confirmed by scanning through hematoxylin and eosin (H&E)-stained serial transverse sections of tail skin. In sections from control mice that show the largest concentration of sebocytes, the sebaceous glands fill the entire field of view (Figure 2C). This was never the case for null mice, where no more than two or three sebocytes were ever observed (Figure 2D).
Epidermal hyperthickening and dysplasia develop with age in the absence of Fgfr2b
As K5-R2b-null mice aged, the hair phenotype became more extreme, such that by 18 months of age, both males and females had extremely sparse pelage hair. H&E staining of longitudinal sections through back skin harvested from control (Figure 3A and A′) and K5-R2b (Figure 3B and B′) mice highlighted the sparse hair follicles in the mutant mice. Somewhat surprisingly, the interfollicular epidermis of K5-R2b-null mice was thicker than that of control mice. Interfollicular epidermis of control mice is extremely thin and although the dermis becomes tougher with age, ordinarily remains little more than one-cell thick (Figure 3A′). In mice lacking epidermal Fgfr2b, not only was the epidermis thicker but also there was an apparent parakeratosis, where keratinocyte nuclei that normally would be lost as the cells commit to terminal differentiation were retained within the epidermis (Figure 3B′). Such differences were even more apparent in the tail, where differences in skin phenotypes are more marked by virtue of its multilayered structure and higher proliferative index. Transverse sections from wild-type tail skin displayed a clearly defined basal layer of keratinocytes, with nuclei obviously becoming less abundant toward the keratinized surface of the skin (Figure 3C). By comparison, the basal layer of keratinocytes in Fgfr2b-null tail skin showed regions of dysplasia while the differentiation process in the epidermis was compromised, with clear evidence of parakeratosis (Figure 3D).
Figure 3.
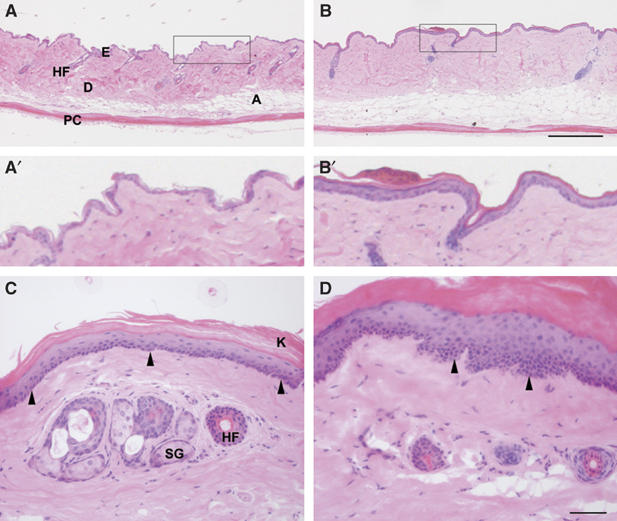
Epidermal hyperthickening in the absence of Fgfr2b. Transverse sections through back and tail skin of 18-month-old mice were stained with H&E. Back skin of control (A and A′) and K5-R2b-null mice (B and B′). In wild-type tail skin (C), the basal layer of keratinocytes is clearly visible as a distinct line of nuclei at the dermo-epidermal junction (arrowheads) and anuclear, keratinized squames can be seen sloughing from the surface. By comparison, Fgfr2b-null tail skin (D) shows regions of dysplasia (arrowheads) and parakeratosis. A, adipose layer; D, dermis; E, epidermis; HF, hair follicle; K, keratinized squames; PC, panniculus carnosus; SG, sebaceous gland. Scale bars, 200 μm (A, B) and 50 μm (C, D).
Defects in the skin were not restricted to elderly mice alone. Thorough macroscopic examination of our mouse colony revealed that K5-R2b-null virgin female mice exhibited a clear hyperproliferative defect in the nipples. Nipples of wild-type 8-month-old virgin female mice were barely discernible by the naked eye (Supplementary Figure 3A). In contrast, those of mice lacking Fgfr2b in the epidermis were very prominent (Supplementary Figure 3B). Transverse sections through the nipples of control (Supplementary Figure 3C) and K5-R2b-null mice (Supplementary Figure 3D) revealed epithelial hyperplasia and the frequent occurrence of keratin-filled cysts in the mutant mice.
Spontaneous papilloma formation in the absence of Fgfr2b
Despite being bred onto a mixed background of 129ola and C57Bl6/J strains, which are relatively tumor resistant (Woodworth et al, 2004), 10% of K5-R2b mice older than 8 months developed spontaneous papillomas (n=6 from 60 mice; one papilloma per mouse). These papillomas exhibited classical exophytic morphology, with a stalk of relatively normal skin leading to a hyperthickened, keratinized papilloma packed with inflammatory cells (Figure 4A). These papillomas also showed upregulation of keratin 6, not normally expressed in the interfollicular epidermis, which is a classical marker of activated keratinocytes (Figure 4B). 5-Bromo-2′-deoxyuridine (BrdU) staining for proliferating cells highlighted the hyperproliferative nature of the papilloma keratinocytes and also revealed that proliferation was not restricted to the basal layer (Figure 4C). Interestingly, the papillomas tended to arise in regions of mechanical stress, such as around the face and in the perianal region.
Figure 4.
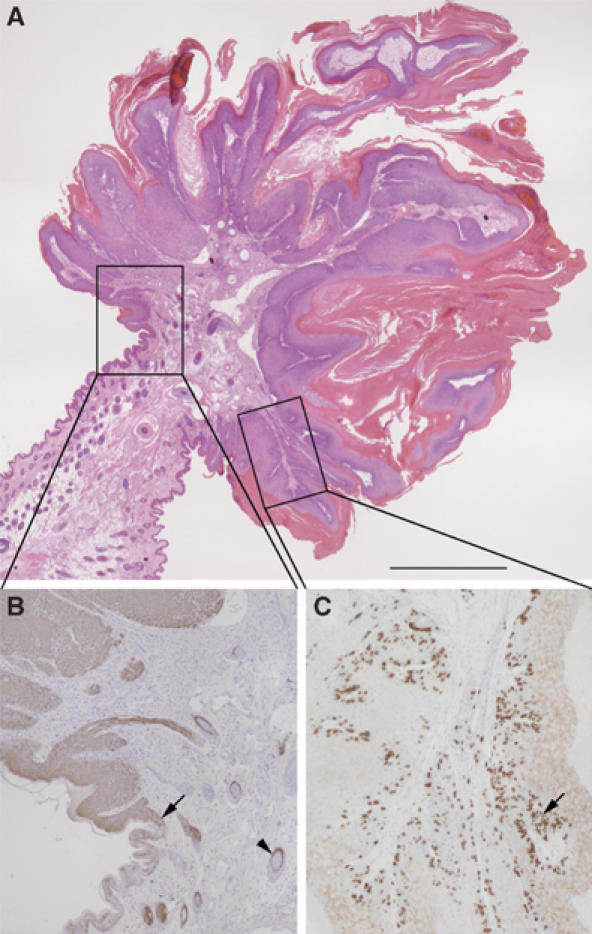
Spontaneous papilloma formation in the absence of Fgfr2b. (A) H&E staining reveals classical exophytic morphology of spontaneous papillomas, with a stalk of relatively normal skin leading to a hyperthickened, keratinized papilloma packed with inflammatory cells. Higher magnification views of the boxed areas in (A) show that papilloma keratinocytes express keratin 6 (brown immunostaining in (B)), normally restricted to hair follicles (arrowhead), from the margin, where the papilloma begins (arrow). BrdU staining for proliferating cells (C) in both basal and suprabasal layers (arrow). Scale bar, 200 μm.
Heightened sensitivity to skin carcinogenesis in mice lacking Fgfr2b in the skin
Having observed the occurrence of spontaneous papillomas and other epidermal defects, we hypothesized that Fgfr2b might be fulfilling a tumor-suppressive role in the skin. Therefore, we assessed the sensitivity of K5-R2b-null mice to classical two-step skin carcinogenesis protocols using DMBA initiation followed by TPA tumor promotion. In an initial study, using only female mice (n=minimum of eight per group) and a twice weekly TPA treatment regime, mice lacking Fgfr2b in the epidermis developed their first papillomas within 8 weeks of DMBA treatment (Figure 5A). This was 3 months earlier than the first papilloma that appeared in K5-R2b-null mice treated with TPA alone. In contrast, control mice treated with DMBA/TPA did not develop papillomas until 34 weeks post-initiation, by which time 100% of the knockout mice had developed papillomas or even carcinomas. Control mice subject to TPA treatment alone never developed papillomas.
Figure 5.
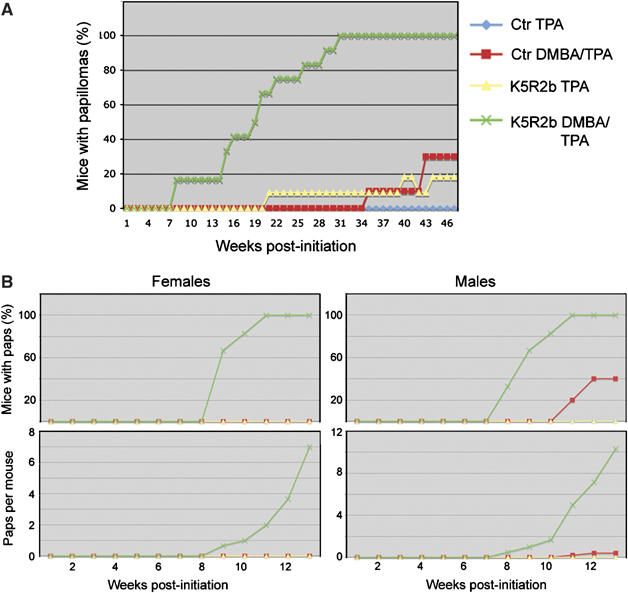
Enhanced sensitivity to skin carcinogenesis in K5-R2b-null mice. Cohorts of female mice were subjected to classical two-step skin carcinogenesis treatment. (A) Female mice were treated with DMBA or solvent acetone alone at 8 weeks of age and then all treated twice weekly with TPA for 15 weeks. In a repeat study (B), the protocol was adjusted slightly, such that male and female mice were treated, giving a total of eight groups (key same as for (A)). The treatment regime differed only in that the mice received once weekly applications of a double dose of TPA.
To confirm the validity of these dramatic findings, the study was repeated with both male and female mice (average n=5). The protocol was adjusted slightly, with the mice receiving once weekly applications of 7.4 μg TPA rather than the twice weekly application of 3.7 μg TPA. Once again, the results were clearcut; even in cases where control mice treated with DMBA developed papillomas, the number of papillomas per mouse was never above two, whereas knockout mice were recorded as developing up to 26 per animal (Figure 5B). Thus, both sets of data show an unequivocally enhanced sensitivity to DMBA-induced skin carcinogenesis in K5-R2b-null mice.
Enhanced progression of skin lesions to squamous cell carcinoma in K5-R2b-null mice
Mutant mice also exhibited changes that suggested that Fgfr2b might play an important role in blocking the progression of benign papillomatous lesions to benign keratoacanthomas (Figure 6A), and more invasive squamous cell carcinomas (Figure 6B). In the present study, on a C57Bl6/Jx129ola background, no lesion on a DMBA-treated control mouse progressed to squamous cell carcinoma (n=3 papillomas from 10 mice >6 months old). Although the mice used in this study were on a mixed background, our observations are in line with published data, which suggest that papillomas do not convert on a C57Bl6/J background (Woodworth et al, 2004). In contrast, K5-R2b-null mice treated with DMBA developed squamous cell carcinomas (n=6 squamous cell carcinomas from 57 papillomas on 12 mice >6 months old; maximum of two carcinomas per mouse).
Figure 6.
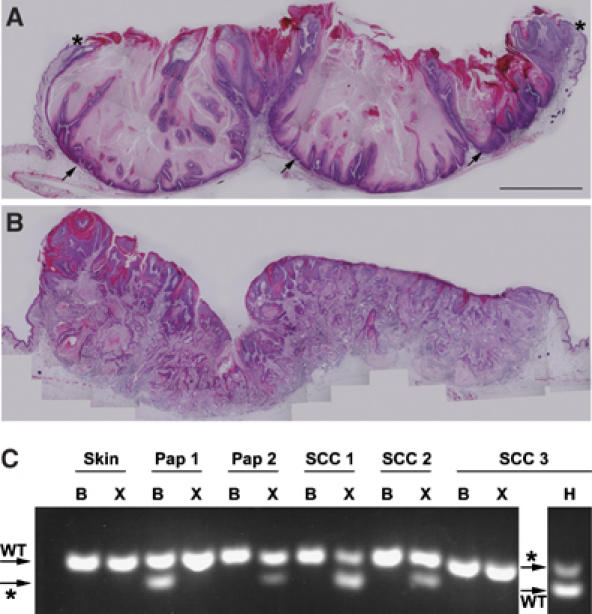
Progression of skin carcinogenesis lesions to squamous cell carcinoma. Transverse sections through lesions from K5-R2b-null mice highlight keratoacanthoma (A) and squamous cell carcinoma (B). Although keratoacanthomas no longer exhibit the exophytic growth of papillomas, with their margins covered with normal skin (asterisks in (A)), they are still non-invasive, with a clearly defined basal layer of keratinocytes (arrows in (A)). In contrast, the lesions that progress to squamous cell carcinoma (B) show no clearly defined basal surface, but rather a mass of invasive keratinocyte projections and a strong recruitment of inflammatory cells. (C) RFLP analysis of mutations in codon 61 of ha-ras. PCR products derived from skin, papilloma (Pap) or squamous cell carcinoma (SCC) digested with BspHI (B), XbaI (X) and Hpy118II (H) were separated on 4% agarose gels. Bands diagnostic of the wild-type (WT) and mutant (*) alleles are indicated (arrows).
Mutations in codon 61 of ha-ras
DNA obtained from frozen skin, papillomas and tumors excised from mice was analyzed by restriction fragment length polymorphism (RFLP) for ha-ras codon 61 mutations. No mutations were detected in 20 uninvolved skin samples taken from K5-R2b mice. Three of the four previously reported ha-ras mutations (Jaworski et al, 2005) were detected in this study (Figure 6C). One tumor arising spontaneously on a K5-R2b-null mouse showed G61L conversion in the absence of DMBA treatment. This activating mutation was found in a further five tumors taken from K5-R2b-null mice treated with DMBA/TPA. Of further mutations in this group, one mouse had two tumors, with each bearing a different activating mutation, G61L and G61K respectively; a tumor from a different mouse harbored the G61K mutation; the G61H mutation was found in two papillomas from another; and, of the additional seven papillomas analyzed from the treated K5-R2b-null mice, all showed G61L mutation although the mutant band was often quite weak, indicating dilution of the sample with non-mutated cells. These comprised either inflammatory cells or non-mutated keratinocytes from the margins of the lesion. Two papillomas arising spontaneously in the K5-R2b mice treated only with TPA showed no detectable ha-ras mutations and, surprisingly, this also was the case for the only two papillomas analyzed from DMBA/TPA-treated control mice. This may reflect a large contribution of non-mutated cells in samples taken from these relatively small lesions masking any mutant polymerase chain reaction (PCR) product.
Differential gene expression in knockout and wild-type skin
To gain further insight into the potential mechanism underlying the enhanced sensitivity of Fgfr2b-null skin to cancer development, we performed gene expression profiling on RNA isolated from back skin samples from 1-year-old control and mutant mice, using the Illumina Sentrix MouseRef-8® microarray platform. Using a two-fold change in expression level as a cutoff, we detected elevated expression levels of several differentiation-specific genes in mutant skin, which most likely reflect the epidermal hyperthickening (data not shown). We also saw elevated expression of the pro-inflammatory cytokine interleukin (IL)-18 (Supplementary Figure 4). Several genes also showed reduced expression in mutant skin, among them Serpin a3b, a member of the serine protease inhibitor family that was reduced dramatically (Supplementary Figure 4). Differential expression levels were confirmed by semiquantitative RT–PCR, using RNA samples taken from 3- and 6-month-old control and mutant mice. These included full-thickness back and tail skin, as well as RNA isolated from only the epidermis of tail skin. For all samples, we confirmed that the starting cDNAs were of equivalent concentration and quality by amplifying the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT). Only products from cycles 30 and 35 are illustrated, but these clearly show the differences between control and mutant tissues, and that the increase in IL-18 expression is seen both in total skin and epidermis of mutant mice (Supplementary Figure 4). This indicates that IL-18 is likely to be expressed by mutant keratinocytes themselves, rather than the levels just reflecting increased inflammatory infiltrate.
Alongside these studies, we also examined gene expression in tumors and papillomas of control (papillomas only) and knockout mice, to look for differences in expression of Fgf ligands and receptors as well as known Fgf target genes (auf dem Keller et al, 2006). However, no significant differences were found in the expression levels of any of the above, or in expression of Igf or Egf ligand or receptor families (data not shown).
Altered γδT-cell features in Fgfr2b-null skin
Previous work has shown that γδT cells resident within the epidermis, also known as dendritic epidermal T-cells, are key sources of Fgfs 7 and 10 during wound repair (Jameson et al, 2002), and also that mice lacking γδT cells show enhanced sensitivity to the two-step model of skin carcinogenesis, as used in our study (Girardi et al, 2001). Therefore, we analyzed the γδT cells resident within the skin of 1-year-old control and K5-R2b-null mice, using immunofluorescent staining of epidermal whole mounts to assess both cell number and morphology. The most appropriate tissue for this purpose is the epidermis of the ear, as it can be dissociated from the dermis in a manner similar to tail epidermis, but is much thinner and the γδT cells can more readily be visualized by immmunostaining. By using ImageJ software to analyze single-channel greyscale confocal images that reflected γδT-cell staining, it was possible to quantitate automatically the number of cells per image and also measure their perimeter. We observed significantly more γδT cells within the epidermis of mutant skin when compared with control skin (38% increase, P<0.01 using the Mann–Whitney test; compare Figure 7A and B, quantitated in Figure 7C) and also saw a significant increase in γδT-cell perimeter in mutant skin (33% increase, P<0.001 using the Mann–Whitney test; quantitated in Figure 7D), reflecting a difference in cell morphology, with cells in the mutant skin exhibiting a more dendritic appearance.
Figure 7.
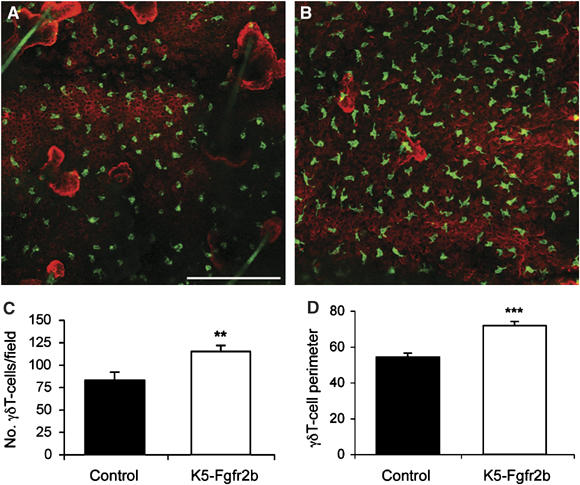
γδT-cell abnormalities in K5-R2b-null epidermis. Whole-mount preparations of ear epidermis from control (A) and K5-R2b-null (B) mice stained with antibodies to γδT cells (green) and keratin 14 (red) highlight the increased γδT-cell density in mutant epidermis (quantitated in (C)). In addition to being present in increased numbers, γδT cells in mutant epidermis display an altered morphology, adopting a more dendritic appearance, reflected by a significantly increased cell perimeter (quantitated in (D)). Scale bar in (A), 50 μm. **P<0.01; ***P<0.001 using the Mann--Whitney test.
Discussion
Our mice, lacking Fgfr2b only in keratinized epithelia, provide the first opportunity to study the specific functions of Fgfr2b in adult skin. Previous studies that have sought to investigate the role of Fgfr2b in the skin have been hampered either by interference with signalling via other receptors that share common ligands in mice overexpressing dominant-negative Fgfr2b in the epidermis (Werner et al, 1994), ligand redundancy in the case of Fgf-7 knockout mice (Guo et al, 1996), or perinatal lethality, and a consequent reliance on skin grafting, in Fgfr2b knockout mice (Petiot et al, 2003).
Defects in skin development in the absence of Fgfr2b
Our initial findings recapitulate those data obtained from Fgfr2b knockout studies where late-stage embryonic skin was grafted onto nude mouse recipients (Petiot et al, 2003). These experiments showed that although keratinocytes could undergo terminal differentiation in the absence of Fgfr2b, they failed to form morphologically normal pelage hairs, and the hair follicles that formed failed to orientate properly, resulting in a chaotic first wave of hair morphogenesis (Petiot et al, 2003). The defects that we observed in K5-R2b-null mice are similar in terms of failed hair patterning, but are less dramatic in terms of follicle orientation. This is entirely consistent with our strategy of using the expression of Cre recombinase under the control of the bovine keratin 5 promoter to delete Fgfr2b, as Cre is not active in the epidermis until around E15.5, with target gene deletion not being attained until late in fetal development (Ramirez et al, 2004), well after follicle patterning has been established. Throughout fetal development, the skin of K5-R2b-null mice, therefore, develops as wild type and, despite there being no detectable Fgfr2b message in neonatal skin of K5-R2b-null mice (data not shown), defects in skin development do not become apparent until the mice are approximately 1 week old. Recent transgenic studies overexpressing dominant-negative Fgfr2b, under the control of the FoxN1 promoter, also have recapitulated the hair patterning defects seen in Fgfr2b-null mice, identifying Igfbp5 as a key modulator of Fgf signalling in the developing hair follicle (Schlake, 2005).
Having confirmed the validity and efficacy of our approach, our study provides the first evidence of the role of Fgfr2b in sebaceous gland development. Fgfr2b is expressed throughout the epidermis, hair follicles and sebaceous glands (Danilenko et al, 1995), and we have observed that the continued presence of Fgfr2b in the skin is a necessary prerequisite for the long-term survival of sebocytes. Whether this is due to a requirement for Fgfr2b to be expressed on sebocytes, or whether keratinocytes produce some kind of survival signal as a consequence of ligand–receptor interaction is the subject of ongoing investigations.
Fgfr2b as a tumor suppressor in the skin
The epidermal hyperthickening that developed as K5-R2b-null mice aged was somewhat surprising, given that Fgfr2-null embryos have thin skin relative to wild-type littermates (Petiot et al, 2003), and a hypertrophic phenotype is not what might be expected when a growth factor receptor is deleted. This led us to investigate whether Fgfr2b might function as a suppressor of growth in the adult.
Fgfr2b has been implicated as a tumor suppressor in the urothelium, with decreased expression correlating with worse prognosis in transitional cell carcinoma (Diez de Medina et al, 1997; Ricol et al, 1999). Given that xenografting studies suggest that this tumor-suppressive effect is due to the C-terminus of Fgfr2b modulating IGF-II signalling (Bernard-Pierrot et al, 2004), the increased tumor cell proliferation and decreased apoptosis seen might well result from elevated IGF-II levels. Assuming similar mechanisms were operative in human disease, they could result in increased aggressiveness of tumors and account for poorer survival of patients with transitional cell carcinoma of the bladder (Diez de Medina et al, 1997; Ricol et al, 1999). However, we saw no changes in the expression of either the ligands Igf-I and Igf -II or their receptors (data not shown), suggesting that this is not the case for our model.
FGFR2 also has been implicated as a tumor suppressor in other cancers. Thus, in human salivary adenocarcinoma, it appears to act to induce differentiation and alter apoptosis of cancer cells (Zhang et al, 2001). In prostate carcinoma, both murine and human, decreased expression of FGFR2 has resulted in overexpression of FGFR1, which is thought to drive uncontrolled proliferation and subsequent tumorigenesis (Jin et al, 2003; Yasumoto et al, 2004). We found no change in Fgfr1 expression in either K5-R2b mutant skin or in neoplastic lesions from carcinogen-treated mice. FGFR2 may well exert its protective effect via different mechanisms dependent on the tissue type, and indeed may not always act as a tumor suppressor. For example, activating mutations in FGFR2 have been described in human gastric cancer (Jang et al, 2001), but our study provides the first evidence of a protective role of Fgfr2b in the skin.
The precise mechanism underlying this protective effect is not yet clear. Fgf-7, signalling via Fgfr2b, has been shown to upregulate a wide array of downstream target genes in keratinocytes (Steiling and Werner, 2003). Among these genes, the Nrf2 transcription factor has been implicated as being an essential regulator of tumor prevention, both acting to detoxify carcinogens and to prevent oxidative damage. Thus, mice expressing a dominant-negative Nrf2 transgene in the epidermis exhibited strikingly enhanced skin tumor development in response to two-stage skin carcinogenesis treatment (auf dem Keller et al, 2006). However, Nrf2 expression was unaffected in K5-R2b-null mice, and no differences in the expression levels of known Nrf2 targets (auf dem Keller et al, 2006) were detected in our microarray experiments, suggesting that this mechanism is not responsible for the observed phenotype.
The absence of Fgfr2b may affect other protective pathways, as we see a large frequency of lesions in K5-R2b-null mice in response to carcinogenic insult, and an unusual range of mutations at the ha-ras codon 61 locus. Classically, topical DMBA treatment elicits a CAA to CTA (G61L) mutation in keratinocytes (Quintanilla et al, 1991), but RFLP analysis identified three independent mutations in lesions of K5-R2b-null mice. This suggests that enhanced genomic instability in the absence of Fgfr2b might underlie the phenotype we observe. This hypothesis is further supported by the finding that FGFR2b-null mice develop papillomas in response to TPA treatment without a preceding carcinogenic insult.
At the level of gene expression, two genes of particular interest showed differential expression between control and mutant skin. IL-18, expression of which is upregulated in RNA samples isolated from mutant skin and epidermis, has been shown to be upregulated both in inflammatory cells and keratinocytes in neoplastic skin lesions (Park et al, 2001; Yamanaka et al, 2006). Thus, upregulation might reflect the increased inflammatory infiltrate in the skin, but the epidermal expression suggests keratinocyte upregulation with subsequent recruitment of inflammatory cells. In contrast, expression of the serine proteinase inhibitor Serpin a3b was markedly reduced in mutant skin and epidermal samples. Very little is known about this molecule, but the related protein Maspin has been shown to act as an epithelial tumor suppressor, and other members of the family have been reported to modulate the activity of the urokinase pathway, a pathway that can drive invasion and metastasis (Lockett et al, 2006). Future studies will address the functions of these and other potential Fgfr2b-dependent genes in the skin.
Recent data supporting the role of inflammatory responses in driving tumor progression (Balkwill, 2004; Balkwill and Coussens, 2004) might suggest that the increased macrophage infiltration observed in K5-R2b-null animals could be acting as a promoting factor. Possibly the lack of sebocytes in such animals leads to drier and more scaly skin, which, together with the enhanced IL-18 expression, causes the accumulation of infiltrating macrophage/monocytes. However, the level of infiltration of these cells, although substantial, may not solely be responsible for the observed changes in response to carcinogen treatment. As indicated, the mice were bred on a carcinogen-resistant background, yet the high frequency of papilloma formation coupled with the conversion to carcinoma was so dramatic as to cause us to wonder whether the level of inflammation per se was the only driving factor.
Previous studies have shown γδT cells, inflammatory cells resident within the epidermis, to play important roles in the skin. As key sources of the Fgfr2b ligands Fgf-7 and Fgf-10, knockout studies showed γδT cells to be crucial for wound re-epithelialization (Jameson et al, 2002). Interestingly, their absence also correlated with enhanced sensitivity to chemical carcinogenesis in the skin (Girardi et al, 2001). Thus, one of their key functions in the skin is to act as sentinels, detecting abnormal keratinocytes, such as those damaged by wounding or by carcinogens. Looking at γδT cells in the epidermis of control and mutant mice, we found significantly increased numbers in the ears of year old null mice. At first sight, this finding seemed paradoxical given the findings already reported on an inverse correlation between γδT-cell levels and carcinogen sensitivity (Girardi et al, 2001). However, the morphology of the cells in mutant skin differed from that in control skin, perhaps reflecting a different state of activation. Activated γδT cells at a wound margin were shown to adopt a more rounded morphology relative to that more distant from the edge (Jameson et al, 2002). Thus, even though there are more γδT cells present in the skin of K5-R2b-null mice, it may be that they are unable to communicate properly with the keratinocytes, resulting in less cell activation and a failure to detect malignant keratinocytes. This possible mechanism is the subject of ongoing investigations. Also, our data thus far only describe differences in ear epidermis, as this is the best site to visualize γδT cells, and it will be important to establish that similar changes occur in back skin.
Although the precise basis for the observed effects on tumor development and progression is yet to be fully understood, our findings provide a potential mechanistic insight into the phenotype we see in K5-R2b-null mice, suggesting that the nature of the inflammatory reaction tips toward a more pro-tumorigenic phenotype. Whatever the basis, the effect of Fgfr2b loss in the skin remains sufficiently severe to suggest that Fgfr2b is a major player in regulating normal skin homeostasis in response to carcinogenic insult and thus represents a potential target for therapeutic intervention or for cancer prevention.
Materials and methods
K5-R2b-null mice
Mice lacking Fgfr2b in the epidermis (K5-R2b null) were generated as follows: female mice were bred to homozygosity for a floxed allele (Fgfr2bflox/flox), with loxP sites inserted into the introns flanking the IIIb exon of the Fgfr2 gene (De Moerlooze et al, 2000). These were crossed with male mice carrying one copy of the Cre recombinase transgene under the control of the keratin 5 promoter (Ramirez et al, 2004). The K5-Cre transgene has been shown previously to give high-efficacy excision of a floxed target gene (Brakebusch et al, 2000). Provided that the cre allele is transmitted paternally, only epithelia derived from keratin 5-expressing cells are targeted for deletion. Maternal transmission results in ubiquitous excision of the floxed sequence caused by Cre expression in the maternal germ line (Ramirez et al, 2004). Mice were maintained on a mixed C57Bl6/J × 129ola background for all studies. Littermate controls (Fgfr2bflox/flox Cre negative) were maintained alongside K5-R2b-null mice under identical husbandry conditions.
RNA isolation and RNase protection assay
RNA isolation (Chomczynski and Sacchi, 1987) and RNase protection assays (on 20 μg total RNA samples) (Werner et al, 1992) were performed as described. As a loading control, 1 μg of each RNA sample was resolved through a 1% agarose gel and stained with ethidium bromide (data not shown). In addition to an antisense RNA probe recognising Fgfr2b, the RNA samples also were hybridized with a probe for the housekeeping gene GAPDH (Zhang et al, 2004).
Preparation of skin sections
Back skin was harvested onto a nitrocellulose membrane (Amersham, UK) and fixed overnight in 1% acetic acid/95% ethanol at 4°C, rinsed in 100% ethanol and processed for paraffin wax embedding. Longitudinal sections (4 μm) were cut along the line of the pelage hair follicles, parallel to the dorsal midline. Tail skin was removed from the bone and laid flat on a nitrocellulose membrane as above and, after wax embedding, transverse sections (4 μm) were cut perpendicular to the line of the hair follicles.
Immunohistochemistry
Wax sections (4 μm) were incubated with the appropriate primary antibody: macrophages/monocytes—1:125 dilution of a rat monoclonal antibody targeted to F4/80 (Serotec, Oxford, UK); keratin 6—1:2000 dilution of a rabbit polyclonal anti-K6 antibody (Covance, Denver, CO, USA), and incubated with biotinylated donkey anti-rat IgG or anti-rabbit IgG (Dianova GMBH, Hamburg, Germany). The VectaStain avidin–biotin–peroxidase complex kit was then used according to the manufacturer's instructions, before peroxidase detection with a diaminobenzidine-peroxidase substrate kit (both from Vector Laboratories, Burlingame, CA, USA). Sections were counterstained with Mayer's hemalum. Macrophage/monocyte infiltration was quantified by counting F4/80-positive cells in 2 × 0.3 mm2 fields from immunostained back skin sections (n=4 mice for each genotype).
Immunofluorescent γδT-cell staining
Ears were cut from mice (12 months old, n=3 mice per genotype) and the skin peeled away from the cartilage using forceps. Skin preparations were incubated in PBS+20 mM EDTA for 4 h at room temperature, and then the epidermis was removed with forceps and washed in PBS before fixation in ice-cold acetone for 20 min at −20°C. After rinsing in PBS, epidermal sheets were blocked for 1 h in 2% BSA in PBS at room temperature and then primary antibodies (keratin 14—1:10 000 dilution of a rabbit polyclonal anti-keratin 14 antibody (Covance, Harrogate, UK); γδT cells—1:100 dilution of GL3, an FITC-conjugated hamster anti-γδT-cell receptor monoclonal antibody (BD Biosciences, Oxford, UK)) were added directly to the blocking solution and incubated overnight at 4°C. The following morning, sheets were washed with PBS and incubated for 90 min at room temperature with a 1:100 dilution of Cy-3-conjugated donkey anti-rabbit secondary antibody (Upstate, Hampshire, UK), washed in PBS and mounted on silane-coated slides in Vectashield mounting medium+DAPI (Vector Labs, Burlingame, CA). Images were acquired on a Zeiss Axiovert 200M Confocal Microscope using the LSM510 Meta software (Zeiss, Welwyn Garden City, UK) and analyzed using ImageJ software (Rasband and Image, 1997–2004).
BrdU labelling
BrdU labelling was performed as described (Werner et al, 1994). Sections (4 μm) were incubated with a peroxidase-conjugated monoclonal antibody directed against BrdU (Roche Diagnostics, Rotkreuz, Switzerland) and stained with a diaminobenzidine-peroxidase substrate kit as above.
Tail epidermis whole mounts
Epidermal whole mounts were prepared as described (Braun et al, 2003) and fixed overnight in 1% acetic acid/95% ethanol at 4°C. After rinsing in 100% ethanol, they were rehydrated through a graded alcohol series to water and transferred to PBS. For Supplementary Figure 2, the specimens were counterstained for 10 s in a 1:4 dilution of Mayer's hemalum/water and mounted in 30% glycerol in PBS. For Figure 2, whole mounts were processed using the VectaStain peroxidase detection kit as described above for immunohistochemistry, dehydrated through alcohols into xylene and mounted in organic mounting medium.
Expression analysis
Total RNA was isolated from skin and papillomas of 1-year-old control and mutant mice, and squamous cell carcinoma from mutant mice only (using TriReagent according to the manufacturer's instructions; Sigma, Poole, UK). Total RNA (250 ng) was fluorescently labelled and samples hybridized to an Illumina Sentrix MouseRef-8® microarray chip according to the manufacturer's protocols (Illumina, San Diego, CA) and a two-fold change in expression level used as a cutoff. Differential expression levels were confirmed by semiquantitative RT–PCR, using RNA samples taken from 12-week-old control and mutant mice. These included full thickness back and tail skin, as well as RNA isolated from only the epidermis of tail skin. For all samples, we ran PCR reactions for 20, 25, 30 and 35 cycles to ensure we were comparing linear amplification products and confirmed that the starting cDNAs were of equivalent concentration and quality by amplifying the housekeeping gene HPRT. Primer details are as follows: IL-18 (forward: acaactttggccgacttcac; reverse: atcttccttttggcaagcaa; TM=60°C; product 388 bp), Serpin a3b (forward: tgctcagatggcacactagg; reverse: tcttcatcccggaagtatgg; TM=60°C; product 707 bp), HPRT (forward: cctgctggattacattaaagcgctg; reverse: gtcaagggcatatccaacaacaaac; TM=60°C; product 351 bp).
Skin carcinogenesis
Cohorts of female control and K5-R2b-null mice were subjected to two-step skin carcinogenesis protocols. In the first experiment, female mice (n=minimum of eight per group) were shaved at 7 weeks of age before treatment with topically applied DMBA (Sigma, UK; 25 μg in 200 μl acetone), or acetone alone, 1 week later. From 9 weeks old, all mice were treated twice weekly with TPA (Sigma, UK; 3.7 μg in 200 μl acetone) for 15 weeks. In a repeat study, the protocol was adapted such that both male and female mice were treated, giving a total of eight groups. The treatment regime differed only in that instead of twice weekly application of 3.7 μg TPA, the mice received 15 once weekly applications of 7.4 μg TPA in 200 μl acetone, in accordance with altered animal licence permission. The group sizes were as follows: females, Ctr TPA n=5, Ctr DMBA/TPA n=5, K5R2b TPA n=5, K5R2b DMBA/TPA n=6; males, Ctr TPA n=5, Ctr DMBA/TPA n=5, K5R2b TPA n=3, K5R2b DMBA/TPA n=3. Mice were monitored on a daily basis and papilloma/tumor counts recorded weekly for up to 46 weeks post-initiation, at which stage the experiment was terminated. If mice appeared sick or the tumor burden reached a predefined limit, they were killed and examined post-mortem. Skin lesions and macroscopically normal skin were harvested and either fixed overnight in 10% buffered formalin, snap frozen in liquid nitrogen or embedded in OCT for frozen sectioning. All experiments with animals were carried out under Home Office licence and according to Institutional guidelines.
Ha-ras mutational analysis
Small pieces (1–2 mm2) of frozen tissues were digested with proteinase K overnight. After heat inactivation, 1 μl of the resultant mixture was amplified by PCR using standard methods to give a 166-bp fragment. Forward and reverse primers were GGGAGACA TGTCTACTGGACATC and TAGCCATAGGTGGCTCACCT, respectively. Skin, papillomas and tumors were analyzed for mutations at ha-ras codon 61 by RFLP analysis (Jaworski et al, 2005). Restriction enzymes used were Hpy118III, TaqI, XbaI and BspHI (New England Biolabs). For further details, see Table I.
Table 1.
Ha-ras RFLP analysis
| Diagnostic restriction sites in Ha-ras PCR product | ||||
|---|---|---|---|---|
| Hpy118III | TaqI | XbaI | BspHI | |
| tCNNga | tCGA | tCTAga | tCATga | |
| WT sequence | ||||
| CAA (Gln) | 1 | 1 | — | — |
| Mutant sequences | ||||
| AAA (Lys) | — | 1 | — | — |
| CGA (Arg) | 1 | 2 | — | — |
| CTA (Leu) | 1 | 1 | 1 | — |
| CAT (His) | 1 | 1 | — | 1 |
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends
Acknowledgments
We thank Professor José Jorcano for keratin-5 Cre transgenic mice, Dr Jessica Strid and Professor Adrian Hayday for advice on γδT-cell biology, Pooja Seedah and George Elia for excellent technical assistance, Dr Charles Mein and Nadiya Mahmud for help with microarray analysis, Dr Gareth Thomas for histopathological advice and Dr Simon Hughes for advice on RFLP analysis. This work was supported by Cancer Research UK (RG, VF, IH and CD), The Royal Society (RG), The Wellcome Trust (RG and A-MC), Bart's and The London Charitable Foundation (MJ) and the Swiss National Science Foundation (grant no. 3100Ao-109340/1 to SW).
References
- auf dem Keller U, Huber M, Beyer TA, Kumin A, Siemes C, Braun S, Bugnon P, Mitropoulos V, Johnson DA, Johnson JA, Hohl D, Werner S (2006) Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol Cell Biol 26: 3773–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F (2004) Cancer and the chemokine network. Nat Rev Cancer 4: 540–550 [DOI] [PubMed] [Google Scholar]
- Balkwill F, Coussens LM (2004) Cancer: an inflammatory link. Nature 431: 405–406 [DOI] [PubMed] [Google Scholar]
- Bernard-Pierrot I, Ricol D, Cassidy A, Graham A, Elvin P, Caillault A, Lair S, Broet P, Thiery JP, Radvanyi F (2004) Inhibition of human bladder tumour cell growth by fibroblast growth factor receptor 2b is independent of its kinase activity. Involvement of the carboxy-terminal region of the receptor. Oncogene 23: 9201–9211 [DOI] [PubMed] [Google Scholar]
- Brakebusch C, Grose R, Quondamatteo F, Ramirez A, Jorcano JL, Pirro A, Svensson M, Herken R, Sasaki T, Timpl R, Werner S, Fassler R (2000) Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J 19: 3990–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun KM, Niemann C, Jensen UB, Sundberg JP, Silva-Vargas V, Watt FM (2003) Manipulation of stem cell proliferation and lineage commitment: visualisation of label-retaining cells in whole mounts of mouse epidermis. Development 130: 5241–5255 [DOI] [PubMed] [Google Scholar]
- Celli G, LaRochelle WJ, Mackem S, Sharp R, Merlino G (1998) Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J 17: 1642–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Danilenko DM, Ring BD, Yanagihara D, Benson W, Wiemann B, Starnes CO, Pierce GF (1995) Keratinocyte growth factor is an important endogenous mediator of hair follicle growth, development, and differentiation. Normalization of the nu/nu follicular differentiation defect and amelioration of chemotherapy-induced alopecia. Am J Pathol 147: 145–154 [PMC free article] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest J, Hajihosseini M, Rosewell I, Dickson C (2000) An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal–epithelial signalling during mouse organogenesis. Development 127: 483–492 [DOI] [PubMed] [Google Scholar]
- Diez de Medina SG, Chopin D, El Marjou A, Delouvee A, LaRochelle WJ, Hoznek A, Abbou C, Aaronson SA, Thiery JP, Radvanyi F (1997) Decreased expression of keratinocyte growth factor receptor in a subset of human transitional cell bladder carcinomas. Oncogene 14: 323–330 [DOI] [PubMed] [Google Scholar]
- Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar RE, Hayday AC (2001) Regulation of cutaneous malignancy by gammadelta T cells. Science 294: 605–609 [DOI] [PubMed] [Google Scholar]
- Grose R, Dickson C (2005) Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev 16: 179–186 [DOI] [PubMed] [Google Scholar]
- Guo L, Degenstein L, Fuchs E (1996) Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev 10: 165–175 [DOI] [PubMed] [Google Scholar]
- Guo L, Yu QC, Fuchs E (1993) Targeting expression of keratinocyte growth factor to keratinocytes elicits striking changes in epithelial differentiation in transgenic mice. EMBO J 12: 973–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson J, Ugarte K, Chen N, Yachi P, Fuchs E, Boismenu R, Havran WL (2002) A role for skin gammadelta T cells in wound repair. Science 296: 747–749 [DOI] [PubMed] [Google Scholar]
- Jang JH, Shin KH, Park JG (2001) Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res 61: 3541–3543 [PubMed] [Google Scholar]
- Jaworski M, Buchmann A, Bauer P, Riess O, Schwarz M (2005) B-raf and Ha-ras mutations in chemically induced mouse liver tumors. Oncogene 24: 1290–1295 [DOI] [PubMed] [Google Scholar]
- Jin C, McKeehan K, Guo W, Jauma S, Ittmann MM, Foster B, Greenberg NM, McKeehan WL, Wang F (2003) Cooperation between ectopic FGFR1 and depression of FGFR2 in induction of prostatic intraepithelial neoplasia in the mouse prostate. Cancer Res 63: 8784–8790 [PubMed] [Google Scholar]
- Lockett J, Yin S, Li X, Meng Y, Sheng S (2006) Tumor suppressive maspin and epithelial homeostasis. J Cell Biochem 97: 651–960 [DOI] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS (1998) Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev 12: 3156–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N (2001) Fibroblast growth factors. Genome Biol 2: REVIEWS3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Byun D, Kim TS, Kim YI, Kang JS, Hahm ES, Kim SH, Lee WJ, Song HK, Yoon DY, Kang CJ, Lee C, Houh D, Kim H, Cho B, Kim Y, Yang YH, Min KH, Cho DH (2001) Enhanced IL-18 expression in common skin tumors. Immunol Lett 79: 215–219 [DOI] [PubMed] [Google Scholar]
- Petiot A, Conti FJ, Grose R, Revest JM, Hodivala-Dilke KM, Dickson C (2003) A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development 130: 5493–5501 [DOI] [PubMed] [Google Scholar]
- Quintanilla M, Haddow S, Jonas D, Jaffe D, Bowden GT, Balmain A (1991) Comparison of ras activation during epidermal carcinogenesis in vitro and in vivo. Carcinogenesis 12: 1875–1881 [DOI] [PubMed] [Google Scholar]
- Ramirez A, Page A, Gandarillas A, Zanet J, Pibre S, Vidal M, Tusell L, Genesca A, Whitaker DA, Melton DW, Jorcano JL (2004) A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis 39: 52–57 [DOI] [PubMed] [Google Scholar]
- Rasband WS, Image J (1997–2004) Bethesda, Maryland, USA: National Institutes of Health, http://rsb.info.nih.gov/ij/
- Revest JM, Spencer-Dene B, Kerr K, De Moerlooze L, Rosewell I, Dickson C (2001) Fibroblast growth factor receptor 2-IIIb acts upstream of Shh and Fgf4 and is required for limb bud maintenance but not for the induction of Fgf8, Fgf10, Msx1, or Bmp4. Dev Biol 231: 47–62 [DOI] [PubMed] [Google Scholar]
- Ricol D, Cappellen D, El Marjou A, Gil-Diez-de-Medina S, Girault JM, Yoshida T, Ferry G, Tucker G, Poupon MF, Chopin D, Thiery JP, Radvanyi F (1999) Tumour suppressive properties of fibroblast growth factor receptor 2-IIIb in human bladder cancer. Oncogene 18: 7234–7243 [DOI] [PubMed] [Google Scholar]
- Schlake T (2005) FGF signals specifically regulate the structure of hair shaft medulla via IGF-binding protein 5. Development 132: 2981–2990 [DOI] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S (1999) Fgf10 is essential for limb and lung formation. Nat Genet 21: 138–141 [DOI] [PubMed] [Google Scholar]
- Steiling H, Werner S (2003) Fibroblast growth factors: key players in epithelial morphogenesis, repair and cytoprotection. Curr Opin Biotechnol 14: 533–537 [DOI] [PubMed] [Google Scholar]
- Sundberg JP, Hogan ME (1994) Hair types and subtypes in the laboratory mouse.In Handbook of Mouse Mutations with Skin and Hair Abnormalities: Animal Models and Biomedical Tools,Sundberg JP (ed) pp 57–68. Bar Harbor, MI: CRC Press [Google Scholar]
- Werner S, Peters KG, Longaker MT, Fuller-Pace F, Banda MJ, Williams LT (1992) Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Natl Acad Sci USA 89: 6896–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Smola H, Liao X, Longaker MT, Krieg T, Hofschneider PH, Williams LT (1994) The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science 266: 819–822 [DOI] [PubMed] [Google Scholar]
- Woodworth CD, Michael E, Smith L, Vijayachandra K, Glick A, Hennings H, Yuspa SH (2004) Strain-dependent differences in malignant conversion of mouse skin tumors is an inherent property of the epidermal keratinocyte. Carcinogenesis 25: 1771–1778 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Clark R, Dowgiert R, Hurwitz D, Shibata M, Rich BE, Hirahara K, Jones DA, Eapen S, Mizutani H, Kupper TS (2006) Expression of interleukin-18 and caspase-1 in cutaneous T-cell lymphoma. Clin Cancer Res 12: 376–382 [DOI] [PubMed] [Google Scholar]
- Yasumoto H, Matsubara A, Mutaguchi K, Usui T, McKeehan WL (2004) Restoration of fibroblast growth factor receptor2 suppresses growth and tumorigenicity of malignant human prostate carcinoma PC-3 cells. Prostate 61: 236–242 [DOI] [PubMed] [Google Scholar]
- Zhang H, Dessimoz J, Beyer TA, Krampert M, Williams LT, Werner S, Grose R (2004) Fibroblast growth factor receptor 1-IIIb is dispensable for skin morphogenesis and wound healing. Eur J Cell Biol 83: 3–11 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Toratani S, Sato JD, Kan M, McKeehan WL, Okamoto T (2001) Growth inhibition by keratinocyte growth factor receptor of human salivary adenocarcinoma cells through induction of differentiation and apoptosis. Proc Natl Acad Sci USA 98: 11336–11340 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends